

Abstract
DNA mismatch repair (MMR) models have proposed that MSH proteins identify DNA polymerase errors while interacting with the DNA replication fork. MLH proteins (primarily Mlh1-Pms1 in baker’s yeast) then survey the genome for lesion-bound MSH proteins. The resulting MSH-MLH complex formed at a DNA lesion initiates downstream steps in repair. MLH proteins act as dimers and contain long (20 – 30 nanometers) unstructured arms that connect two terminal globular domains. These arms can vary between 100 to 300 amino acids in length, are highly divergent between organisms, and are resistant to amino acid substitutions. To test the roles of the linker arms in MMR, we engineered a protease cleavage site into the Mlh1 linker arm domain of baker’s yeast Mlh1-Pms1. Cleavage of the Mlh1 linker arm in vitro resulted in a defect in Mlh1-Pms1 DNA binding activity, and in vivo proteolytic cleavage resulted in a complete defect in MMR. We then generated a series of truncation mutants bearing Mlh1 and Pms1 linker arms of varying lengths. This work revealed that MMR is greatly compromised when portions of the Mlh1 linker are removed, whereas repair is less sensitive to truncation of the Pms1 linker arm. Purified complexes containing truncations in Mlh1 and Pms1 linker arms were analyzed and found to have differential defects in DNA binding that also correlated with the ability to form a ternary complex with Msh2-Msh6 and mismatch DNA. These observations are consistent with the unstructured linker domains of MLH proteins providing distinct interactions with DNA during MMR.
Keywords: mismatch repair, Mlh1-Pms1, linker arms, DNA binding, mutator phenotype
Introduction
DNA mismatch repair (MMR) is a conserved pathway that corrects misincorporation and slippage errors introduced by DNA polymerase during DNA replication. MMR in eukaryotes initiates with the binding of MSH proteins (Msh2-Msh6 or Msh2-Msh3) to base-base mismatches and loop mismatches up to 17 nt in size.1,2 This interaction results in the recruitment of MLH proteins (primarily Mlh1-Pms1 in baker’s yeast) followed by the initiation of downstream excision and resynthesis steps that maintain template strand information.3–5 Mutations in the MSH and MLH MMR genes result in large increases in mutation rate and are associated with hereditary non-polyposis colorectal cancer.6 Interactions between MMR factors (MSH, MLH) and components of the replication machinery such as the processivity clamp PCNA have led to the idea that MSH proteins rapidly scan behind the replication fork to identify DNA polymerase errors.1,7–12 Studies showing that MSH proteins act as sliding clamps and co-localize with replication components in S-phase are consistent with replication tracking models in which the identification of mismatches on DNA by MSH proteins coincides with transient nucleosome disruption by the passing replication machinery.11,13–19
Recently Hombauer et al.11 showed in baker’s yeast that Mlh1 -Pms1 form nuclear foci whose appearances are dependent on MSH complexes and the frequency of DNA mismatches in the genome. These observations suggest that MLH interactions with MSH proteins are temporally distinct from the initial binding of MSH proteins to mismatch DNA. The work of Hombauer et al.,11 coupled with in vitro studies showing that MLH proteins can bind and diffuse along DNA, suggest that Mlh1-Pms1 interacts with DNA during MMR.12,20–22
Structural studies have revealed important insights into how MLH proteins interact with each other and with the nucleotide cofactor ATP.23–27 The MLH proteins contain N- and C-terminal domains that are connected by linker arms. The N-terminal domains (NTDs) of MLH family members are highly conserved and contain an ATP binding site that belongs to the GHKL family of ATPases.28,29 The structurally conserved C-terminal domains (CTDs) are essential for dimerization.21 Linker arms, ~150 amino acids for Mlh1 and ~250 amino acids for Pms1, connect the N-terminal and C-terminal globular domains of MLH proteins. These arms are variable in length between MLH family members, resistant to amino acid substitution, and highly divergent in sequence context.26,29,30 Consistent with these properties, Argueso et al.30 performed an alanine-scan mutagenesis of yeast MLH1 and found that very few mutations in the linker arm region conferred defects in MMR. Secondary structure prediction analysis suggest that the linker arms are random coils that are highly disordered in solution.26 A fully extended ring structure of Mlh1-Pms1, with 20–nanometer (nm) and 30– nm arms, respectively, could be similar in size to cohesin ring complexes that connect sister chromatids.12,29,31 However, large conformational changes affecting the linker arms of the S. cerevisiae Mlh1-Pms1 complex were observed in atomic force microscopy and proteolysis analyses that appear dependent on nucleotide occupancy (ADP or ATP) in the individual ATP binding domains.29 In fact these conformational changes were proposed to modulate the opening and closing of an Mlh1-Pms1 ring.29 In addition, single-molecule analysis suggested that the yeast Mlh1-Pms1 complex adopts a ring-like configuration capable of encircling DNA and bypassing barriers such as nucleosomes while using a rapid hopping/stepping diffusion mechanism.12,26 The role that the linker arms play during the diffusion of Mlh1-Pms1 along DNA remain unknown, although they could act as either passive tethers that just link the N- and C-terminal domains, or the linkers themselves might transiently interact with the DNA and thus provide additional DNA-binding surfaces that could participate in the diffusive motion.
To gain a clearer understanding of the mechanism by which Mlh1-Pms1 interacts with DNA, we created a series of deletions within the linker arm domains of both Mlh1 and Pms1. In mutator assays, we show that the linker arm of Mlh1 is more sensitive to deletion than Pms1. Proteolytic cleavage of the linker arm of Mlh1 leads to a loss of MMR activity in vivo and loss of DNA binding activity in vitro. Purified complexes containing deletions in Mlh1-Pms1 linker arms were analyzed and found to have different defects in DNA binding. Together these observations are consistent with the unstructured linker domains of MLH proteins having distinct interactions with DNA that are important for early steps in MMR.
Results
Cleavage of the Mlh1 linker arm in vivo impairs MMR
Previous structural and single molecule studies suggested that Mlh1-Pms1 adopts a ring-like structure that can wrap around DNA.12,21,24,26,27,29 In support of this idea our groups showed in single molecule analysis that Mlh1-Pms1 did not dissociate upon encountering anchored DNA ends or the apex of looped DNA, but dissociated from free ends of “single-tethered” DNA.12 We also found that proteolytic cleavage of the Mlh1 linker arm weakened Mlh1-Pms1 DNA-binding activity in bulk DNA binding assays.
