

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-sensitive transcription factor which is responsible for most 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicities. Without ligand, the AhR complex is cytoplasmic and contains p23. Our objective was to investigate whether the wild type p23 levels are important for the AhR function. We generated eight p23-specific knockdown stable cell lines via either electroporation or lentiviral infection. Five of these stable cell lines were generated from a mouse hepatoma cell line (Hepa1c1c7) and three were from human hepatoma and cervical cell lines (Hep3B and HeLa). All of them expressed lower AhR protein levels, leading to reduced ligand-induced, DRE-driven downstream activity. The AhR protein levels in p23-specific knockdown stable cells were reversed back to wild type levels after exogenous p23 was introduced. Reduction of the AhR protein levels in these stable cells was caused by a decrease in the AhR message levels and an increase of the AhR protein degradation in the absence of ligand. This ligand-independent degradation of AhR was insensitive to MG132, suggesting that the 26S proteasome was not responsible for the degradation. In addition, MG132 could not protect AhR from the ligand-induced degradation in both mouse and human p23-knockdown stable cells.
Keywords: p23 co-chaperone, aryl hydrocarbon receptor, AhR, protein degradation, dioxin
1. Introduction
p23 was first discovered in the unactivated avian progesterone receptor complex [1] and the human homolog was subsequently cloned and characterized [2]. The p23 protein has been found in most mouse tissues, namely liver, kidney, lung, thymus, ovary, spleen, and stomach [3]. It is now known that p23 is present in many Hsp90 chaperone complexes. Results from immunoadsorption studies showed that p23 is involved in the in vitro assembly of many active complexes containing progesterone receptor [4, 5], glucocorticoid receptor [6], and telomerase [7]. Interestingly, p23 plays a role in disassembling some nuclear receptor/enhancer complexes [8] and synthesizing prostaglandin E2 [9].
The aryl hydrocarbon receptor (AhR) is a xenobiotic-sensitive transcription factor which is responsible for most dioxin toxicities [10]. The unliganded AhR resides in the cytoplasm as a complex containing a dimer of Hsp90, one molecule of p23, and one molecule of XAP2 (aka ARA9 and AIP) [11–13]. Once the liganded AhR complex is in the nucleus, the AhR nuclear translocator (Arnt) causes the dissociation of the AhR complex and heterodimerizes with AhR [14]. Recently researchers have been interested in the role of AhR in immune response [15, 16] and cancer [17–20]. With regard to the AhR signaling pathway, the ligand-dependent dissociation of Hsp90 from AhR requires p23, suggesting that p23 is important for the ligand responsiveness of AhR [14]. Binding of p23 with the AhR/Hsp90 complex is essential for the ligand-dependent nuclear import of AhR [21]. Interestingly, p23 appears to confer the ligand-dependent formation of the AhR/Arnt/DRE complex using Hepa-1 C4 cytosol and the rabbit reticulocyte lysate-expressed Arnt [14], consistent with the ligand responsive role of p23 for the receptor. In addition, formation of the stable complex of Hsp90 and p23 is essential for the XAP2-mediated cytoplasmic retention of AhR [22]. The involvement of p23 in the AhR signaling has also been implicated in a yeast system by deletion studies [23]; p23 appears to stabilize the AhR complex by inhibiting the Hsp90 ATPase activity in yeast [24]. Our gel shift data showed that p23 promotes the formation of the AhR/Arnt/DRE complex in vitro using baculovirus expressed human AhR and Arnt proteins [25]. Contrary to a body of evidence suggesting that p23 is involved in the AhR signaling pathway, p23 may not be necessary for the AhR function in vivo [26]. Here we provide evidence supporting that p23 controls the AhR levels in human and mouse cell lines and the wild type cellular levels of p23 are important for AhR function. In addition, our data have revealed a distinct mechanism for AhR degradation: this degradation likely involves an unidentified p23-controlled, labile protease which degrades AhR via a non-proteasome-mediated mechanism.
2. Material and methods
2.1. Reagents
3-methylcholanthrene, benzo[a]pyrene, actinomycin-D, and cell culture media were purchased from Sigma (St. Louis, MO). Cycloheximide was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). MG132 was purchased from Cayman Chemical (Ann Arbor, MI). Cell culture reagents, if not specified, were purchased from Invitrogen (Carlsbad, CA). All other chemicals, if not specified, were purchased from Sigma (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). Oligonucleotides were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from Tissue Culture Biologicals (Tulare, CA) or Thermo Scientific (Rockford, IL). Hepa1c1c7, Hep3B, HeLa, and the stable cells generated from these cell lines were grown in DMEM supplemented with 10% fetal bovine serum, 2mM GlutaMAX-I, 10 U/ml of penicllin, and 10 μg/ml of streptomycin. All cells were maintained at 37 °C and 5% CO2. Anti-AhR polyclonal goat IgG (N-19), anti-Arnt polyclonal rabbit IgG (H-172), anti-CYP1A1 monoclonal mouse IgG (B-4), anti-lamin A/C polyclonal goat IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-AhR SA210 polyclonal rabbit IgG was purchased from Enzo Life Sciences (Farmingdale, NY). Anti-p23 monoclonal mouse IgG MA3-414 was purchased from Affinity Bioreagents (Golden, CO). Anti-GAPDH rabbit polyclonal IgG G9545 was purchased from Sigma (St. Louis, MO). Anti-β-actin monoclonal mouse IgG was purchased from Ambion (Austin, TX). All secondary IgGs conjugated with IRDye 800CW or 680 were purchased from LI-COR Bioscience (Lincoln, NE). p23-specific SureSilencing shRNA plasmids (clones 1–4, NM_019766) were purchased from SA Biosciences (Frederick, MD). pLKO.1 p23-specific shRNA plasmid set (clones 1–5, NM_006601) were purchased from Thermo Scientific (Rockford, IL). psPAX, pMD2.g, and pLKO.1 shRNA scramble plasmids for lentivirus generation were purchased from Addgene (Cambridge, MA).
2.2. Generation of p23-specific knockdown Hepa1c1c7 stable cells via electroporation
SureSilencing shRNA plasmids (#1–4) were used to generate knockdown stable cells. Electroporation using a BTX ECM830 electroporator (Harvard Apparatus, Holliston, MA) was performed to introduce a shRNA plasmid (20 μg) into Hepa1c1c7 cells (4 × 106 cells) using the following setting: 400 μl, LV Mode, 180V, 70 ms, one pulse. After a 5-min incubation before and after electroporation, cells (5 μl) were diluted in fresh medium (1 ml) and seeded into individual wells of a 12-well plate. Drug selection using puromycin (Sigma, St. Louis, MO, 5 μg/ml) was initiated on the third day. Cells were fed with the complete media containing puromycin every three days and eventually reached confluence in a 75-cm2 flask after 6–8 weeks.
2.3. Generation of p23-specific knockdown Hep3B and HeLa stable cells using lentiviruses
Lentiviruses containing either a p23-specific or nonspecific shRNA were prepared as follows: AD-293 cells (Agilent Technologies, Santa Clara, CA, 4×105) in 1ml of growth media (10% fetal bovine serum, 2mM GlutaMAX-I, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin in DMEM) were seeded in individual wells of a 6-well plate. On the next day, cells were transfected with X-tremeGENE 9 (Roche Applied Sciences, Indianapolis, IN) containing various plasmids (1 μg of pLKO.1 shRNA or pLKO.1 p23-specific shRNA #1–5, 759 ng of psPAX2, and 250 ng of pMD2.G) at a 3:1 liposome:DNA ratio. Fresh media (3 ml) was exchanged 15 h after transfection. After 24 h, the media containing virus was collected and another 3 ml of fresh media was added. After another 24 h, the media was collected and combined with the previously collected media. The collected media was filtered through a 0.45 μm syringe filter and the filtered media was used for infection. Stable cells were then generated as follows: Hepa1c1c7, Hep3B or HeLa cells (3×105) in 1ml of growth media in individual wells of a 6-well plate were infected with 200 μl of the filtered media containing lentiviruses which expressed shRNA (p23 specific or nonspecific). After 48 h, the media was changed to 2 ml of fresh media containing 4 μg/ml of puromycin for stable cell selection. Cells were subsequently passed and reached 100% confluence in a 75cm2 flask in 3–4 weeks. p23 knockdown stable Hepa1c1c7 cells were generated using p23-specific shRNA #2 and #3; p23 knockdown stable Hep3B cells were generated using p23-specific shRNA #3 and 4; p23 knockdown stable HeLa cells were generated using p23-specific shRNA #3.
2.4. DRE-driven luciferase expression
Transient transfection was performed as described previously [27]. In brief, cells (5×104) were seeded in individual wells of a 24-well plate. After 24 h, a plasmid:FuGene HD complex (450 ng/0.9 μl) was added to each well. Eighteen hours after transfection, cells were exposed to 0.1% DMSO with or without 1 μM 3-methylcholanthrene for 6 h, followed by the luciferase assay using the Dual-light galacton-plus reagents (Invitrogen, Carlsbad, CA). The luciferase activities were normalized by internal β-galactosidase activities.
2.5. EROD assay
Hepa1c1c7, Hep3B or HeLa cells (2×105) were seeded in individual wells of a 12-well plate. On the next day, the medium was changed to 0.5 ml of fresh medium containing DMSO or 1 μM 3-methylcholanthrene. For HeLa cells, the medium also contained 10 mM sodium butyrate during induction since sodium butyrate restores the ligand-dependent induction of the CYP1A1 message levels in HeLa cells which express higher levels of AhRR [28]. After 6 h for Hepa1c1c7 and Hep3B cells or 16 h for HeLa cells, the cells were washed once with 0.5 ml of fresh media, followed by incubation with 0.5 ml of fresh media containing 5 μM resorufin ethyl ether (Sigma, St. Louis, MO) and 10 μM dicumarol (Sigma, St. Louis, MO). After 30 min, 200 μl of the media was transferred from each well into a 96-well plate and the fluorescence was measured using a Berthold Tri-Star LB941 plate reader (excitation at 544nm and emission at 590nm). Cell lysis was performed by adding 150 μl of lysis buffer (100mM potassium phosphate, pH 7.8, 1% Triton X-100) into each well, followed by platform rotation for 10 min at 4°C. Following centrifugation at 16,000g for about 10 min at 4 °C, the resulting supernatants were obtained and used for protein content determination by BCA assay (Thermo Scientific, Rockford, IL). The fluorescence values were normalized by the corresponding protein concentrations.
2.6. Quantitative Western analysis
Nitrocellulose membranes (0.22 μm, LI-COR Bioscience, Lincoln, NE) with transferred proteins were blocked in 5% BSA (Santa Cruz Biotechnology, Santa Cruz, CA) in PBS containing 0.1% Tween-20 for 1 h, followed by the primary antibody incubation in the same blocking buffer overnight. Dilutions for antibodies were as follows: 1:200 for AhR (N-19), p23, and Arnt; 1:2,000 for AhR (SA210); 1:100 for lamin A/C and CYP1A1; 1:10,000 for β-actin and GAPDH. After (primary and secondary) antibody incubation, membranes were washed for 5 min four times with PBS containing 0.1% Tween-20. Incubation of the donkey secondary antibody conjugated with IRDye (1:10,000) was performed in PBS buffer containing 5% BSA and 0.1% Tween-20 for 1 h. Anti-goat IgG and anti-rabbit IgG conjugated with IRDye 800CW were used to detect AhR and Arnt; anti-mouse IgG conjugated with IRDye 680 was used to detect p23, CYP1A1, and β-actin; anti-rabbit IgG and anti-goat IgG conjugated with IRDye 680 were used to detect GAPDH and lamin A/C. Membranes were scanned and quantified using an Odyssey infrared imaging system (LI-COR Bioscience, Lincoln, NE).
2.7. Preparation of whole cell extract
Lysis buffer (25 mM HEPES, pH 7.4, 0.4 M KCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 1 mM PMSF, and 2 μg/ml of leupeptin) was used to harvest cells from a 75 cm2 flask (300 μl) or wells from a 6-well plate (50 μl/well). After three cycles of freeze/thaw, lysates were kept on ice for 30 min and then centrifuged at 14,000g for 10 min at 4 °C. The supernatants were defined as whole cell extract.