To further investigate the role of the unstructured linker domains, we tested whether cleavage of the Mlh1 linker arm abolished MMR functions in vivo. We integrated MLH1 alleles containing TEV cleavage sites into a strain background containing the TEV protease gene with a nuclear localization signal under the galactose inducible promoter32 (T ables 1 and 2; Materials and Methods). There are no proteins in S. cerevisiae that contain the canonical TEV cleavage site, and TEV expression does not have any discernable effects on growth and proliferation.32,33 These strains also contain the lys2::insE-A14 frameshift allele to measure MMR function. 34 Using this Lys+ reversion assay, we tested the effects of TEV protease cleavage at two different sites (after amino acid T448 or Y499) in the Mlh1 linker arm (Fig. 1). Strains bearing these two alleles displayed mutation rates similar to mlh1Δ in the presence of galactose, but were otherwise functional for MMR in the presence of the non-inducing carbon source sucrose (Table 3). In strains lacking TEV protease, these alleles fully complemented the mlh1Δ mutator phenotype (Table 3, data not shown). Together these and previous in vitro studies12 show that an intact Mlh1 linker arm is required in MMR. However, we note that weakening of DNA binding by cleaving the Mlh1 linker domain may not be the only reason for the MMR defect because Mlh1 and Pms1 linker arms appear to change dramatically upon nucleotide binding.29
Fig. 1.
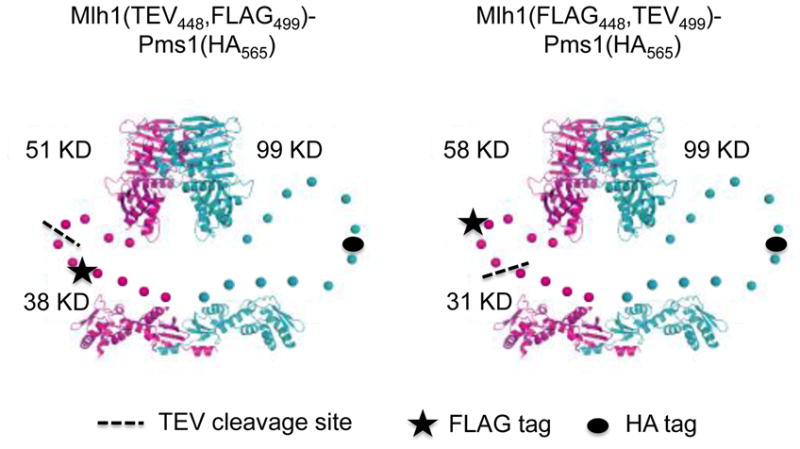
Location of TEV cleavage sites in Mlh1-Pms1 linker arms. Cartoon of predicted structures of TEV-containing Mlh1-Pms1 constructs, based on structural and biochemical data.26,27 Mlh1 is in magenta and Pms1 is in blue. Linker arms are illustrated by a series of unconnected dots. Approximate positions of the TEV cleavage site (black dashed line), FLAG-tag (star) and HA-tag (filled circle) are shown. The exact position of each tag is described in the Materials and Methods.
Table 3.
TEV protease cleavage of Mlh1 confers a mutator phenotype in vivo
| Relevant genotype | n | Mutation rate (10−7), (95% confidence interval) | Relative to wild-type | ||
|---|---|---|---|---|---|
| Sucrose | Galactose | Sucrose | Galactose | ||
| Wild-type | 15 | 4.9 (4.1–5.6) | 7.8 (3.2–30) | 1.0 | 1.6 |
| mlh1Δ | 15 | 15,900 (10,400–27,700) | 30,100 (11,900–55,000) | 3,250 | 6,140 |
| MLH1 (FLAG448) | 15 | 4.7 (3.7–6.8) | 76 (6.7–157) | 0.96 | 16 |
| MLH1 (TEV448, FLAG499) | 15 | 13 (9–122) | 16,700 (4,860–37,400) | 2.7 | 3,410 |
| MLH1 (FLAG448, TEV499) | 15 | 5.6 (3.9–25) | 28,000 (13,600–43,300) | 1.1 | 5,710 |
The indicated alleles were integrated into the strain EAY2576 and tested in the lys2::insE-A14 mutator assay. TEV protease was expressed under the galactose promoter, allowing for carbon source-dependent cleavage of MLH1 constructs containing consensus TEV protease cleavage sites within the unstructured linker arm region of the protein. Lys+ reversion rates under un-induced (sucrose) and induced (galactose) conditions for TEV protease expression are indicated. n = number of independent measurements. Mutation rates were normalized to Wild-type grown in sucrose. Strains used in these experiments were: Wild-type-EAY3099; mlh1Δ-EAY3098; MLH1(FLAG448)-EAY3100; MLH1(TEV448, FLAG499)-EAY3102; MLH1(FLAG448, TEV499)-EAY3101.
We tested whether cleavage of Mlh1 in vivo conferred a dominant negative phenotype by transforming EAY3102 (MLH1(TEV448, FLAG499)) with pEAA109 (MLH1,ARS CEN ) and pRS415 (ARS CEN). As expected, the rate of reversion to Lys+ was similar to mlh1Δ in EAY3102 containing pRS415 grown in sucrose and galactose. In contrast, the reversion rate in EAY3102 containing pEAA109 was indistinguishable when cells were grown in sucrose compared to sucrose and galactose (0.7 × 10−6 (0.5 – 0.8 × 10−6, 95% C.I.) vs. 1.0 × 10−6 (0.6 – 2.9 × 10−6, 95% C.I.)). These results indicate that cleaved Mlh1 does not confer a dominant negative phenotype in vivo, consistent with the idea that stable DNA-binding is a prerequisite for association with Msh2-Msh6 and subsequent MMR steps.
The results obtained above can be explained by TEV protease cleavage causing the entire Mlh1-Pms1 complex to fall apart, or the Mlh1 NTD could dissociate, leaving just the Mlh1 CTD bound to the full-length Pms1. Alternatively, both the Mlh1 NTD and CTD could remain associated with intact Pms1 upon TEV cleavage. We attempted to distinguish these possibilities by examining the integrity of Mlh1-Pms1 complexes in MLH1(TEV,FLAG) strains grown in galactose to induce TEV protease expression. Western blot analysis was then performed on the induced cultures to detect the presence of the C-terminal Mlh1 cleavage product using anti-FLAG antibody. However, only a small amount of cleavage product (estimated to be less than 10% of full length Mlh1) was observed in cultures grown for fours in galactose (data not shown). One explanation consistent with this observation and our genetic analysis is that there are two populations of Mlh1 in the cell, a small population (~10%) that participates in MMR and is TEV sensitive, and a second larger population (~90%) that is TEV resistant but does not act in MMR.
Because we did not detect efficient cleavage of Mlh1(TEV) in vivo, we tested the integrity of the Mlh1-Pms1 complex after TEV protease cleavage in vitro, followed by immunoprecipitation with an anti-HA antibody specific to the Pms1(HA) subunit (Fig. 1, 2a). This analysis showed that the Mlh1 N-terminal and C-terminal domains remained associated with Pms1(HA) after the Mlh1 linker arm was cleaved with TEV protease (Fig. 2b). Control reactions performed with Mlh1(TEV448, FLAG499)-Pms1 lacking an HA tag showed that immunoprecipitation was specific. Similar results were obtained in reactions containing or lacking large molar excesses of 40 bp duplex DNA substrate, DNAseI or apyrase, suggesting that DNA or ATP were not required for the integrity of the TEV-cleaved complex (data not shown). These results provide further evidence that the integrity of the linker arms is essential for MMR (see Discussion).
Fig. 2. Mlh1(TEV)-Pms1 complexes remain intact after TEV cleavage.

(a). Wild-type and the indicated Mlh1-Pms1 complexes wereexpressed and purified from S. cerevisiae (see Materials and Methods). 0.5 μg of each complex were loaded into each lane. Proteins were electrophoresed in 8% SDS-PAGE and the gel was stained with Coomassie blue. The sizes of the relevant molecular weight standards (M) are indicated. (b). Immunoprecipitation (IP) of the Mlh1(TEV448, FLAG499)-Pms1(HA565) complex using an anti-HA antibody. Mlh1-Pms1 was untreated or treated with TEV protease prior to IP (Materials and Methods). Input lanes show TEV untreated and treated complexes prior to IP. Control reactions were performed in parallel with Mlh1(TEV448, FLAG499)-Pms1 lacking an HA tag on Pms1. Bands arising from BSA (**) and IgG (*) present in the IP reactions are indicated.
Deletions in the Mlh1-Pms1 unstructured linker arms confer differential MMR defects
Our previous work12 and the above TEV protease cleavage experiments encouraged us to test if shortening of the linker arm domains in MLH proteins would alter both DNA binding and MMR functions. To examine this we created a variety of deletions along the predicted linker arm domains of each protein (Fig. 3). The limits of the linker arm domains (336–480 in Mlh1, 390–634 in Pms1; A. Guarne personal communication) were conservatively chosen to decrease the possibility of disrupting the N or C -terminal globular domains. MLH1 alleles contain a FLAG-epitope tag in a position downstream of the linker arm domain (after amino acid Y499) that was previously shown to not disrupt Mlh1 MMR function.12,30 All PMS1 alleles, except pms1Δ390-610, contain a HA-epitope tag within the linker arm domain (after amino acid D565) that was also previously shown not to affect MMR.12
Fig. 3.

Schematic diagram of Mlh1 and Pms1 linker arm deletion series. (a). Outline of amino acid deletions (ΔX-Y) created within the Mlh1 linker arm domain. The location of the FLAG epitope tag, after Y499 in Mlh1, is indicated by the red bar. Equal amounts of crude cellular extracts (20 μg) from strains bearing the indicated MLH1 allele were loaded onto and separated in 8% SDS-PAGE and then probed with an anti-FLAG antibody. (b). Outline of amino acid deletions (ΔX-Y) created within the Pms1 linker arm domain. The location of the HA-epitope tag, after D565 in Pms1, is indicated by the blue bar. Equal amounts of crude cellular extracts (20 μg) from strains expressing the indicated PMS1 allele were loaded onto and separated in 8% SDS-PAGE and then probed with anti-HA antibody. For Panels (a) and (b), MMR function, as assayed in lys2A14 mutator assays, is described as similar to wild-type (+++), a weak mutator (++), or a null phenotype (−). The domains of Mlh1 and Pms1 are not drawn to scale.
Individual mlh1 and pms1 mutant alleles were over-expressed using the galactose inducible promoter to assess protein stability. After induction, crude extracts were collected and mutant proteins were identified by western blot analysis using antibodies specific to the relevant epitope-tags. Using an anti-FLAG antibody, each mlh1 linker arm deletion polypeptide was detected at levels equivalent to those seen in extracts containing full length Mlh1(FLAG) (Fig. 3). The pms1 linker arm deletions polypeptides were detected using an anti-HA antibody. With the exception of pms1Δ600-625, which showed reduced expression levels, pms1 linker arm deletions displayed expression levels similar to Pms1(HA). pms1Δ390-610 expression level was not tested because this construct did not contain an HA-tag; however as shown in Fig. 2a, MLH complexes containing this pms1 truncation could be purified with yields similar to Mlh1-Pms1. Thus on the whole Mlh1 and Pms1 linker arm deletions are expressed at roughly wild-type levels.
Individual linker domain mutants were tested in the lys2::insE-A14 reversion assay in the presence of their wild-type heterodimeric partner (Table 4). The complete deletion of the linker arm in either MLH1 or PMS1 conferred a null phenotype for MMR. With the exception of the 25 amino acid deletions mlh1Δ348-373 and mlh1Δ445-470, all of the mlh1 mutants displayed mutation rates similar to an mlh1Δ strain. mlh1Δ348-373 displayed a low to intermediate mutation rate and mlh1Δ445-470 showed a rate similar to wild-type. In contrast, most of the pms1 linker domain mutants showed mutation rates similar to wild-type. 25 (pms1Δ600-625) and 50 (pms1Δ584-634) amino acid deletions conferred weak mutator phenotypes and the complete deletion (pms1Δ390-610) conferred a null phenotype. A possible explanation for the weak mutator phenotype seen in pms1Δ600-625 is reduced protein expression/stability (Fig. 3). Together these results suggest that the Mlh1 linker arm is more sensitive to deletion than the Pms1 linker arm.
Table 4.