2.8. Preparation of nuclear and cytoplasmic extracts
Cells were grown in a 75 cm2 flask and treated with 3-methylcholanthrene (1 μM) or DMSO for 1 h before nuclear and cytoplasmic extraction as follows: cells were resuspended into 200 μl of the resuspension buffer (25 mM HEPES, pH 7.4, 5 mM KCl, 0.5 mM MgCl2, 1 mM DTT, 0.5% NP-40, 1 mM PMSF, and 2 μg/ml of leupeptin), followed by tumbling rotation at 4 °C for 15 min. After centrifugation at 3,000g for 2 min at 4 °C, the supernatant was defined as the cytoplasmic extract. The pellet was then resuspended into 100 μl of the nuclear extraction buffer (25 mM HEPES, pH 7.4, 350 mM NaCl, 10% sucrose, 0.05% NP-40, 1 mM DTT, 1 mM PMSF, and 2 μg/ml of leupeptin), followed by tumbling rotation at 4 °C for 1 h. After centrifugation at 14,000g for 10 min at 4 °C, the supernatant was defined as the nuclear extract. All extracts were stored at −80 °C and their protein contents were determined by the BCA assay.
2.9. Real-time qPCR
To quantify the AhR message, we performed real-time qPCR using primers corresponding to the mouse AhR. Primer sequences were obtained from the Primer Bank [29] (AhR: forward, GCCCTTCCCGCAAGATGTTAT and reverse, CAGGGGTGGACTTTAATGCAA). RNA extraction and cDNA synthesis were performed as described previously [30]. Real-time qPCR was performed using the Bio-Rad SYBR green supermix (Hercules, CA) on a Bio-Rad iCycler. PCR conditions (40 cycles) were as follows: 90 °C for 10 sec and 60 °C (annealing and extension) for 1 min. SYBR Green fluorescence readings were taken at 60 °C when the fluorescence intensity corresponded solely to the PCR product of interest. Normalized fold increase of the endogenous transcript was determined by the 2−ΔΔCT method [31] using β-actin (forward, CCACACTGTGCCCATCTAGG and reverse, AGGATCTTCATGAGGTAGTCAGTCAG) as the internal standard.
2.10. Gel shift assay
Cells were treated with 1 μM 3-methylcholanthrene or DMSO for 1 h before harvest. The protocol for preparing nuclear extracts for the gel shift assay was different from Section 2.8 because this protocol was optimized for the native AhR gel shift complex formation. Nuclear extracts were prepared as follows: cells from a 90% confluent 75 cm2 flask were resuspended into 200 μl of the lysis buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, 10% glycerol, 1 mM PMSF, and 2 μg/ml of leupeptin). After 3 cycles of freeze/thaw, the lysates were centrifuged at 16,000g for 10 min at 4 °C. The pellets were resuspended into 100 μl of lysis buffer containing 0.4 M KCl. After on ice for 30 min, the suspensions were centrifuged at 14,000g for 10 min at 4 °C. Supernatants were immediately diluted with lysis buffer to 0.1 M final concentration of KCl and were defined as nuclear extracts for gel shift assay. For gel shift assays, nuclear extracts were normalized by either protein content (5 μg) or the amount of AhR. Samples (adjusted with lysis buffer containing 0.1 M KCl to a final volume of 13 μl) were incubated with poly-dIdC (EMD Millipore Chemicals, Darmstadt, Germany, 3 μg in 1 μl) for 10 min at room temperature. After that, the annealed DRE conjugated with IRDye 680 (0.5 pmol in 2 μl) was added (OL439, IRD680-TCGAGTAGATCACGCAATGGGCCCAGC; OL440, IRD680-TCGAGCTGGGCCCATTGCGTGATCTAC) (Integrated DNA Technologies, San Diego, CA). After a 10-min incubation at room temperature, 10X orange loading dye (LI-COR, Lincoln, NE) was added to each sample, followed by electrophoresis on a native 5% acrylamide TBE gel at 185 volts for 2 h at 4 °C. The gel, which was still in the glass plates, was analyzed directly using a LI-COR Odyssey infrared imaging system.
2.11. Chromatin immunoprecipitation assay
Cells (80–90% confluent on a 60 mm plate) were treated with either 1 μM 3-methylcholanthrene or DMSO in FBS-free DMEM media for 1 h at 37 °C. After that, cells were cross-linked with 1% formaldehyde in FBS-free DMEM for 10 min at room temperature. Glycine (125 mM final concentration) was added to quench the reaction. After washing the cells three times with cold PBS, the cell pellet was resuspended in the lysis buffer (150 μl, 50 mM Tris, pH 8.1, 10 mM EDTA, and 1% SDS), followed by sonication in a circulating ice water bath using a Misonix S4000 sonicator equipped with a cuphorn (40% output, 10 sec on/15 sec off for 10 times). The sonicated cell lysate was centrifuged at 16,000g for 10 min at 4 °C. The resulting supernatant was diluted ten times into the dilution buffer (20 mM Tris, pH 8.1, 2 mM EDTA, 1% Triton X-100, and 150 mM NaCl), ready as the sample for chromatin co-immunoprecipitation assay. A sample aliquot (75 μl) was set aside as the input. ChIP assay was performed according to published protocol [32] with modification: each sample (1.5 ml) was pre-cleared with Dynabeads Protein G (Invitrogen, Carlsbad, CA, 3 μl) containing sheared salmon sperm DNA (Invitrogen, Carlsbad, CA, 1 μg) and goat serum (Invitrogen, Carlsbad, CA, 2.5 μl) by rotating for 2 h at 4 °C. The pre-cleared supernatant was transferred to a tube containing Dynabeads Protein G (10 μl) pre-incubated with anti-AhR IgG (1.5 μg, SA210) for 1 h at 4 °C. After an overnight incubation at 4 °C, the pellet was obtained using a DynaMag-2 magnet (Invitrogen, Carlsbad, CA) and then washed sequentially with buffer I (dilution buffer plus 0.1% SDS), buffer II (Buffer I except 500 mM NaCl), buffer III (10 mM Tris, pH 8.1, 1 mM EDTA, 250 mM LiCl, 1% NP40, and 1% deoxycholate), and finally two times with 10 mM Tris, pH 8, 1 mM EDTA. Each wash involved a 10-min incubation at 4 °C. The washed pellet was resuspended into the elution buffer (75 μl, 0.1 M NaHCO3 and 1% SDS). The resuspended sample (and also the input) was heated at 65 °C for 7 h to reverse cross-linking. The resulting sample was treated with RNase A (Invitrogen, Carlsbad, CA, 0.75 μl of 10 mg/ml) for 30 min at 37 °C, followed by the protease treatment (0.225 μl of 50 mg/ml of proteinase K (Epicentre, Madison, WI), 0.975 μl of 0.5 M EDTA, and 1.95 μl of 1 M Tris, pH 6.5) for 2 h at 45 °C. DNA was then purified using a Promega Wizard SV gel clean-up kit. PCR was performed using an DRE-specific primer set (forward primer: OL525, CACGCGAGACAGCAGGAG; reverse primer: OL526, TTGTGAGTTGGGTAGCTGGG) and a Perkin Elmer GeneAmp 2400 cycler with the following protocol: 94 °C for 2 min, followed by 30 cycles of 94 °C for 30 sec, 56 °C for 30 sec, and 72 °C for 30 sec. PCR products were visualized and quantified using a LI-COR Odyssey infrared imaging system after staining the DNA with SYTO 60 (Invitrogen, Carlsbad, CA).
2.12. Preparation of microsomes
To 90% confluent cells on a 10 cm plate, 7.5 ml of 0.1M sodium phosphate buffer, pH 7.4, was used to resuspend the cells. The resuspended cells were sonicated for 30 sec twice, with a 30 sec pause in between. The sonicated cells were centrifuged at 10,000g for 20 min 4 °C. Supernatants were then centrifuged at 100,000g for 1 h at 4 °C. The pellets were resuspended into 250 μl of cold microsome buffer (10 mM Tris, pH 7.4, 150 mM KCl, and 20% glycerol) to become microsomes.
2.13. Statistical analysis
We performed one-way (Fig. 1A–C, 5C, 6A left, 6D, and 7A) and two-way (Fig. 2A–C, 3D–E, 4B, 5A, B, E, and S3 in supplementary material) ANOVA followed by post hoc Bonferroni’s multiple comparison test to determine the statistical significance. The slopes (Fig. 6A–B) were analyzed using ANCOVA. All statistics were performed using the GraphPad Prism 5 software. Statistical significance is indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001; ***p < 0.0001; †p > 0.05 (not significant). The n values indicate the numbers of replicates in a particular experiment.
Fig. 1.
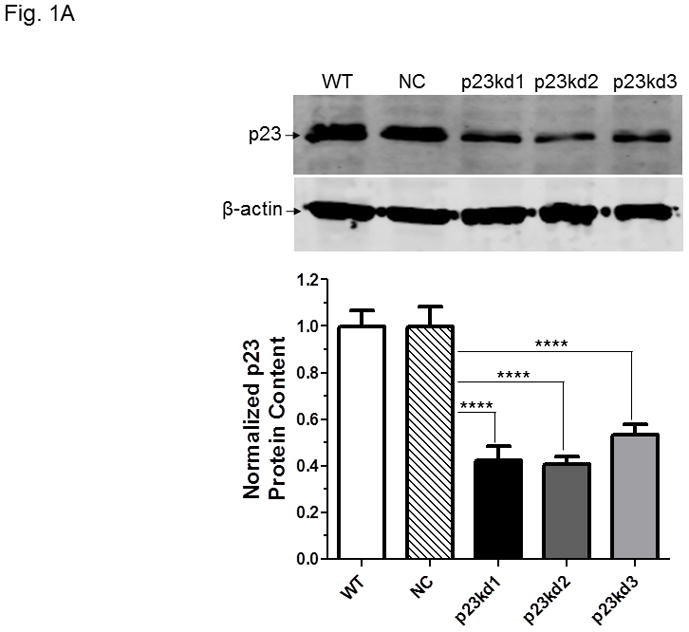
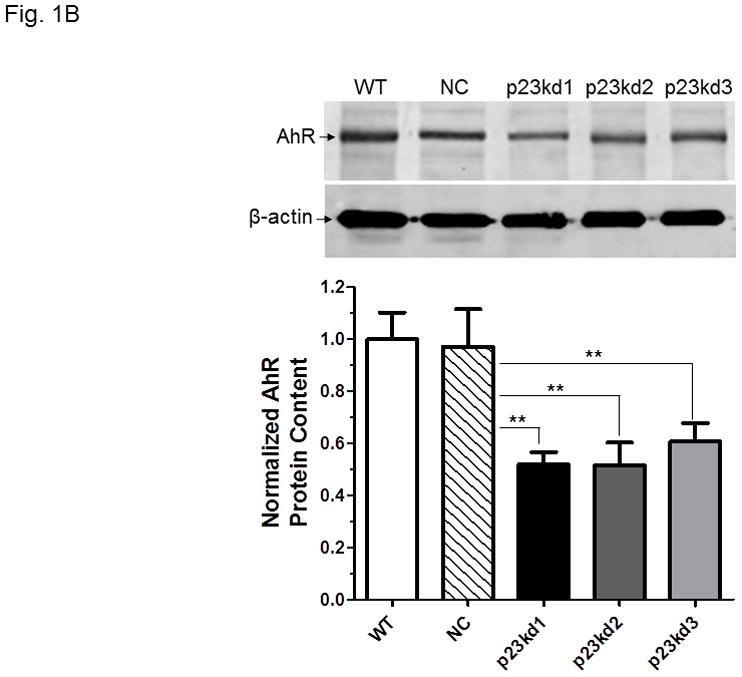
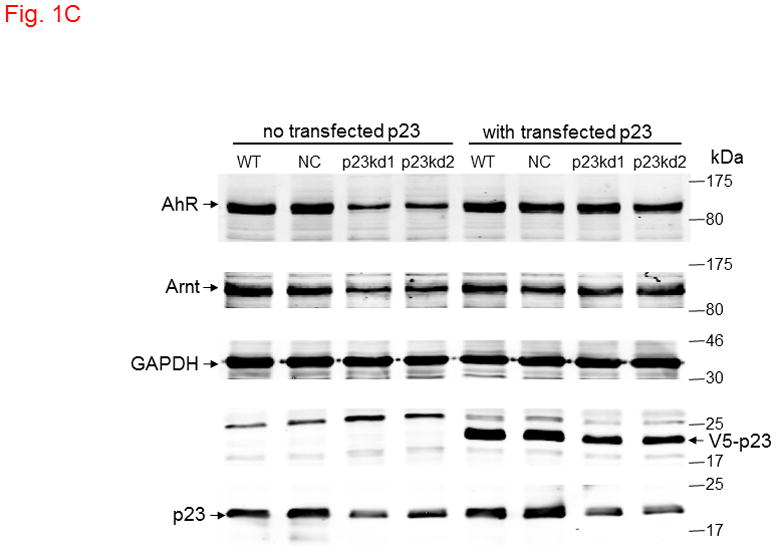
p23 and AhR protein levels in p23-specific knockdown stable Hepa1c1c7 cells generated by electroporation. Five cell lines examined: wild type (WT), negative control knockdown stable (NC), and p23 knockdown stable p23kd1-3. Western results showing p23 (A) and AhR (B) levels. Protein levels in WT are arbitrarily set as 1. Each lane contained 19–27 μg of whole cell extract and was normalized by β-actin. N-19 was used to detect AhR. The plot (bottom) is the quantified data showing the means with error bars (mean ± SD, n = 3). The images (above) are a representation of the triplicate data. C, Western results showing that V5-p23 restored the AhR levels in p23kd1-2 cells. Cells (3×106 in 0.4 ml) were transfected with either pcDNA-V5 or pcDNA-V5-p23 plasmid (30 μg in 30 μl) by electroporation (190 V, 70 ms). After incubation at 37°C for 60 h, the cells were harvested and whole cell extract was prepared for Western analysis. Each lane contained 20 μg of whole cell extract. GAPDH was used for normalization. SA210 was used to detect AhR.