mlh1 and pms1 linker arm deletions confer differential mutator phenotypes
| Relevant genotype | n | Mutation rate (10−7), (95% C.I.) | Relative to wild-type |
|---|---|---|---|
| MLH1 | 20 | 7.5 (3.5–18) | 1.0 |
| MLH1 (FLAG499) | 20 | 7.7 (5.2–25) | 1.0 |
| mlh1Δ | 20 | 45,100 (23,000–255,000) | 6,000 |
| mlh1Δ396-421 (FLAG499) | 20 | 89,700 (15,900–180,000) | 12,000 |
| mlh1Δ348-373 (FLAG499) | 20 | 323 (132–1,080) | 43 |
| mlh1Δ445-470 (FLAG499) | 20 | 7.5 (3.3–13) | 1.0 |
| mlh1Δ359-409 (FLAG499) | 20 | 15,800 (10,000–37,400) | 2,100 |
| mlh1Δ407-457 (FLAG499) | 20 | 49,400 (14,700–127,000) | 6,600 |
| mlh1Δ357-457 (FLAG499) | 20 | 49,500 (11,300–169,000) | 6,600 |
| mlh1Δ336-480 (FLAG499) | 20 | 53,000 (22,900–70,600) | 7,070 |
| PMS1 | 15 | 1.5 (0.9–2.4) | 1.0 |
| PMS1 (HA565) | 15 | 5.3 (4.1–17) | 3.5 |
| pms1Δ | 15 | 23,100 (14,000–76,100) | 15,400 |
| pms1Δ450-475 (HA565) | 15 | 7.1 (5.2–9.0) | 4.7 |
| pms1Δ600-625 (HA565) | 15 | 489 (127–916) | 326 |
| pms1Δ437-487 (HA565) | 15 | 9.5 (6.6–21) | 6.3 |
| pms1Δ511-561 (HA565) | 15 | 16 (6.9–24) | 10.7 |
| pms1Δ584-634 (HA565) | 15 | 415 (152–839) | 277 |
| pms1Δ450-550 (HA565) | 15 | 10 (6.4–84) | 6.7 |
| pms1Δ390-610 | 15 | 14,300 (8,230–23,000) | 9,500 |
| wild-type (MLH1, PMS1) | 15 | 2.1 (0.8–5.8) | 1.0 |
| MLH1 (FLAG499), PMS1 (HA565) | 15 | 14 (8–28) | 6.7 |
| mlh1Δ pms1Δ | 15 | 13,800 (10,800–26,000) | 6,570 |
| mlh1Δ445-470, pms1Δ450-475 | 15 | 7.9 (4.5–31) | 3.8 |
| mlh1Δ445-470, pms1Δ600-625 | 15 | 285 (135–672) | 136 |
| mlh1Δ445-470, pms1Δ437-487 | 15 | 8.0 (6.7–16) | 3.8 |
| mlh1Δ445-470, pms1Δ511-561 | 15 | 22 (12–47) | 10.5 |
| mlh1Δ445-470, pms1Δ584-634 | 15 | 205 (132–343) | 98 |
| mlh1Δ445-470, pms1Δ450-550 | 15 | 25 (10–77) | 12 |
| mlh1Δ348-373, pms1Δ450-475 | 15 | 563 (195–928) | 268 |
| mlh1Δ348-373, pms1Δ600-625 | 15 | 16,100 (3,440–137,000) | 7,670 |
| mlh1Δ348-373, pms1Δ437-487 | 15 | 511 (407–658) | 243 |
| mlh1Δ348-373, pms1Δ511-561 | 15 | 917 (494–1,770) | 437 |
| mlh1Δ348-373, pms1Δ584-634 | 15 | 6,760 (4,780–11,800) | 3,220 |
| mlh1Δ348-373, pms1Δ450-550 | 15 | 835 (585–1,740) | 398 |
The indicated mlh1 and pms1 alleles listed were tested in the lys2::insE-A14 mutator assay and Lys+ reversion rates (C. I., confidence interval) were calculated as described in the Materials and Methods. n = number of independent measurements. For each strain (Table 1), the relevant allele was expressed from an ARS-CEN plasmid under the native promoter of the corresponding wild-type gene. The following plasmids (Table 2) were transformed into EAY1366 (mlh1Δ): pEAA213 (MLH1), pEAA375 (MLH1(FLAG499)), pRS415 (dummy vector) and pEAA526-532 (mlh1 linker arm deletions). The following plasmids were transformed into EAY3097 (pms1Δ): pEAA238 (PMS1), pEAA517 (PMS1(HA565)), pRS413 (dummy vector), and pEAA544-550 (pms1 linker arm deletions). Combinations of the above plasmids were transformed into EAY1365 (mlh1Δ, pms1Δ) to analyze the genotypes shown.
To test for synthetic defects involving mlh1 and pms1 alleles, mutant alleles of mlh1 and pms1 that showed wild-type or intermediate MMR defects were tested in combination (Table 4; Fig. 4). Double mutants involving mlh1Δ445-470 and pms1 alleles recapitulated the mutator phenotype of the individual pms1 allele. This was not surprising because strains containing mlh1Δ445-470 displayed a MMR phenotype that was indistinguishable from wild-type. Interestingly, double mutants involving the intermediate allele mlh1Δ348-373 and pms1 alleles showed synergistic increases in mutation rate, suggesting that a mild defect seen in an individual linker mutant is exacerbated when combined with a partner that has a shortened linker arm. One interpretation of this result is that the shortening of the linker arm domains of Mlh1-Pms1 confers a defect in MMR by disrupting protein-DNA interactions within the complex or with other MMR components (Discussion). A second possibility is that the MMR defect is due to a smaller size of the Mlh1-Pms1 ring. Lastly, it is possible that the mutations disrupt structural transitions in Mlh1-Pms1 reported by Sacho et al..29
Fig. 4.

Mlh1 and Pms1 linker arm deletion mutants display synergistic defects in MMR. Mutation rates of mlh1Δ348-378, pms1Δ584-634 and pms1Δ600 -625 single and double mutant strainswere determined in the lys2A14 assay as described in the Materials and Methods. Rates are shown as a percentage of the corresponding null (Table 4).
Deletions of Mlh1-Pms1 linker arms have differential effects on DNA binding activity
To better characterize the MMR defect created by shortening the Mlh1 and Pms1 linker arms we purified complexes containing a complete deletion of one linker arm (Mlh1-pms1Δ390-610, mlh1-Δ336-480-Pms1). We also purified a complex (mlh1Δ348-373-pms1Δ584-634) in which the mlh1 and pms1 mutations displayed a synergistic defect in MMR, resulting in a null-like phenotype. All complexes could be purified at levels similar to wild-type (Fig. 2a).
Previous work showed that Mlh1-Pms1 binds to DNA through non-specific backbone contacts; it displays no specificity for mismatch DNA.12,20 Based on our single molecule analysis of intact and TEV-cleaved MLH complexes,12 we suspected that the MMR defect observed for linker deletions in the lys2A14 reversion assay was due at least in part to an impairment of DNA binding activity. To test this, we performed electromobility shift assays (EMSA) with short (40-bp) radio-labeled oligonucleotides and wild-type and mutant MLH complexes (Fig. 5, Fig. S1). We were unable to detect DNA binding by Mlh1-pms1Δ390-610 at protein concentrations up to 500 nM. However, both mlh1Δ336-480-Pms1 and mlh1Δ348-373-pms1Δ584-634 displayed DNA binding affinities that appeared similar to the Mlh1-Pms1 complex. These observations indicate that the Pms1 linker arm appears more important than the Mlh1 linker arm for the binding of Mlh1-Pms1 to DNA. More sensitive DNA binding assays will need to be performed to determine if the mlh1Δ336-480-Pms1 and mlh1Δ348-373-pms1Δ584-634 mutant complexes display subtle difference in DNA binding relative to Mlh1-Pms1.
Fig. 5.

mlh1 and pms1 linker arm deletions display altered DNA binding affinities. EMSA was performed as described in Materials and Methods. All reactions contained 60 nM 40-bp homoduplex substrate. Titration reactions contained the indicated amounts of Mlh1-Pms1, Mlh1-pms1Δ390-610, mlh1Δ336-480-Pms1, and mlh1Δ348-373-pms1Δ584-634 complexes. Free and bound substrates are indicated. % bound was calculated using ImageJ software as the amount bound divided by the total (bound + free) and is indicated below each lane.