Fig. 5.
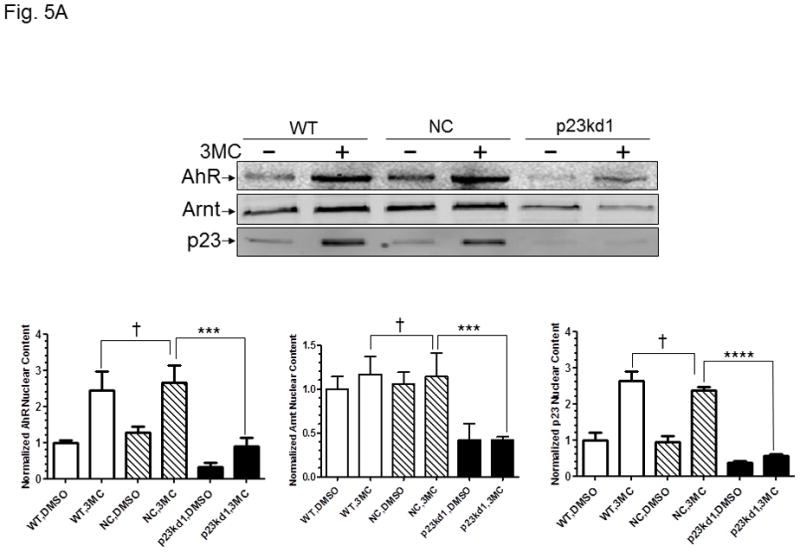
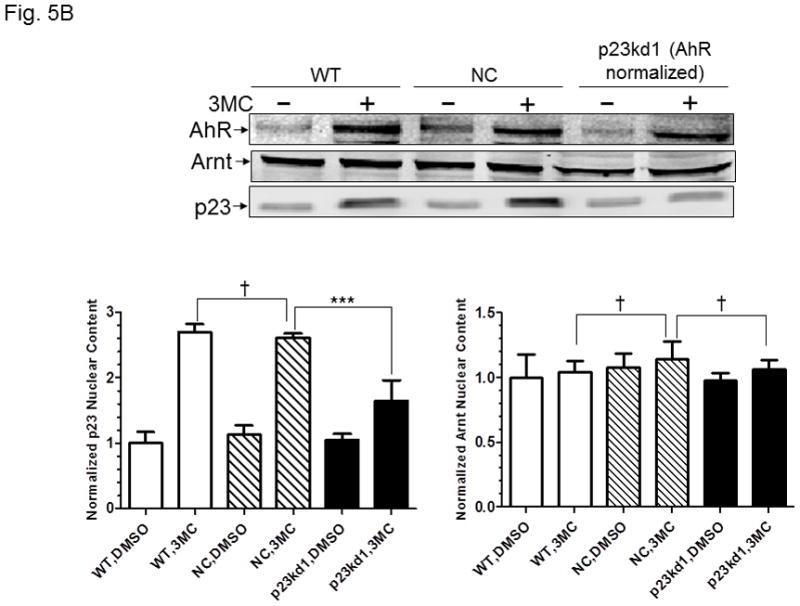
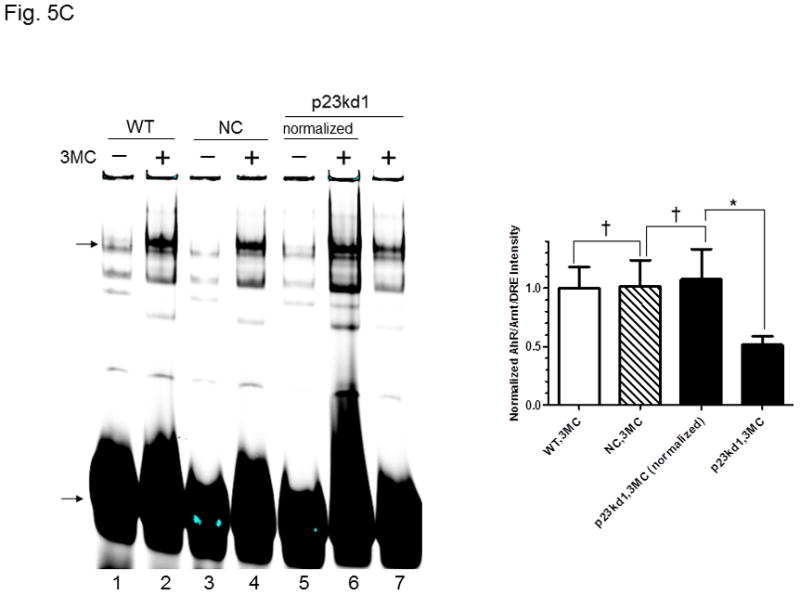
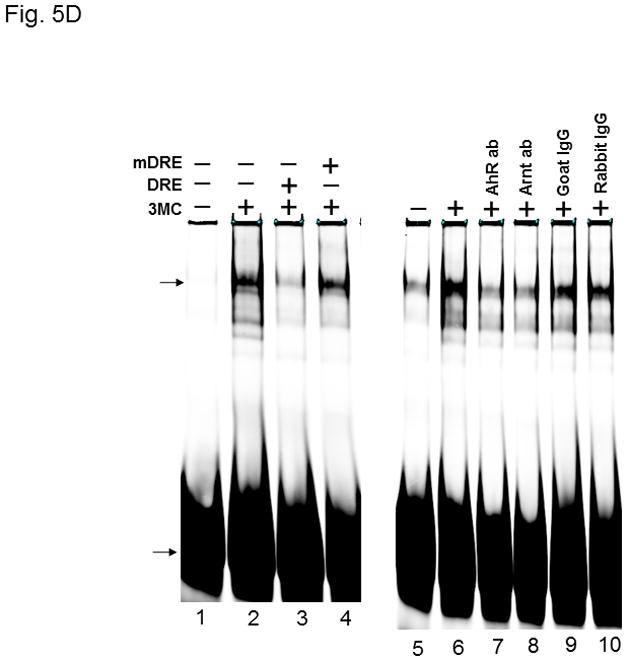
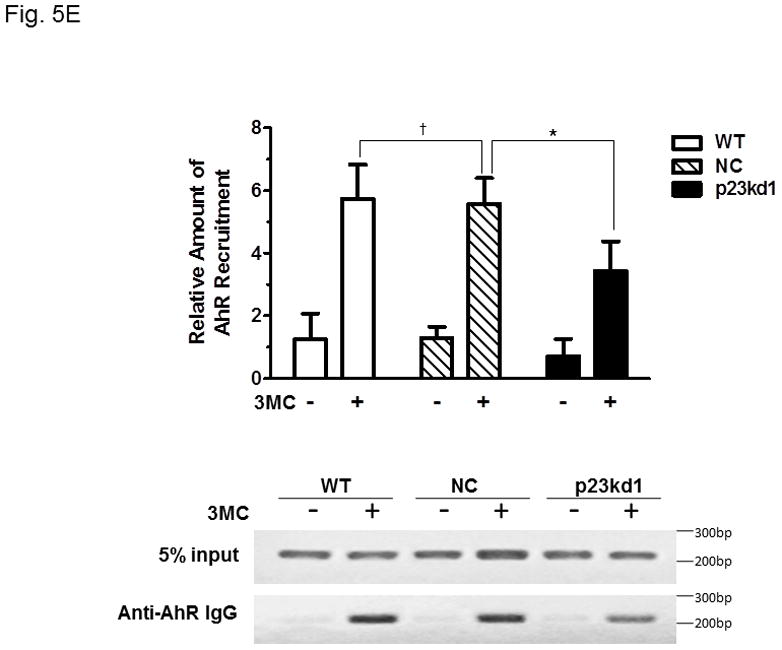
AhR/Arnt/DRE complex formation in p23kd1 Hepa1c1c7 nuclear extracts. A, Western results showing the amount of AhR, Arnt, and p23 in gel shift nuclear extracts with or without 3-methylcholanthrene (3MC) treatment (top). Each lane contained 20 μg of protein. The plot below shows the AhR (left), Arnt (middle) and p23 (right) levels with error bars (mean ± SD, n = 3). The images above are a presentation of the triplicate data. B, Western results showing the levels of Arnt and p23 in gel shift nuclear extracts when the AhR levels after 3-methylcholanthrene (3MC) treatment were normalized among the WT, NC, p23kd1 gel shift nuclear extract. More protein was loaded for p23kd1 nuclear extracts (+/− 3-methylcholanthrene). The plots below show the p23 (left) and Arnt (right) levels with error bars (mean ± SD, n = 3). The images above are a presentation of the triplicate data. N-19 was used to detect AhR in A and B. C, gel shift results of nuclear extracts from cells either treated with 3-methylcholanthrene (3MC) or vehicle DMSO alone for 1 h before harvest. Lanes 2, 4, 6 contained normalized amount of AhR whereas lanes 1–4, 7 contained normalized amount of protein (5 μg). The upper arrow indicates the AhR/Arnt/DRE complex whereas the lower arrow indicates the free probe. The plot (right) shows the means with error bars (mean ± SD, n = 3). The gel shift image (left) is a presentation of the triplicate data. D, controls to validate the AhR/Arnt/DRE complex. Each lane contained 5 μg of protein. Lanes 1–4 and 5–10 were from two separate gels. Lanes 2–4, 6–10 were 3-methylcholanthrene (3MC)-treated WT gel shift nuclear extract whereas lanes 1 and 5 were DMSO-treated WT extract. DRE, 5 pmol (10X) of unlabelled DRE; mDRE, 5pmol (10X) of unlabelled mutated DRE. 6 μg (3 μl) of IgG was added to samples at the end for an additional 20 m at room temperature (lanes 7–10): AhR ab, anti-AhR goat IgG (N-19); Arnt ab, anti-Arnt rabbit IgG (H172); goat IgG or rabbit IgG was negative controls. E. ChIP data showing the 3-methylcholanthrene (3MC)-mediated AhR recruitment at the cyp1a1 promoter of WT, NC, and p23kd1 cells. The images below are a representative of agarose gel images showing the amplified DRE region of the cyp1a1 promoter (214bp). 5% input represents the PCR product using 5% of the starting lysate as the template. This experiment was repeated three times with similar results. The error bars represent mean ± SD, n = 3.
Fig. 6.
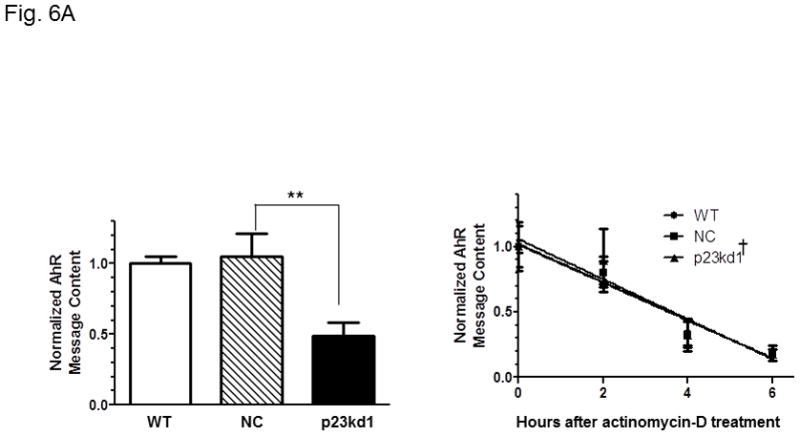
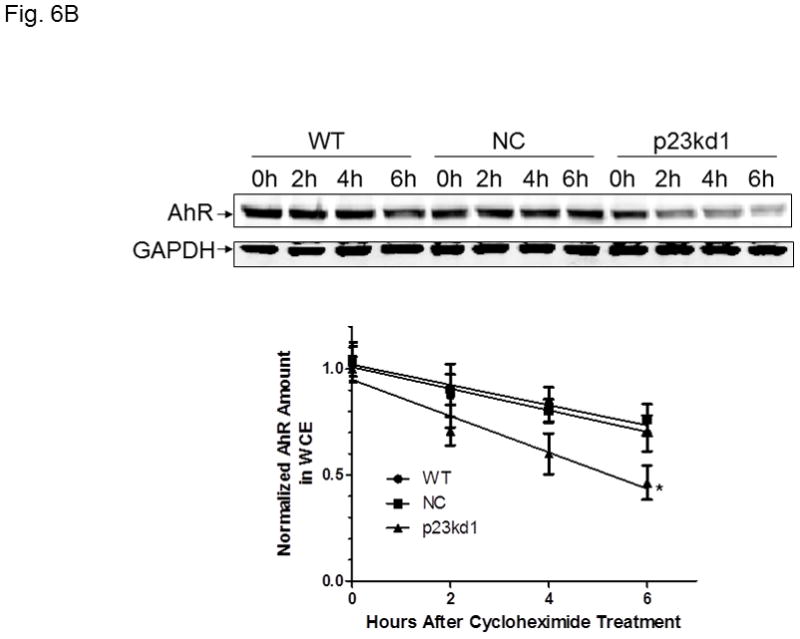
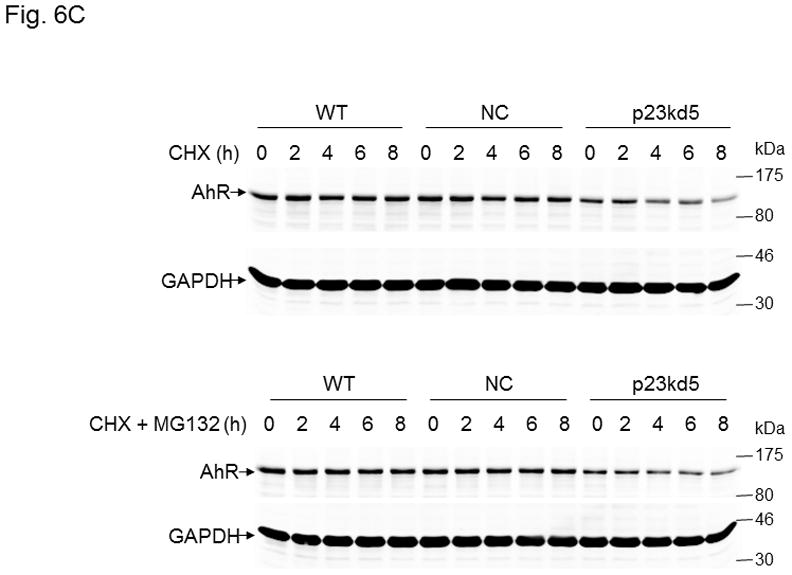
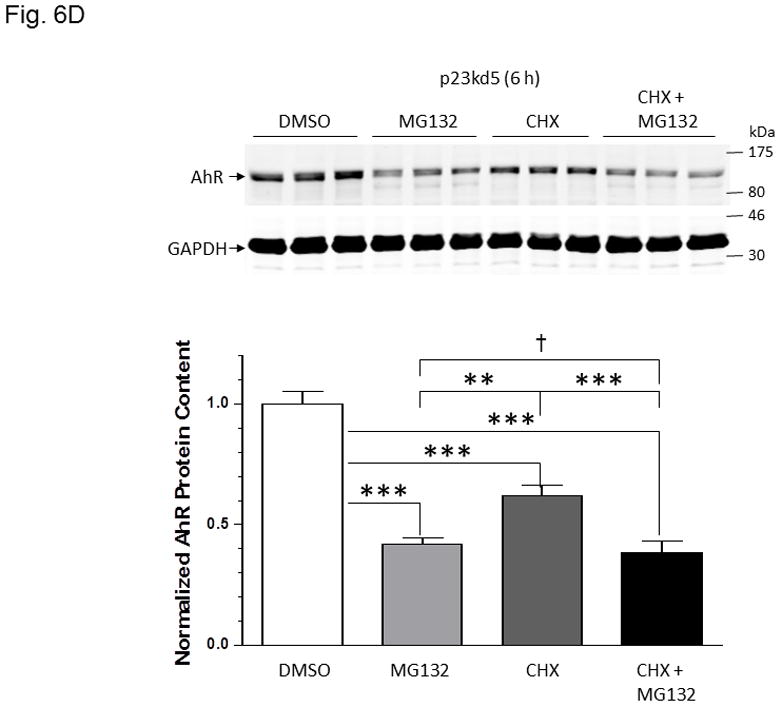
Effect of p23 on AhR transcription and protein stability in p23kd1 and p23kd5 cells. WT, wild type Hepa1c1c7; NC, negative control knockdown stable Hepa1c1c7; p23kd1 and p23kd5, p23-specific knockdown stable Hepa1c1c7. GAPDH was used for Western normalization. A. real-time qPCR results showing the amount of the AhR message after actinomycin-D treatment. Cells (5×105) were treated with actinomycin-D (5 μg/ml) for 0–6 h. The β-actin message was used for normalization. The graph (left) shows the amount of AhR message right after cycloheximide treatment. The time-dependent amount of AhR message after cycloheximide treatment was plotted with zero time point arbitrarily set as one to focus on the degradation rate. The plots show the means with error bars (mean ± SD, n = 3). This experiment was repeated once with similar results. The slopes of the figure (right) are not statistically different. B, Western results showing the amount of AhR after cycloheximide treatment. Cells (5×105) were treated with cycloheximide (50 μg/ml) for 0–6 h one day after seeding and then whole cell extract was obtained. Each lane contained 50 μg of protein. The amount at zero time point was arbitrarily set as one. The plot below shows the means with error bars (mean ± SD, n = 3). The images above are a presentation of the triplicate data. This experiment was repeated once with similar results. The slope of p23kd1 is significantly different. C, same as B except that p23kd5 was used instead of p23kd1. Treatment with cycloheximide (CHX) (50 μg/ml) alone (top) or cyclohexmide (50 μg/ml) and 10 μM MG132 (bottom) was up to 8 h. D, p23kd5 cells were treated for 6 h with either DMSO, 10 μM MG132, cyclohexmide (CHX) (50 μg/ml), or both. This Western was repeated two times and plotted (mean ± SD, n = 3). SA210 was used to detect AhR in B–D.
Fig. 7.
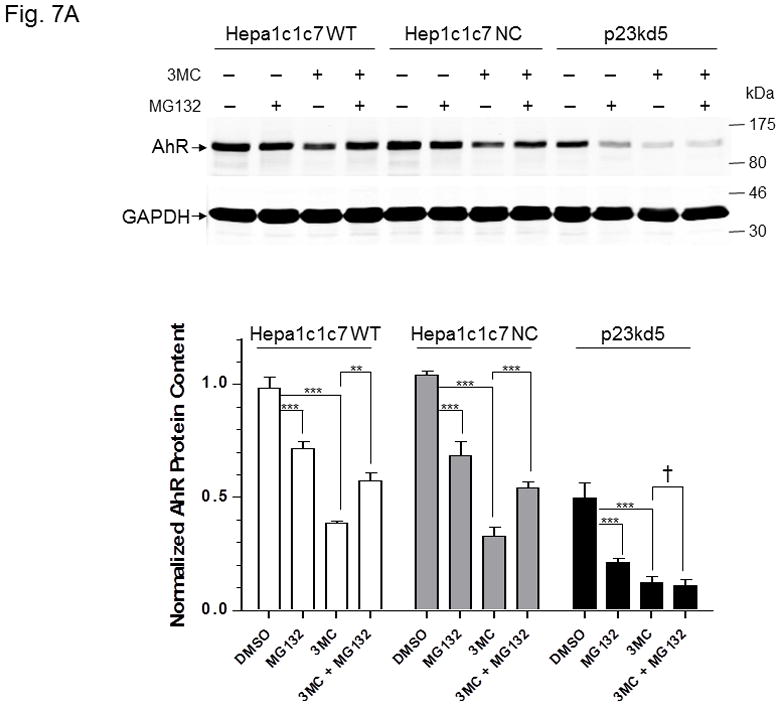
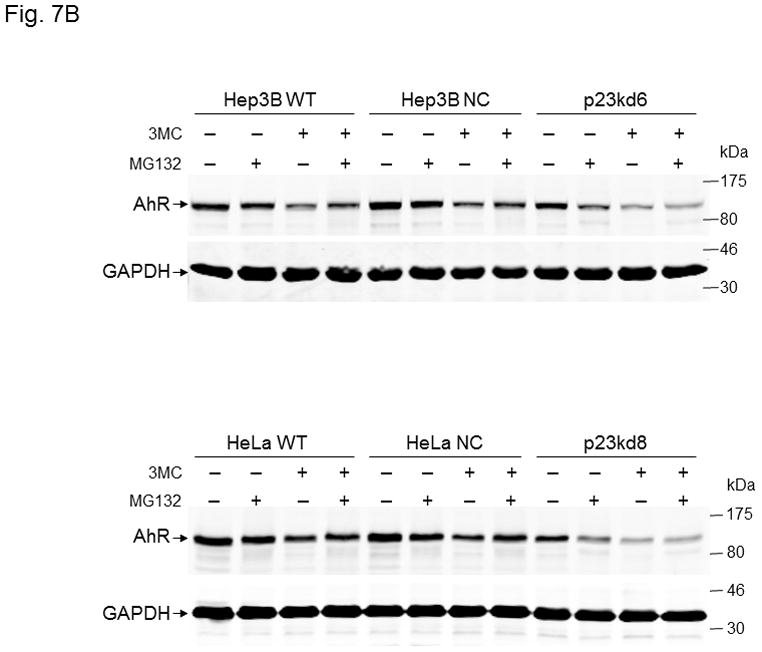
MG132 effect on 3-methylcholanthrene-induced AhR degradation in p23-specific knockdown stable Hepa1c1c7, Hep3B, and HeLa cells. SA210 was used to detect AhR. Wild type (WT), negative control knockdown stable (NC), and p23-specific knockdown stable p23kd5 Hepa1c1c7 (A), p23kd6 Hep3B (B, top) p23kd8 HeLa (B, bottom) were used. Cells were treated for 6 h with either DMSO, 1 μM 3-methylcholanthrene (3MC), 10 μM MG132, or both. Each lane contained 20 μg of whole cell extract. GAPDH was used for normalization. Western in A was repeated twice and plotted (mean ± SD, n = 3).
Fig. 2.