Mlh1-Pms1 and Msh2-Msh6 form a ternary complex on mismatch DNA. This interaction requires ATP and is thought to serve as an intermediate to signal downstream effectors to complete repair.35 Although the conserved connector domain II of Msh2-Msh6 was shown to be important for interactions between Msh and Mlh proteins, it is not known which region(s) of Mlh1-Pms1 is required for this association.5 EMSA was used to rule out the possibility that deletions within the linker domain of Mlh1 and Pms1 disrupted association with Msh2-Msh6 at a DNA mismatch (Fig. 6). In these assays, Mlh1-Pms1 complexes were present at a concentration below detectable DNA binding in the absence of Msh2-Msh6 to eliminate the possibility that they block Msh2-Msh6 access to the mismatch site. Mutant complexes that displayed DNA binding activity (mlh1Δ336-480-PMS1 and mlh1Δ348-373-pms1Δ584-634; Figure 5) displayed ternary complexes with Msh2-Msh6 and mismatch DNA. MLH1-pms1Δ390-610 was defective in ternary complex formation, suggesting that DNA-binding is a prerequisite for association with Msh2-Msh6.
Fig. 6.

mlh1-pms1 complexes that bind to DNA also form ternary complexes with Msh2-Msh6 at a DNA mismatch. EMSA was performed as described in the Materials and Methods. Binding reactions contained 60 nM 40-bp (+1) mismatch substrate and 1 mM ATP. 150 nM Msh2-Msh6, and 100 nM Mlh1-Pms1 or mutant derivatives were included as indicated.
Discussion
In this study we showed that the unstructured linker arms of Mlh1 and Pms1 are important for Mlh1-Pms1 DNA binding activity and truncations or proteolytic cleavage of these linker arms impair MMR functions in vivo. Our analysis revealed that Mlh1 is more sensitive to linker arm deletion but that the Pms1 linker arm appears more important for Mlh1-Pms1 binding to DNA. Previously we showed that TEV cleavage in the linker arm of Mlh1 disrupted Mlh1-Pms1 binding to DNA in vitro.12 In contrast to this study, isolated NTDs from Mlh1 and Pms1, isolated CTDs from E. coli MutL, and Mlh1 in the absence of a partner MLH protein were shown to bind stably to DNA.21,25,36 One explanation for our results is that associations remain between cleaved Mlh1-Pms1 complexes that inhibit DNA binding through unknown mechanisms. In support of this, co-immunoprecipitation experiments performed on Mlh1(TEV)-Pms1 following TEV cleavage showed that the two resulting fragments of Mlh1 still interact with full-length Pms1, suggesting that the complex is not destroyed (Fig. 2b). These observations also support a role for a ring-like structure for Mlh1-Pms1 in MMR.12 Interactions between different domains of Mlh1 and Pms1 (e.g. as shown for the Mlh1 and Pms1 NTDs in the ATP hydrolysis cycle by Sacho et al.29) may be required for coordination of DNA binding.21,25 The fact that Mlh1-Pms1 has multiple DNA binding sites that map to both subunits might necessitate such coordination.21 Nonetheless, the finding that TEV cleavage of the linker arm of Mlh1 in yeast cells resulted in an elevated mutation rate implies that the DNA binding activity of the intact heterodimer is important for mismatch correction.
Our data, which show that the N- and C-terminal domains of TEV cleaved Mlh1 remain associated with Pms1 in the absence of DNA, are in contrast with observations obtained from Sacho et al. 29 who reported that in the absence of nucleotide cofactors such as ATP yeast Mlh1-Pms1 (yMutLalpha) is “predominately in an open and “v-shaped” extended conformation, in which a large compact central domain is connected to two smaller domains by flexible arms.” Based on their observations one would have not have expected stoichiometric recovery of the N- and C-terminal domains of TEV cleaved Mlh1 after immunoprecipitation with an antibody specific to Pms1. However, our data are consistent with work from Gorman et al.12 who found in single molecule studies that Mlh1–Pms1 has properties consistent with ring-like architecture in the absence of ATP. More specifically, Gorman et al.12 found that when hydrodynamic force was used to push Mlh1–Pms1, most complexes (>95%) did not dissociate upon encountering anchored ends, nor did Mlh1–Pms1 dissociate from the apex of looped DNA. In contrast, Mlh1–Pms1 immediately dissociated from free ends of ‘single-tethered’ DNA. At present we do not have a good explanation for the different observations obtained from the two studies.
Deletion analysis of the Mlh1 and Pms1 linker arms showed that the length of the Mlh1 arm was more critical for MMR function than the length of the Pms1 arm. Our findings for Pms1 are analogous to those seen when truncations were made in the linker arm of E. coli MutL.26 In that study they found that deletions up to one-third the size of the linker arm in MutL did not disrupt DNA binding activity or MMR function. The fact that the linker arm of Pms1 is twice the length of the Mlh1 linker arm may allow for larger truncations to be made in Pms1 without compromising its activity. Alternatively, there may be important residues along the Mlh1 linker arm that are critical for protein function. In support of this, an alanine-scanning mutagenesis screen identified a MMR defective allele, mlh1-31, that is mutated at residues R401 and D403 and overlaps with several of our non-functional deletion constructs.30 Interestingly, mlh1-31 was still able to associate with Pms1 and form a ternary complex with Msh2-Msh6 at a mismatch, suggesting it has a defect in downstream repair functions that are possibly associated with other protein-protein interactions. It is also worth mentioning that not all of our deletion constructs that conferred a null-phenotype removed these key residues, indicating the presence of additional important amino acids within the linker arms.
Expression analysis ruled out the possibility that null-phenotypes seen for the linker arm deletions constructs were a consequence of protein instability or due to a lack of protein expression. This finding allowed us to purify and test complexes for DNA binding activity and Msh2-Msh6 interactions. In contrast to our results for MMR function, it appears that the linker arm of Pms1 is more important for the DNA binding function of this complex than the Mlh1 arm. Consistent with this, deletion of the linker arm in Pms1 impaired the ability of Mlh1-pms1Δ390-610 to form a ternary complex with Msh2-Msh6 at a DNA mismatch but the corresponding linker arm deletion in Mlh1, mlh1-Δ336-480-Pms1, did not affect this interaction (Fig. 6). We attempted to purify a complex containing deletions in both linker arms but could not obtain high enough yields of protein.
Combinatorial analysis of intermediate linker arm deletion mutations showed that they conferred synergistic effects on mutation rates. These effects were only seen with the intermediate mlh1 allele (mlh1Δ348-373), whereas combinations with the wild-type functioning mlh1 allele (mlh1Δ445-47) displayed the individual phenotype of the pms1 allele tested. Our results indicate that combinations of weakened alleles result in significant defects in protein function. One possibility is that there are threshold linker arm sizes in both Mlh1 and Pms1 that are required for the complex to form a functional ring that can bind to DNA. Another possibility is that the ability of Mlh1-Pms1 to diffuse along DNA while searching for targets is compromised when the linker arms of either protein are truncated. In the future it will be important to directly test these hypotheses.
It is not surprising that deletion mutations in the linker arms of Mlh1 and Pms1 display differential effects on DNA binding and MMR. Liskay and colleagues37,38 observed differential requirements for the ATPase motifs of Mlh1 and Pms1 in MMR, and Hargreaves et al.39 found that Msh2-Msh6 interactions with Mlh1-Pms1 at a mismatch site requires ATP occupancy by only the Msh6 subunit. The observed asymmetries are likely to be important in promoting repair specificity at different stages in the MMR reaction. Also, Sacho et al.29 hypothesized that conformational changes involving the linker arms of Mlh1-Pms1 promote essential interactions with other MMR components. Mlh1-Pms1 interactions with PCNA and Exo1 are of particular interest because they are thought to be critical for stimulating and completing excision steps in MMR.40–42 Mlh1-Pms1 has been shown to harbor a latent endonuclease activity that is attributed to the C-terminal domain of Pms1.42 Activation of this activity is thought to be triggered by interactions with other MMR components to displace a regulatory subdomain that blocks access toDNA. 22 Thus understanding how Mlh1-Pms1 interacts with DNA through linker arms will likely provide important clues on how it interacts with downstream repair factors.
Materials and Methods
Strains and plasmids
Yeast strains (Table 1) were grown in yeast extract/peptone/dextrose (YPD), minimal complete, or minimal selective media.43 Plasmids used in this study are listed in Table 2. For PMS1 constructs the HA epitope was inserted after amino acid D565 and is shown as Pms1(HA565). For MLH1 constructs, the FLAG epitope was inserted after T448 or Y499 in Mlh1(FLAG448 or FLAG499). TEV protease cleavage sites were inserted into Mlh1 after T448 in Mlh1(FLAG499) and after amino acid 499 in Mlh1(FLAG448).12 Full details of plasmid and strain constructions are available upon request.
Table 1.
Strains used in this study
| K9872 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), MLH1 |
| EAY3098 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), mlh1Δ::KanMX4, lys2::insE-A14 |
| EAY3099 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), MLH1::KanMX4, lys2::insE-A14 |
| EAY3100 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), MLH1(FLAG448)::KanMX4, lys2::insE-A14 |
| EAY3101 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), MLH1(FLAG448, TEV499)::KanMX4, lys2::insE-A14 |
| EAY3102 | MATa, ura3, leu2-3, 112, omns GAL-NLS-myc9-TEV protease-NLS2::TRP1(10-fold integrant by southern), MLH1 (TEV448, FLAG499)::KanMX4, lys2::insE-A14 |
| EAY1269 | MATa, ura3-52, leu2Δ1, trp1Δ63, lys2::insE-A14 |
| EAY3097 | MATa, ura3-52, leu2Δ1, trp1Δ63, his3Δ200, lys2::insE-A14, pms1Δ::KanMX4 |
| EAY1366 | MATa, ura3-52, leu2Δ1, trp1Δ63, his3Δ200, lys2::insE-A14, mlh1Δ::KanMX4 |
| EAY1365 | MATa, ura3-52, leu2Δ1, trp1Δ63, his3Δ200, lys2::insE-A14, mlh1Δ::KanMX4, pms1Δ::KanMX4 |
| BJ2168 | MATa, ura3-52, leu2-3, 112, trp1-289, prb1-1122, prc1-407, pep4-3 |
Table 2.
Plasmids used in this study
| Plasmids | Relevant genotype | Vector type |
|---|---|---|
| pEAI160 | mlh1Δ::KanMX4 | Integration |
| pRS413 | ARS-CEN, HIS3 | |
| pRS415 | ARS-CEN, LEU2 | |
| pEAA213 | MLH1 | ARS-CEN, LEU2 |
| pEAA373 | MLH1 (FLAG448) | ARS-CEN, LEU2 |
| pEAA375 | MLH1 (FLAG499) | ARS-CEN, LEU2 |
| pEAA515 | MLH1 (FLAG448-TEV499) | ARS-CEN, LEU2 |
| pEAA516 | MLH1 (TEV448-FLAG499) | ARS-CEN, LEU2 |
| pEAA526 | mlh1Δ348-373 (FLAG499) | ARS-CEN, LEU2 |
| pEAA527 | mlh1Δ445-470 (FLAG499) | ARS-CEN, LEU2 |
| pEAA528 | mlh1Δ359-409 (FLAG499) | ARS-CEN, LEU2 |
| pEAA529 | mlh1Δ407-457 (FLAG499) | ARS-CEN, LEU2 |
| pEAA530 | mlh1Δ357-457 (FLAG499) | ARS-CEN, LEU2 |
| pEAA531 | mlh1Δ336-480 (FLAG499) | ARS-CEN, LEU2 |
| pEAA532 | mlh1Δ396-421 (FLAG499) | ARS-CEN, LEU2 |
| pEAA238 | PMS1 | ARS-CEN, HIS3 |
| pEAA517 | PMS1 (HA565) | ARS-CEN, HIS3 |
| pEAA544 | pms1Δ450-475 (HA565) | ARS-CEN, HIS3 |
| pEAA545 | pms1Δ600-625 (HA565) | ARS-CEN, HIS3 |
| pEAA546 | pms1Δ437-487 (HA565) | ARS-CEN, HIS3 |
| pEAA547 | pms1Δ511-561 (HA565) | ARS-CEN, HIS3 |
| pEAA548 | pms1Δ584-634 (HA565) | ARS-CEN, HIS3 |
| pEAA549 | pms1Δ450-550 (HA565) | ARS-CEN, HIS3 |
| pEAA550 | pms1Δ390-610 | ARS-CEN, HIS3 |
| pMH1 | GAL1-MLH1-VMA1-CBD | 2μ, TRP1 |
| pEAE269 | GAL1-MLH1 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE308 | GAL1-mlh1Δ348-373 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE309 | GAL1-mlh1Δ445-470 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE310 | GAL1-mlh1Δ359-409 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE311 | GAL1-mlh1Δ407-457 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE312 | GAL1-mlh1Δ357-457 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE313 | GAL1-mlh1Δ336-480 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pEAE314 | GAL1-mlh1Δ396-421 (FLAG499)-VMA1-CBD | 2μ, TRP1 |
| pMH8 | GAL10-PMS1 | 2μ, LEU2 |
| pEAE296 | GAL10-PMS1 (HA565) | 2μ, LEU2 |
| pEAE298 | GAL10-pms1Δ450-475 (HA565) | 2μ, LEU2 |
| pEAE299 | GAL10-pms1Δ600-625 (HA565) | 2μ, LEU2 |
| pEAE300 | GAL10-pms1Δ437-487 (HA565) | 2μ, LEU2 |
| pEAE301 | GAL10-pms1Δ511-561 (HA565) | 2μ, LEU2 |
| pEAE302 | GAL10-pms1Δ584-634 (HA565) | 2μ, LEU2 |
| pEAE303 | GAL10-pms1Δ450-550 (HA565) | 2μ, LEU2 |
| pEAE304 | GAL10-pms1Δ390-610 | 2μ, LEU2 |
Linker arm deletion series construction
Vectors were created to test each MLH1 and PMS1 linker arm deletion in complementation (ARS CEN) and over-expression (GAL1/10, 2μ) assays (Table 2). mlh1 linker arm deletion complementation vectors were derived from pEAA213 which expresses MLH1 from its native promoter.44 pms1 linker arm deletion complementation vectors were derivatives of the pEAA238, which expresses PMS1 from its native promoter.44 Expression vectors were derived from pMH1 (GAL1-MLH1-VMA-CBD, 2μ, TRP1) and pMH8 (GAL10-PMS1, 2μ, LEU2).45 Each deletion was constructed by overlap-extension PCR to remove the portion of the corresponding protein.46 DNA fragments containing the relevant linker arm deletion were inserted into pEAA213, pEAA238, pMH1, and pMH8 and confirmed by DNA sequencing (Cornell BioResource Center).