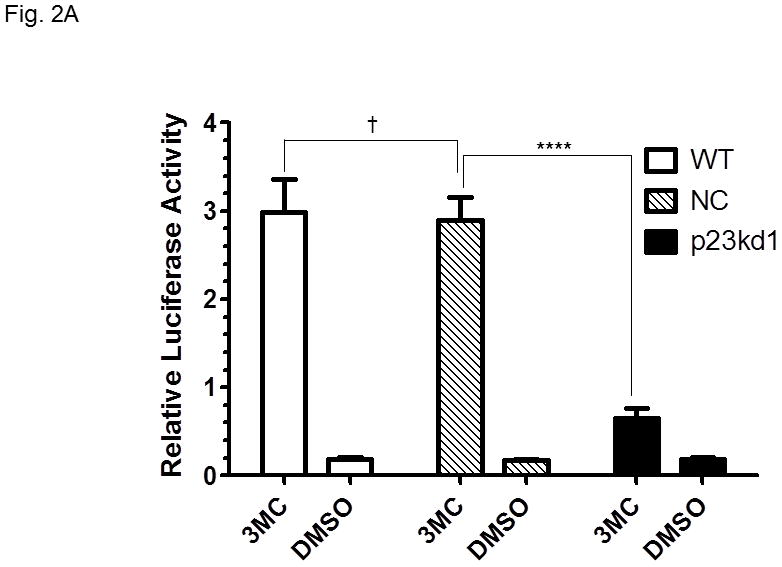
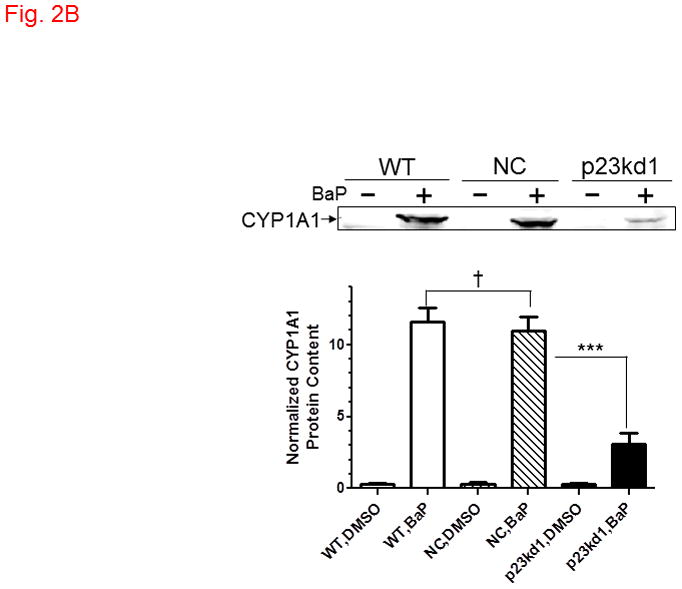
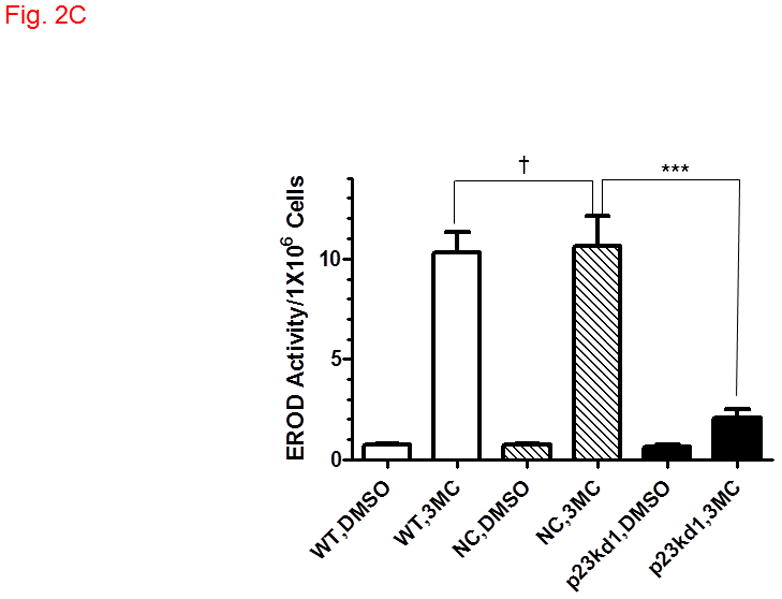
AhR downstream gene activation in p23kd1 cells. WT, wild type Hepa1c1c7; NC, negative control knockdown stable Hepa1c1c7; p23kd1, p23-specific knockdown stable Hepa1c1c7. A, DRE-driven luciferase activity in the presence of 1 μM 3-methylcholanthrene (3MC) or vehicle DMSO for 6 h. Cells were transfected with pGudLuc1.1 (400 ng) and β-galactosidase (50 ng) plasmids. The plot represents mean ± SD (n = 3). Luciferase activities were normalized by corresponding β-galactosidase activities. This experiment was repeated three times with similar results. B, CYP1A1 induction by 10 μM benzo[a]pyrene (BaP) for 12 h. Each lane contained 100 μg of microsomes for Western analysis. The plot below shows the means with error bars (mean ± SD, n = 3). The images above are a representation of the triplicate data. This experiment was repeated two times with similar results. C, a plot showing the EROD activity of CYP1A1. Induction was performed in a rotating cell suspension. The plot represents the means with error bars (mean ± SD, n = 3). Normalized EROD activity represents fluorescence units obtained from 1×106 WT, NC, or p23kd1 cells with or without 3-methylcholanthrene (3MC) treatment. This experiment was also performed in a modified monolayer microplate format two times with similar results.
Fig. 3.
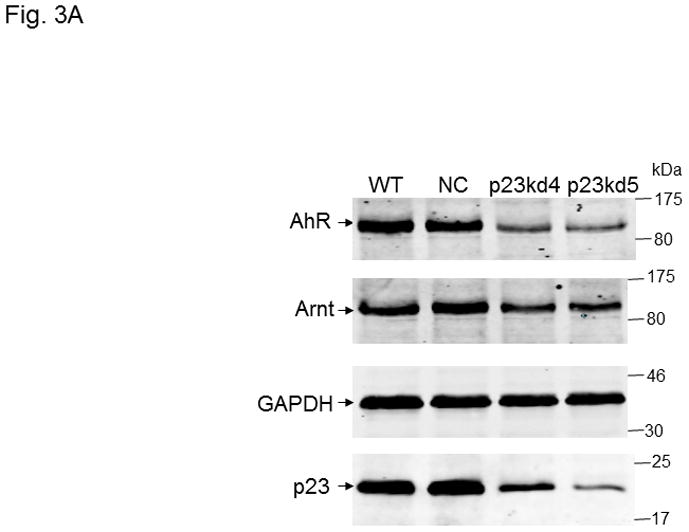
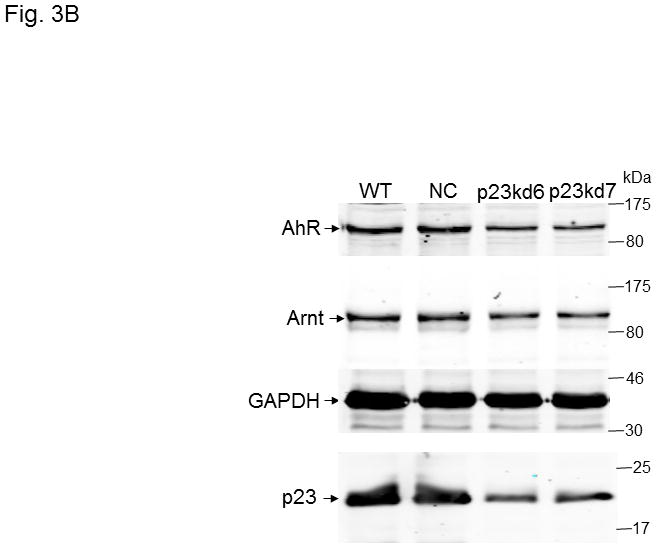
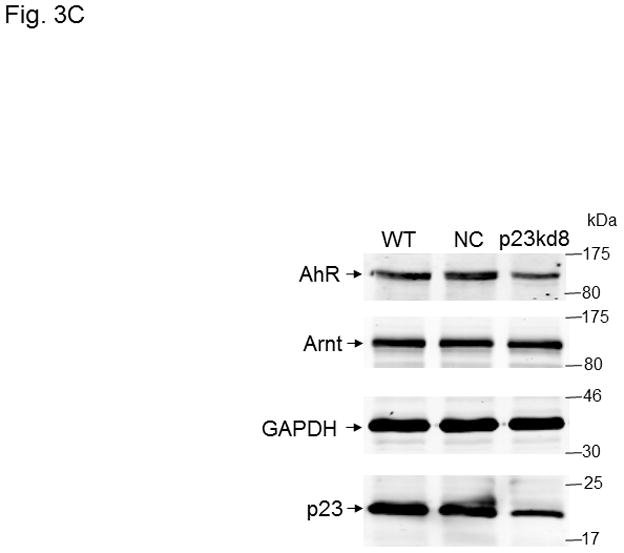
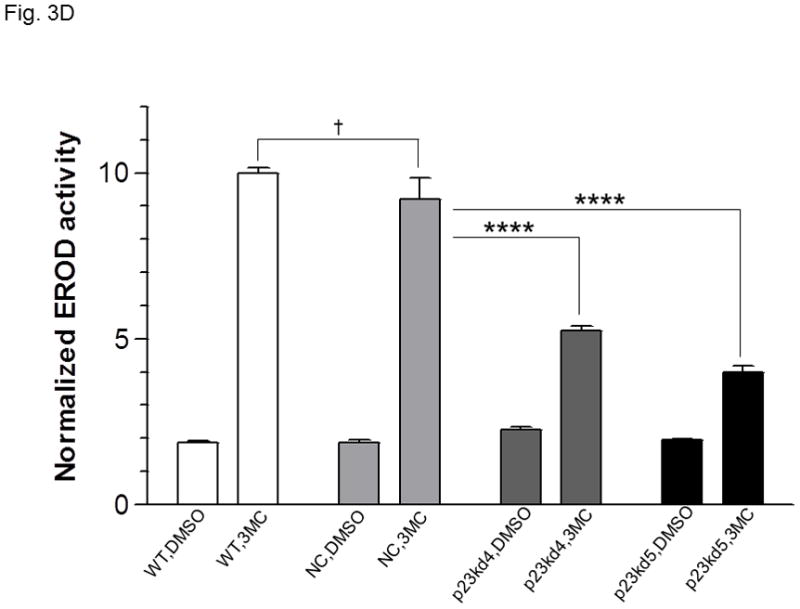
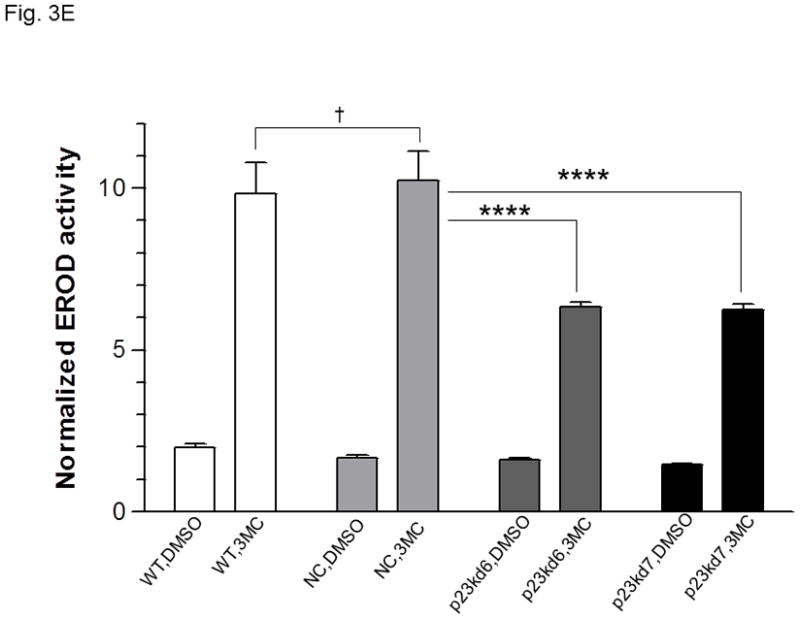
p23, AhR, and Arnt protein levels in p23-specific knockdown stable Hepa1c1c7 cells generated by lentiviral infection. Western results showing AhR, Arnt, and p23 levels in whole cell extract. Each lane contained 20 μg of whole cell extract and was normalized by GAPDH. A, wild type (WT), negative control knockdown stable (NC), and p23 knockdown stable p23kd4/5 Hepa1c1c7 cells. B, wild type (WT), negative control knockdown stable (NC), and p23 knockdown stable p23kd6/7 Hep3B cells. C, wild type (WT), negative control knockdown stable (NC), and p23 knockdown stable p23kd8 HeLa cells. SA210 was used to detect AhR. D and E, EROD activities of the lentivirus generated p23-specific knockdown stable cell lines Hepa1c1c7 (D, n = 3) and Hep3B (E, n = 4) with their corresponding wild type (WT) and negative control (NC) +/− 3-methylcholanthrene (3MC) treatment. The error bars represent means ± SD.
Fig. 4.
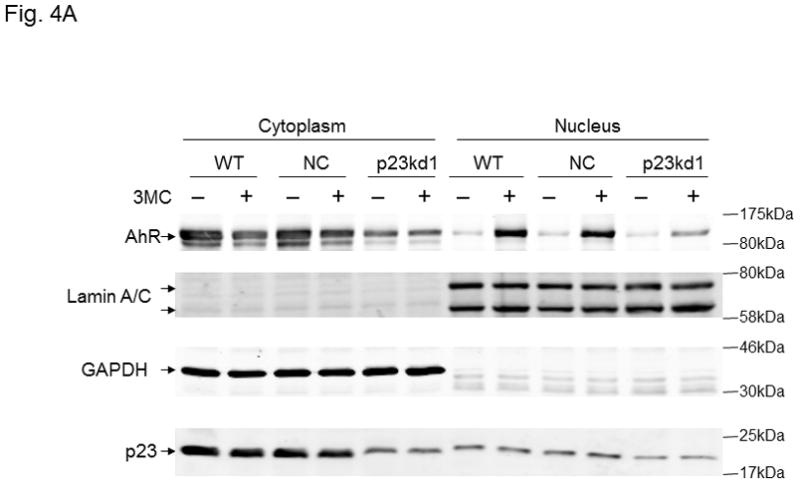
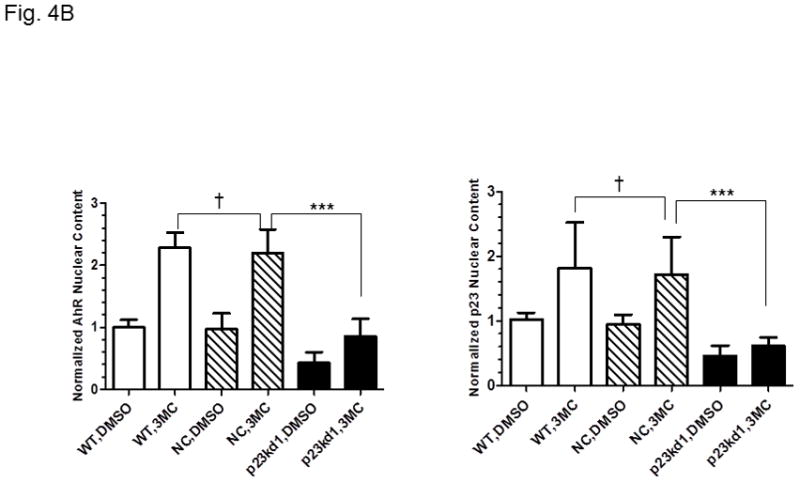
Cytoplasmic and nuclear levels of p23 and AhR in p23kd1 cells. WT, wild type Hepa1c1c7; NC, negative control knockdown stable Hepa1c1c7; p23kd1, p23-specific knockdown stable Hepa1c1c7. Cells were treated with 1 μM 3-methylcholanthrene (3MC) or vehicle DMSO alone for 1 h before fractionation. Each lane contained 20 μg of protein. GADPH and lamin A/C were marker controls for normalization for cytoplasmic and nuclear extracts, respectively. The Western images (A) are a representation of the replicate data. SA210 was used to detect AhR. Plots (B) showing the means with error bars (mean ± SD, n = 3 for left panel and n = 5 for right panel).
3. Results
3.1. p23 knockdown stable Hepa1c1c7 cells generated by electroporation contained 50% or less of the wild type p23 levels
We used five p23-specific shRNAs targeting the mouse and human p23 gene (NM_019766, NM_006601) to generate the p23 knockdown stable cell lines (Table I). Among the four SureSilencing shRNA plasmids used in electroporation to generate stable cell lines, shRNA #3 and #4 gave significant p23 knockdown at the protein level. We generated three clones of p23 knockdown stable cells from three separate transfection experiments: p23kd1 and p23kd2 cells were generated using shRNA #3 whereas shRNA #4 was used to generate p23kd3 cells. All three knockdown stable cells contained 40–50% of the wild type (WT) p23 protein levels (Fig. 1A). A heterogeneous clone of the negative control knockdown stable (NC) Hepa1c1c7 cells was generated using the control shRNA plasmid to see whether the stable cell selection procedure would affect the p23 levels. We observed that the amount of p23 between the WT and NC cells was essentially identical (Fig. 1A), suggesting that the reduced amount of p23 observed in p23 knockdown stable cells was caused by the p23-specific, shRNA-mediated mechanism.
Table I.
A summary of p23-specific knockdown Hepa1c1c7, Hep3B, and HeLa stable cell lines p23kd1-8. The shRNA plasmids used to generate the stable cell lines and the shRNA sequences are included. The percentages indicate the amount of p23 present in each stable cell line.
| shRNA plasmid | shRNA sequences | Hepa1c1c7 | Hep3B | HeLa |
|---|---|---|---|---|
| SureSilencing shRNA plasmid #3 | TAGTGGATGCCCTAACTTAGATG | p23kd1 (40–50%) p23kd2 (40–50%) |
||
| SureSilencing shRNA plasmid #4 | TTGCTTGAGAGTATGTACATTTT | p23kd3 (40–50%) | ||
| pLKO.1 p23-specific shRNA plasmid #2 | CCAGAAGTAGATGGAGCAGAT | p23kd4 (40–50%) | ||
| pLKO.1 p23-specific shRNA plasmid #3 | CCAAATGATTCCAAGCATAAA | p23kd5 (20–25%) | p23kd6 (30–40%) | p23kd8 (40–50%) |
| pLKO.1 p23-specific shRNA plasmid #4 | GAAGACAGTAAGGATGTTAAT | p23kd7 (40–50%) |
3.2. p23 knockdown stable Hepa1c1c7 cells contained less AhR and Arnt
We examined whether knockdown of p23 would alter the AhR and Arnt levels in Hepa1c1c7 cells. The total AhR protein levels in whole cell extract were reduced in p23kd1, p23kd2, and p23kd3 cells by about 40–50%, as compared to the controls (Fig. 1B). Similar percent of reduction was observed when we used GAPDH instead of β-actin as the marker for normalization (data not shown). Interestingly, the Arnt levels in whole cell extract were also suppressed in all three p23 knockdown stable Hepa1c1c7 cells by about 40–50% (Fig. S1 in supplementary material). To prove that the reduction of AhR and Arnt was caused by reduction of the p23 content, we transiently transfected a plasmid expressing the V5 fusion of p23 into two of our p23 knockdown stable cells and observed that the levels of AhR and Arnt returned to the levels in wild type and NC cells (Fig. 1C). Further increase of the p23 levels in V5-p23 transfected WT and NC Hepa1c1c7 cells, however, did not change the AhR and Arnt contents. Since p23kd1-3 cells were similar in p23 expression, most of the subsequent experiments were performed using p23kd1 cells to further examine how reduction of p23 would affect the AhR signaling.
3.3. 3-methylcholanthrene-dependent, DRE-driven luciferase expression was less in p23kd1 cells
Since the AhR and Arnt contents were reduced in p23 knockdown stable Hepa1c1c7 cells (p23kd1-3), we examined whether activation of gene transcription would be affected in p23kd1 cells. Results from our transient transfection studies showed that only 22% of the 3-methylcholanthrene-activated, DRE-driven luciferase activity of the WT and NC Hepa1c1c7 cells was observed in p23kd1 cells (Fig. 2A). We observed about 14-fold induction of the luciferase activity by 1 μM 3-methylcholanthrene in both the WT and NC cells whereas only 3-fold induction in p23kd1 cells. We also examined this transcriptional activity using another AhR ligand (β-napthoflavone) and found similar extent of reduction of the luciferase protein expression in p23kd1 cells (Fig. S2 in supplementary material), confirming that activation of the AhR-dependent gene transcription in p23kd1 cells was hampered. Expression of the luciferase protein was 12-, 10- and 3-fold induced by 10 μM β-napthoflavone, respectively, in WT, NC, and p23kd1 Hepa1c1c7 cells and the amount of the β-napthoflavone-activated luciferase protein expression in p23kd1 cells was only 12% of the amount expressed in the two control cells. We measured the amount of the luciferase protein by Western analysis rather than performing the luciferase assay because β-napthoflavone inhibits the luciferase activity (our unpublished observation, [33]).
3.4. Benzo[a]pyrene-mediated CYP1A1 induction was less in p23kd1 cells
Next, we examine whether endogenous target gene induction would be compromised in p23kd1 cells. We observed a 30-fold induction of the CYP1A1 protein production after treatment with 10 μM benzo[a]pyrene for 12 h in both WT and NC Hepa1c1c7 cells; however, only 10-fold induction was observed in p23kd1 cells (Fig. 2B). The amount of the induced CYP1A1 protein in p23kd1 cells was about 30% of the induced controls. When we measured the EROD activity corresponding to the CYP1A1 function in these cells, we observed that the EROD activity was 14-fold induced after treatment with 1 μM 3-methylcholanthrene for 6 h in both WT and NC Hepa1c1c7 cells whereas only about 3-fold increase was observed in p23kd1 cells (Fig. 2C). About 20% of the WT 3-methylcholanthrene-induced activity was observed in p23kd1 cells; this is consistent with the reduced amount of the CYP1A1 protein induced by another AhR ligand (benzo[a]pyrene) in p23kd1 cells.
3.5. Knockdown of p23 in Hep1c1c7, Hep3B, and HeLa cells using lentiviruses showed reduced AhR levels and EROD activity
In an effort to validate whether reduced p23 levels would lead to reduction of the AhR and Arnt cellular contents, we examined the effect of p23 knockdown in other cell lines. We were also concerned about the nonspecific effects caused by our electroporation method to generate stable cell lines. Thus, we used lentiviruses to generate p23 knockdown stable cell lines from Hepa1c1c7, Hep3B, and HeLa cells to address whether reduction of the cellular AhR and Arnt levels are indeed p23-specific. All the p23 knockdown stable cells generated from Hepa1c1c7, Hep3B, and HeLa cells via lentiviral infection showed reduced AhR levels (Table I, Fig 3A–C) and reduced EROD activity (Fig. 3D–E and S3 in supplementary material), strongly suggesting that wild type p23 levels is required to sustain the cellular levels of AhR. However, we were not able to detect any significant reduction of the Arnt levels in all the lentivirus-generated p23 knockdown stable cells. It appeared that reduction of the Arnt protein levels was only observed in Hepa1c1c7 stable cells generated by electroporation; therefore, the observed effect on Arnt might not be caused by compromised p23 levels.
3.6. p23kd1 cells contained less nuclear AhR after ligand activation
Next, we performed a subcellular fractionation experiment to address whether knockdown of p23 would reduce the nuclear AhR levels after ligand treatment. We used GAPDH and lamin A/C as marker proteins to ensure the integrity of the cytoplasmic and nuclear fractions. After treating the cells with 1 μM 3-methylcholanthrene for 1 h, we observed nuclear localization of AhR in all Hepa1c1c7 cells (p23kd1, NC, and WT) by Western analysis (Fig. 4A). Although the percent nuclear translocation of AhR in p23kd1 cells appeared to be comparable to the two controls, the total amount of the nuclear AhR in p23kd1 cells was about 40% of the controls (Fig. 4B, left). Realizing that the total AhR levels in p23kd1 cells was about 50% of the WT levels, it appeared that reduction of the nuclear AhR levels observed in p23kd1 cells was a direct result of the reduced amount of the total AhR in p23kd1 cells, whereas 3-methylcholanthrene activated a similar extent of the AhR nuclear entry in all three cell lines (p23kd1, NC, and WT). Although p23 did not appear to translocate into nucleus upon 3-methylcholanthrene treatment, reduction of the cellular p23 levels limited the amount of p23 in the nucleus (Fig. 4A and B, right).