lys2::insE-A14 reversion assay
pEAA213 (MLH1), pEAA238 (PMS1) and derivative plasmids were transformed into EAY1366 (mlh1Δ, lys2::insE-A14) and EAY3097 (pms1Δ, lys2::insE-A14), respectively, using standard methods.47 Plasmids were maintained by growing strains in minimal selective (leucine dropout for pEAA213 and derivatives, histidine dropout for pEAA238 and derivatives) media. When tested in combination, pEAA213 and pEAA238 and derivatives were co-transformed into EAY1365 (mlh1Δ pms1Δ, lys2::insE-A14). TEV assays were preformed in strains EAY3098-EAY3102 maintained in minimal media containing either 4% sucrose as the sole carbon source or 2% sucrose + 2% galactose as carbon sources. Each strain was sequenced to confirm integrations and to verify lys2::insE-A14 integrity. Rates of lys2::insE-A14 reversion were calculated as μ = f/ln(N·μ), where f is reversion frequency and N is the total number of revertants in the culture.37 For each strain, 15–20 independent cultures, obtained from two to three independent transformants bearing a unique allele, were assayed to determine the mutation rate. 95% confidence intervals and all computer aided rate calculations were performed as previously described.37,48
Mlh1-Pms1 expression and purification
Mlh1–Pms1 was expressed and purified from six liters of galactose-induced cell cultures of S. cerevisiae BJ216845 containing pMH1 (GAL1-MLH1-VMA-CBD, 2μ, TRP1 ) and pMH8 (GAL10-PMS1, 2μ, LEU2 ). Mlh1–Pms1 linker arm deletion complexes were purified from BJ2168 containing the relevant pMH1 and pMH8 derivatives (Table 2 and Fig. 2a). Western blot analysis was performed on cell lysates collected after galactose induction (Fig. 3). Cells were pelleted, washed with chitin buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM EDTA), repelleted, and resuspended in SDS–protein loading buffer. 20 μg of each sample were loaded onto each lane of an 8% SDS–PAGE gel. After separating the proteins by electrophoresis, samples were transferred to nitrocellulose membrane. Membranes were blocked with 4% milk overnight and probed with a 1:2000 dilution of 12CA5 (αHA, Roche) or a 1:1000 dilution of M2 (αFLAG, Sigma) antibody, followed by incubation with a 1:5000 dilution of α-mouse IgG secondary antibody (Jackson Immunoresearch). Proteins were visualized by the ECL detection method (Amersham/GE).
Immunoprecipitation assays
12 μg of Mlh1-Pms1 was cut with 0.12 μg of TEV protease (gift from Ailong Ke), and 4 μl of anti-HA antibody (Roche) was added to each sample (±TEV cleavage) along with 285 μl of binding buffer (300 mM NaCl, 20 mM Tris pH 7.5, 1 mM EDTA, 10 mM β-mercaptoethanol, 1 mM PMSF, 0.1% NP-40, 1 mg per ml BSA). When indicated, DNAseI (2 units) and apyrase were preincubated with Mlh1-Pms1 in buffers recommended by the manufacturer (New England Biolabs). All subsequent steps were performed at 4°C. After a one-hour incubation on an oscillation rocker, 20 μl of protein A sepharose (GE Healthcare) suspended at 1:1 (v/v) in incubation buffer (200 mM NaCl, 50 mM Tris pH 7.5, 1 mM EDTA, 10 mM β-mercaptoethanol, 1 mM PMSF, 1% NP-40) was added to each reaction. Following a one-hour incubation on an oscillating rocker, samples were centrifuged at 3000 RPM for 20 seconds. The supernatant was removed and the protein A sepharose beads were washed three times with 200 μl per wash of primary wash buffer (300 mM NaCl, 50 mM Tris pH 7.5, 1 mM EDTA, 10 mM β-mercaptoethanol, 1 mM PMSF, 0.1% NP-40) followed by two washes with 200 μl per wash of secondary wash buffer (50 mM NaCl, 50 mM Tris pH 7.5, 1 mM EDTA, 10 mM β-mercaptoethanol, 1 mM PMSF, 0.1% NP-40). 16 μl of 3X SDS-loading buffer was added to each reaction and samples were boiled for 3 minutes, and analyzed by 10% SDS-PAGE and stained with Coomassie blue (Fig. 2b).