3.7. Formation of the 3-methylcholanthrene-dependent AhR/Arnt/DRE gel shift complex was less in p23kd1 cells
The nuclear extraction protocol used to obtain nuclear extracts for gel shift assays was different from the subcellular fractionation experiment in order to optimize the native AhR complex formation. Nonetheless, the p23kd1 nuclear extract contained significantly less p23 (Fig. 5A), similar to what we observed in the subcellular fractionation studies (Fig. 4A). In addition, it contained significantly less AhR upon 3-methylcholanthrene treatment. The Arnt levels in the p23kd1 nuclear extract were also reduced to about 35% of the control levels. When we equalized the AhR levels of all 3-methylcholanthrene-treated nuclear extracts, we observed that all the Arnt levels were similar but the p23 levels of the p23kd1 nuclear extract remained low (60% of the controls) (Fig. 5B). We performed gel shift assays to explore whether this reduced amount of the nuclear AhR would cause less AhR/Arnt/DRE complex formation. When the 3-methylcholanthrene-treated nuclear extracts were normalized by the protein content, 50% of the gel shift intensity of the WT and NC Hepa1c1c7 cells was observed in p23kd1 cells (Fig. 5C). When the nuclear extracts were normalized by the amount of AhR, the gel shift intensities were similar among all three 3-methylcholanthrene-treated nuclear extracts, suggesting that less gel shift complex formed in the p23kd1 nuclear extract was likely caused by a lower amount of AhR translocated into the nucleus. We noticed that nonspecific bands in lane 6 were more pronounced because there were more proteins included in this sample in order to normalize the amount of AhR for comparison. The specificity of the AhR/Arnt/DRE gel shift complex was confirmed by the use of a 10-fold excess of DRE or mutated DRE; the AhR- and Arnt-specific antibodies, but not the control IgGs, abolished the gel shift complex (Fig. 5D).
3.8. 3-methylcholanthrene-activated recruitment of AhR to the cyp1a1 promoter was less in p23Kkd1 cells
Next, we examined whether knockdown of p23 would affect the AhR recruitment to the DRE-containing promoter by 3-methylcholanthrene since less amounts of the nuclear AhR was observed in p23kd1 cells after 3-methylcholanthrene treatment. One hour after 3-methylcholanthrene treatment, AhR was recruited to the cyp1a1 promoter to a similar extent in WT and NC cells. However, only 60% of the WT AhR amount was recruited to the p23kd1 cyp1a1 promoter (Fig. 5E).
3.9. p23kd1 cells expressed less AhR messages
We examined whether reduction of the AhR protein levels in p23kd1 cells could be explained by lower amounts of the AhR message expressed. Real-time qPCR data showed that the AhR message was indeed less – about 50% of the WT and NC AhR levels (Fig. 6A left). Next, we addressed whether the reduced amount of the AhR message observed in p23kd1 cells could be caused by an increase of mRNA degradation. After treatment with actinomycin-D to inhibit transcription, cells were harvested at different times to determine the message stability. We observed that the AhR message was degraded at a similar rate in all three Hepa1c1c7 cell lines (p23kd1, NC, and WT), suggesting that mRNA stability control was not the mechanism in altering the AhR message levels in p23kd1 cells (Fig. 6A right).
3.10. Protein degradation rate of AhR was increased in p23kd1 cells
Next, we examined whether AhR degradation could play a role in the reduced AhR levels in p23-knockdown stable Hepa1c1c7 cells in the absence of ligand. We observed that the AhR levels of NC and WT Hepa1c1c7 cells dropped to about 75% after cells were treated with cycloheximide (50 μg/ml), a protein synthesis inhibitor, for 6 h (Fig. 6B). However, the degradation rate of AhR was significantly faster in p23kd1 cells than the two controls, suggesting that reduction of the AhR protein content in p23kd1 cells is partly caused by the increase of its degradation. The enhanced AhR degradation should be p23-specific since p23kd5 – another p23 knockdown stable Hepa1c1c7 cell line which was generated by lentiviral infection rather than electroporation – also showed an increase rate of AhR degradation (Fig. 6C, top). Interestingly, this degradation was still observed in the presence of 10μM MG132, a proteasome inhibitor, for up to 8 h (Fig. 6C, bottom). We further investigated this MG132 effect at the 6-hour time point and observed that the AhR levels were suppressed to 42, 62, and 38% when p23kd5 cells were treated for 6 h with MG132 alone, cycloheximide alone, and both, respectively (Fig. 6D). Since treatment with cycloheximide did not cause a significant decrease of the AhR levels in MG132-treated cells, AhR degradation appeared to be more important in determining the AhR levels than AhR synthesis. Collectively, our data suggested that a labile protease, which accumulates in the presence of MG132, might be responsible for degradation of AhR when the p23 levels are compromised.
3.11. MG132 could not protect AhR from ligand-induced degradation in p23 knockdown stable cell lines
It is known that AhR degradation occurs after ligand activation [34]. Since MG132 did not prevent AhR from degradation in p23 knockdown stable Hepa1c1c7 cells, we further examined whether the normal ligand-triggered AhR degradation could be affected when the p23 levels are suppressed. We treated different Hepa1c1c7 cell lines with 1 μM 3-methylcholanthrene for 6 h and observed that in all cases (WT, NC, and p23kd5), the AhR levels were suppressed to 24–39% of the pretreated levels (Fig. 7A). Treatment with 10 μM MG132 for 6 h also suppressed the AhR levels to 43–73%. Co-treatment with MG132 and 3-methylcholanthrene significantly reversed the ligand-triggered degradation in WT and NC cells but not in p23kd5 cells. Next, we examined whether this phenomenon occurs in cell lines other than Hepa1c1c7 cells. Similarly, we observed that 3-methylcholanthrene triggered degradation of AhR in Hep3B and HeLa cell lines. MG132 reversed the degradation in wild type and negative control knockdown stable but not the p23-specific knockdown stable Hep3B and HeLa cells (Fig. 7B). Collectively, our data supported that although the proteasomal degradation of AhR upon ligand treatment is blocked by MG132 in cells with wild type p23 levels, compromised levels of p23 appear to favor the 26S proteasome-independent degradation of the AhR in the presence or absence of ligand.
4. Discussion
Reducing the p23 levels in a cell line could be a means to provide insights on how p23 affects AhR signaling. In this study, we generated p23 knockdown stable cells from mouse and human cell lines – mouse hepatoma Hepa1c1c7, human hepatoma Hep3B, and human cervical HeLa cells. These three cell lines were used since they have been widely used for studying AhR function. In an effort to address p23-specific effects rather than artificial effects due to cell line manipulation and nonspecific knockdown, we performed the following experiments: (1) we generated knockdown stable cell lines using five p23-specific and two scramble shRNAs to confirm that the observed effects are specific to the knockdown of p23; (2) we compared negative control knockdown stable cells with the wild type cells to determine that the stable cell selection process per se does not cause the observed effects; (3) we generated knockdown stable cell lines using two different methods (electroporation and lentiviral infection) to show that the observed effects are not a result of a particular manipulation; (4) we generated knockdown stable cells from different cell lines (Hepa1c1c7, Hep3B, and HeLa) to ensure that the observed effects are not artificial effects of a particular cell line and (5) we reversed the suppression of the AhR levels in p23-specific knockdown stable cells by expressing exogenous p23 in these cells, validating that the reduction of the AhR levels is indeed p23-specific.
It has been reported that hepatic tissues of p23 heterozygous mice have normal induced levels of cyp1a1 and cyp1a2 messages after TCDD treatment [26]. In addition, AhR appears to bind ligand and induce the expression of the cyp1a1 message normally in late stage p23 null and heterozygous mouse embryos. Taken together, these data argued that p23 is not necessary for the AhR function in vivo. Since these genetically manipulated mouse embryos and mice expressed either no or much less p23, these authors suggested that compensatory mechanisms might have been triggered to allow normal AhR function. For example, it is known that tsp23 is a human p23 homolog with 44% amino acid identity [3]. Therefore, p23-like proteins (such as tsp23) could conceivably be expressed to preserve the normal AhR function in the absence of p23. Our p23-specific knockdown stable cells express relatively higher amounts (20–50%) of the wild type p23 levels and suppress the ligand-activated AhR function. This apparent discrepancy might be explained by the differences in the p23 levels: the extent of p23 reduction in our stable cells might not trigger the compensatory mechanisms as observed in the p23 heterozygous mice and late stage p23 null and heterozygous mouse embryos. In addition, it is conceivable that cell lines might have lost the compensatory ability to restore the AhR levels. Another possibility is that p23 might affect AhR differently in cancer cells than in normal cells, since we used immortalized cancer cell lines rather than normal cells for our study. Nonetheless, our data have led us to speculate that any endogenous event that reduces 50% of wild type p23 levels may potentially inhibit the AhR function by lowering the AhR levels in cancer cells.
Our data revealed that wild type p23 levels maintain the cellular levels of AhR in part by favoring the AhR gene transcription. p23 has been shown to modulate binding of steroid receptors – such as thyroid hormone receptor [8], estrogen receptor [35], and glucocorticoid receptor [8, 36] – to their enhancers which in turn regulates the corresponding target gene transcription. It has been reported that multiple Sp1 enhancers are found upstream to the transcription start site of the human AhR gene [37]. Down-regulation of the AhR expression may be caused by reduced binding of Sp1 to the hypermethylated AhR promoter in human acute lymphoblastic leukemia cells [38]. Interestingly, recruitment of Hsp90 by Sp1 is part of the transcriptional activation process which involves the Sp1 stability control [39, 40]. Realizing that p23 is part of the Hsp90 chaperone complex, it would be interesting to explore whether p23 is involved in the Sp1-mediated activation of the AhR gene transcription.
Degradation of AhR has been intensely studied by many groups; dissociation of p23 from the AhR complex has been linked to both stabilization and degradation of AhR. It is well known that geldanamycin dissociates p23 from the Hsp90 complex [5] and triggers degradation of AhR in Hepa1c1c7 cells [41]. Overexpression of XAP2 also dissociates p23 from the AhR complex [42] and suppresses the CHIP-mediated degradation of AhR [43, 44], although the physiological significance of this degradation has been challenged [45]. There might be an immediate effect on the AhR stability when p23 dissociates from the AhR complex, and the effect may vary depending on the trigger of the dissociation. However, we believe that a sustained lowering of the cellular p23 levels favors degradation of AhR.
Interestingly, our data revealed that AhR is prone to non-proteasome-mediated degradation when the p23 levels are compromised. It has been reported that the proteasome inhibitor MG132, at 8 μM concentration, increases the Arnt levels which in turn triggers nuclear translocation of AhR in mouse embryo primary fibroblasts [46]. On the contrary, it has been shown that AhR translocates into the nucleus of Hepa1 cells after treating the cells with 10 μM MG132 for 8 h; the Arnt levels, however, were unaffected [47]. Nevertheless, it was not clear whether MG132 could affect the total AhR levels by conventional Western analysis. Since MG132 did not reverse the ligand-dependent degradation of AhR in p23-compromised cells, we speculated that a protease, which is degraded by 26S proteasome, might be responsible for AhR degradation. This protease may also degrade AhR but at a very small extent when the p23 levels are at wild type levels: treatment with MG132 (10–25 μM) caused marginally noticeable reduction of the AhR protein levels by Western analysis in Hepa1c1c7 cells [47, 48]. We postulated that p23 might protect AhR from degradation by its presence in the unliganded AhR complex, or p23 might interact directly with the protease and in turn regulate the proteolysis of AhR.
Silencing of the HDAC6 gene results in hyperacetylation of Hsp90, which in turn causes dissociation of p23 from AhR and also inhibition of the AhR ligand binding, leading to failure of the ligand-mediated nuclear translocation of AhR [49]. One might then postulate that reduction of p23 might cause problems in AhR ligand binding. However our data indicated otherwise. We observed that the extent of the AhR response to a ligand in p23-specific knockdown stable cells is consistent with its 50% of the wild type AhR levels: upon ligand activation, about 50% of the wild type levels of AhR translocates into the nucleus and then bind to the DRE as an AhR/Arnt/DRE complex.
p23 has been shown to enhance binding of ER to ERE-containing promoters, leading to activation of target gene transcription [35]. In addition, we previously observed that p23 is capable of promoting the formation of the ligand-activated AhR/Arnt/DRE complex in vitro [25], suggesting that there may be a yet unidentified nuclear role of p23 in the AhR function. Therefore, we examined the ligand-dependent formation of the AhR/Arnt/DRE complex in the wild type and stable cells. We observed that the p23kd1 nuclear extract gave a much weaker gel shift complex as compared to the intensities observed in wild type and negative control knockdown stable nuclear extracts. But after normalizing the amount of AhR and Arnt in all nuclear extracts, the gel shift complex intensities were similar, suggesting that weaker intensity in the p23kd1 nuclear extract was caused by lesser amounts of AhR. When we normalized the AhR content in nuclear extracts, we noticed that there was still a significant reduction of p23 in the p23kd1 nuclear extract. Although the nuclear p23 levels were reduced to 60%, it did not affect the gel shift complex formation. However, we cannot rule out the possibility that p23 is necessary for the formation of the ligand-dependent AhR/Arnt/DRE complex, since 60% of its wild type p23 levels might have been sufficient to cause the formation of the complex.
Supplementary Material
Acknowledgments
We thank Dr. Mike Denison (UC Davis) for providing us the reporter luciferase plasmid pGudluc1.1. This work is supported by a grant from the National Institutes of Health (WKC, R01 ES014050)
Abbreviations
- WT
wild type
- NC
negative control knockdown stable
- p23kd1-8
p23 knockdown stable 1-8
- AhR
aryl hydrocarbon receptor
- Arnt
aryl hydrocarbon receptor nuclear translocator
- DRE
dioxin response element
Footnotes
Conflict of interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith DF, Faber LE, Toft DO. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J Biol Chem. 1990;265:3996–4003. [PubMed] [Google Scholar]
- 2.Johnson JL, Beito TG, Krco CJ, Toft DO. Characterization of a novel 23-kilodalton protein of unactive progesterone receptor complexes. Mol Cell Biol. 1994;14:1956–63. doi: 10.1128/mcb.14.3.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman BC, Felts SJ, Toft DO, Yamamoto KR. The p23 molecular chaperones act at a late step in intracellular receptor action to differentially affect ligand efficacies. Genes Dev. 2000;14:422–34. [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson JL, Toft DO. A novel chaperone complex for steroid receptors involving heat shock proteins, immunophilins, and p23. J Biol Chem. 1994;269:24989–93. [PubMed] [Google Scholar]
- 5.Johnson JL, Toft DO. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol Endocrinol. 1995;9:670–8. doi: 10.1210/mend.9.6.8592513. [DOI] [PubMed] [Google Scholar]
- 6.Hutchison KA, Stancato LF, Owens-Grillo JK, Johnson JL, Krishna P, Toft DO, et al. The 23-kDa acidic protein in reticulocyte lysate is the weakly bound component of the hsp foldosome that is required for assembly of the glucocorticoid receptor into a functional heterocomplex with hsp90. J Biol Chem. 1995;270:18841–7. doi: 10.1074/jbc.270.32.18841. [DOI] [PubMed] [Google Scholar]
- 7.Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13:817–26. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–5. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- 9.Tanioka T, Nakatani Y, Kobayashi T, Tsujimoto M, Oh-ishi S, Murakami M, et al. Regulation of cytosolic prostaglandin E2 synthase by 90-kDa heat shock protein. Biochem Biophys Res Commun. 2003;303:1018–23. doi: 10.1016/s0006-291x(03)00470-4. [DOI] [PubMed] [Google Scholar]
- 10.Poland A, Glover E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: segregation of toxicity with the Ah locus. Mol Pharmacol. 1980;17:86–94. [PubMed] [Google Scholar]
- 11.Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J Biol Chem. 1997;272:11452–6. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- 12.Ma Q, Whitlock JP., Jr A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 1997;272:8878–84. [PubMed] [Google Scholar]
- 13.Petrulis JR, Kusnadi A, Ramadoss P, Hollingshead B, Perdew G. The hsp90 co-chaperone XAP2 alters importin beta recognition of the bipartitie nuclear localization signal of the Ah receptor and represses transcriptional activity. J Biol Chem. 2003;278:2677–85. doi: 10.1074/jbc.M209331200. [DOI] [PubMed] [Google Scholar]
- 14.Kazlauskas A, Poellinger L, Pongratz I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J Biol Chem. 1999;274:13519–24. doi: 10.1074/jbc.274.19.13519. [DOI] [PubMed] [Google Scholar]
- 15.Ramirez JM, Brembilla NC, Sorg O, Chicheportiche R, Matthes T, Dayer JM, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol. 2010;40:2450–9. doi: 10.1002/eji.201040461. [DOI] [PubMed] [Google Scholar]
- 16.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 17.Bunaciu RP, Yen A. Activation of the aryl hydrocarbon receptor AhR promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 2011;71:2371–80. doi: 10.1158/0008-5472.CAN-10-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinatale BC, Schroeder JC, Perdew GH. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol Carcinog. 2011;50:173–83. doi: 10.1002/mc.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gluschnaider U, Hidas G, Cojocaru G, Yutkin V, Ben-Neriah Y, Pikarsky E. beta-TrCP inhibition reduces prostate cancer cell growth via upregulation of the aryl hydrocarbon receptor. PLoS One. 2010;5:e9060. doi: 10.1371/journal.pone.0009060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol. 2010;24:359–69. doi: 10.1210/me.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazlauskas A, Sundstrom S, Poellinger L, Pongratz I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol Cell Biol. 2001;21:2594–607. doi: 10.1128/MCB.21.7.2594-2607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazlauskas A, Poellinger L, Pongratz I. The immunophilin-like protein XAP2 regulates ubiquitination and subcellular localization of the dioxin receptor. J Biol Chem. 2000;275:41317–24. doi: 10.1074/jbc.M007765200. [DOI] [PubMed] [Google Scholar]
- 23.Cox MB, Miller CA., 3rd The p23 co-chaperone facilitates dioxin receptor signaling in a yeast model system. Toxicol Lett. 2002;129:13–21. doi: 10.1016/s0378-4274(01)00465-9. [DOI] [PubMed] [Google Scholar]
- 24.Cox MB, Miller CA., 3rd Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones. 2004;9:4–20. doi: 10.1379/460.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shetty PV, Bhagwat BY, Chan WK. P23 enhances the formation of the aryl hydrocarbon receptor-DNA complex. Biochem Pharmacol. 2003;65:941–8. doi: 10.1016/s0006-2952(02)01650-7. [DOI] [PubMed] [Google Scholar]
- 26.Flaveny C, Perdew GH, Miller CA., 3rd The Aryl-hydrocarbon receptor does not require the p23 co-chaperone for ligand binding and target gene expression in vivo. Toxicol Lett. 2009;189:57–62. doi: 10.1016/j.toxlet.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park MS, Chu F, Xie J, Wang Y, Bhattacharya P, Chan WK. Identification of cyclophilin-40 interacting proteins reveals potential cellular function of cyclophilin-40. Anal Biochem. 2010;410:257–65. doi: 10.1016/j.ab.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haarmann-Stemmann T, Bothe H, Kohli A, Sydlik U, Abel J, Fritsche E. Analysis of the transcriptional regulation and molecular function of the aryl hydrocarbon receptor repressor in human cell lines. Drug Metab Dispos. 2007;35:2262–9. doi: 10.1124/dmd.107.016253. [DOI] [PubMed] [Google Scholar]
- 29.Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–9. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luu TC, Bhattacharya P, Chan WK. Cyclophilin-40 has a cellular role in the aryl hydrocarbon receptor signaling. FEBS Lett. 2008;582:3167–73. doi: 10.1016/j.febslet.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Li Y, Zhang T, Chan WK. The aryl hydrocarbon receptor nuclear translocator-interacting protein 2 suppresses the estrogen receptor signaling via an Arnt-dependent mechanism. Arch Biochem Biophys. 2010;502:121–9. doi: 10.1016/j.abb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang TT. beta-naphthoflavone, an inducer of xenobiotic metabolizing enzymes, inhibits firefly luciferase activity. Anal Biochem. 2002;304:122–6. doi: 10.1006/abio.2001.5562. [DOI] [PubMed] [Google Scholar]
- 34.Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem. 1999;274:28708–15. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- 35.Oxelmark E, Roth JM, Brooks PC, Braunstein SE, Schneider RJ, Garabedian MJ. The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol Cell Biol. 2006;26:5205–13. doi: 10.1128/MCB.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol. 2004;24:2682–97. doi: 10.1128/MCB.24.7.2682-2697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eguchi H, Hayashi S, Watanabe J, Gotoh O, Kawajiri K. Molecular cloning of the human AH receptor gene promoter. Biochem Biophys Res Commun. 1994;203:615–22. doi: 10.1006/bbrc.1994.2227. [DOI] [PubMed] [Google Scholar]
- 38.Mulero-Navarro S, Carvajal-Gonzalez JM, Herranz M, Ballestar E, Fraga MF, Ropero S, et al. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis. 2006;27:1099–104. doi: 10.1093/carcin/bgi344. [DOI] [PubMed] [Google Scholar]
- 39.Hung JJ, Wu CY, Liao PC, Chang WC. Hsp90alpha recruited by Sp1 is important for transcription of 12(S)-lipoxygenase in A431 cells. J Biol Chem. 2005;280:36283–92. doi: 10.1074/jbc.M504904200. [DOI] [PubMed] [Google Scholar]
- 40.Wang SA, Chuang JY, Yeh SH, Wang YT, Liu YW, Chang WC, et al. Heat shock protein 90 is important for Sp1 stability during mitosis. J Mol Biol. 2009;387:1106–19. doi: 10.1016/j.jmb.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 41.Chen HS, Singh SS, Perdew GH. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch Biochem Biophys. 1997;348:190–8. doi: 10.1006/abbi.1997.0398. [DOI] [PubMed] [Google Scholar]
- 42.Hollingshead BD, Petrulis JR, Perdew GH. The aryl hydrocarbon (Ah) receptor transcriptional regulator hepatitis B virus X-associated protein 2 antagonizes p23 binding to Ah receptor-Hsp90 complexes and is dispensable for receptor function. J Biol Chem. 2004;279:45652–61. doi: 10.1074/jbc.M407840200. [DOI] [PubMed] [Google Scholar]
- 43.Lees MJ, Peet DJ, Whitelaw ML. Defining the role for XAP2 in stabilization of the dioxin receptor. J Biol Chem. 2003;278:35878–88. doi: 10.1074/jbc.M302430200. [DOI] [PubMed] [Google Scholar]
- 44.Morales JL, Perdew GH. Carboxyl terminus of hsc70-interacting protein (CHIP) can remodel mature aryl hydrocarbon receptor (AhR) complexes and mediate ubiquitination of both the AhR and the 90 kDa heat-shock protein (hsp90) in vitro. Biochemistry. 2007;46:610–21. doi: 10.1021/bi062165b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pollenz RS, Dougherty EJ. Redefining the role of the endogenous XAP2 and C-terminal hsp70-interacting protein on the endogenous Ah receptors expressed in mouse and rat cell lines. J Biol Chem. 2005;280:33346–56. doi: 10.1074/jbc.M506619200. [DOI] [PubMed] [Google Scholar]
- 46.Santiago-Josefat B, Fernandez-Salguero PM. Proteasome inhibition induces nuclear translocation of the dioxin receptor through an Sp1 and protein kinase C-dependent pathway. J Mol Biol. 2003;333:249–60. doi: 10.1016/j.jmb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 47.Song Z, Pollenz RS. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Mol Pharmacol. 2002;62:806–16. doi: 10.1124/mol.62.4.806. [DOI] [PubMed] [Google Scholar]
- 48.Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275:8432–8. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 49.Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436–45. doi: 10.1074/jbc.M808999200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.