Electromobility shift assays
Protein concentrations were determined using the Bradford49 assay with BSA serving as a standard. EMSA with oligonucleotide substrates were performed as described.50 Briefly, Mlh1-Pms1 titrations were assembled on ice in 15 μl reactions containing 60 nM (5′-32P)-end labeled 40-bp homoduplex substrate, 25 mM Hepes pH 7.6, 40 μg/ml BSA, 1 mM DTT, 50 mM NaCl, and 8% Sucrose (w/v). Mlh1-Pms1 constructs (0–300 nM) were added last, followed by a 5 minute incubation at room temperature (RT). After incubation, samples were loaded on 4% (w/v) non-denaturing polyacrylamide gels containing 0.5X TBE and electrophoresed at 130 V for 1 hour at RT. Gels were dried on 3MM Whatman paper and visualized by PhosphorImaging. Kinetic analysis was done using ImageJ. The 40-bp substrate homoduplex substrate was formed by annealing S1 (5′ dACCGAATTCTGACTTGCTAGGACATCTTTGCCCACGTTGA) and S2 (5′ dTCAACGTGGGCAAAGATGTCCTAGCAAGTCAGAATTCGGT). The electromobility shift assays shown in Fig. 5 were performed ten times with the same trend for DNA binding seen in each assay. A representative assay is shown. For ternary complex assays (Fig. 6)50, 15 μl binding reactions were performed that contained 60 nM (5′-32P)-end labeled 40-bp +1 substrate, 25 mM Hepes pH 7.6, 40 μg/ml BSA, 1 mM DTT, 50 mM NaCl, 1 mM ATP, and 8% sucrose (w/v). Mlh1-Pms1 and mutant derivatives (100 nM) and Msh2-Msh6 (150 nM; purified as described in Alani 1996) were added last, followed by a 5 minute incubation at room temperature (RT). After incubation, samples were analyzed by EMSA as described above. The 40-bp (+1) mismatch substrate was created by annealing S6 (5′ dACCGAATTCTGACTTGCTAGAGACATCTTTGCCCACGTTGA) and S2 (Integrated DNA Technologies).51
Supplementary Material
Two repetitions (a, b) of EMSA assays are shown for the indicated Mlh complexes (see Materials and Methods and Fig. 5).
Highlights.
MLH proteins initiate mismatch repair of DNA polymerase errors by surveying the genome for lesion-bound MSH proteins.
MLH proteins act as dimers and contain unstructured linker arms that connect two terminal globular domains.
MMR is more sensitive to Mlh1 linker arm deletions.
Our observations are consistent with the unstructured linker domains of MLH proteins providing distinct interactions with DNA during MMR.
Acknowledgments
We are grateful to members of the Alani lab for helpful discussions, Alba Guarne for help determining the limits of the unstructured linker arm domains of Mlh1-Pms1, Kim Nasmyth for providing the TEV-integrated strain K9872, Pei Xin Lim for constructing EAY3097, and Alex Wang for conducting preliminary experiments. E.A. and M. V. R. were supported by NIH grant GM053085, and A. J. P. by a SUNY fellowship. E. C. G. was supported by HHMI, an NSF PECASE Award, and NIH grant GM082848. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Abbreviations
- MMR
mismatch repair
- MSH
MutS homolog
- MLH
MutL homolog
- PCNA
proliferating cell nuclear antigen
- NTD
N-terminal domain
- CTD
C-terminal domain
- IP
immunoprecipitation
- EMSA
electromobility shift assays
- TEV
Tobacco Etch Virus
- EDTA
ethylenediaminetetraacetic acid
- YPD
yeast extract/peptone/dextrose
- C. I
confidence interval
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 3.Habraken Y, Sung P, Prakash L, Prakash S. ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1- PMS1 protein complexes. J Biol Chem. 1998;273:9837–9841. doi: 10.1074/jbc.273.16.9837. [DOI] [PubMed] [Google Scholar]
- 4.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendillo ML, Hargreaves VV, Jamison JW, Mo AO, Li S, Putnam CD, Woods VL, Kolodner RD. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc Natl Acad Sci U S A. 2009;106:22223–22228. doi: 10.1073/pnas.0912250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275:36498–501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- 8.Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- 9.Lau PJ, Kolodner RD. Transfer of the MSH2.MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. J Biol Chem. 2003;278:14–17. doi: 10.1074/jbc.C200627200. [DOI] [PubMed] [Google Scholar]
- 10.Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011;147:1040–1053. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorman J, Plys AJ, Visnapuu ML, Alani E, Greene EC. Visualizing one- dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol. 2010;17:932–938. doi: 10.1038/nsmb.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acharya S, Foster PI, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 14.Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 15.Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 16.Gradia S, Subramanian D, Wilson T, Acharya S, Makhov A, Griffith J, Fishel R. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3:255–61. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 17.Gorman J, Chowdhury AA, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong C, Cho WK, Song KM, Cook C, Yoon TY, Ban C, Fishel R, Lee JB. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat Struct Mol Biol. 2011;18:379–385. doi: 10.1038/nsmb.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Hall MC, Wang H, Erie DA, Kunkel TA. High affinity cooperative DNA binding by the yeast Mlh1-Pms1 heterodimer. J Mol Biol. 2001;312:637–647. doi: 10.1006/jmbi.2001.4958. [DOI] [PubMed] [Google Scholar]
- 21.Hall MC, Shcherbakova PV, Fortune JM, Borchers CH, Dial JM, Tomer KB, Kunkel TA. DNA binding by yeast Mlh1 and Pms1: implications for DNA mismatch repair. Nucleic Acids Res. 2003;31:2025–2034. doi: 10.1093/nar/gkg324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, Modrich P, Walker GC, Simmons LA, Friedhoff P, Guarné A. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell. 2010;39:145–151. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 24.Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 25.Guarne A, Junop MS, Yang W. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. EMBO J. 2001;20:5521–5531. doi: 10.1093/emboj/20.19.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, Yang W. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 2004;23:4134–4145. doi: 10.1038/sj.emboj.7600412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosinski J, Steindorf I, Bujnicki JM, Giron-Monzon L, Friedhoff P. Analysis of the quaternary structure of the MutL C-terminal domain. J Mol Biol. 2005;351:895–909. doi: 10.1016/j.jmb.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 28.Dutta R, Inoye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 29.Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine- nucleotide-induced conformational changes in MutL alpha. Mol Cell. 2008;29:112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Argueso JL, Kijas AW, Sarin S, Heck J, Waase M, Alani E. Systematic mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol Cell Biol. 2003;23:873–886. doi: 10.1128/MCB.23.3.873-886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters J-M, Tedeschi A, Schmitz J. The cohesion complex and its roles in chromosome biology. Genes Dev. 2008;22:3089–3114. doi: 10.1101/gad.1724308. [DOI] [PubMed] [Google Scholar]
- 32.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 33.Kohler F. A yeast-based growth assay for the analysis of site-specific proteases. Nucl Acids Res. 2003;31:e16. doi: 10.1093/nar/gng016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–65. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habraken Y, Sung P, Prakash L, Prakash S. ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1- PMS1 protein complexes. J Biol Chem. 1998;273:9837–9841. doi: 10.1074/jbc.273.16.9837. [DOI] [PubMed] [Google Scholar]
- 36.Arana ME, Holmes SF, Fortune JM, Moon AF, Pedersen LC, Kunkel TA. Functional residues on the surface of the N-terminal domain of yeast Pms1. DNA Repair. 2010;9:448–457. doi: 10.1016/j.dnarep.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran PT, Liskay RM. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLalpha. Mol Cell Biol. 2000;20:6390–6398. doi: 10.1128/mcb.20.17.6390-6398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson JR, Erdeniz N, Nguyen M, Dudley S, Liskay RM. Conservation of functional asymmetry in the mammalian MutLα ATPase. DNA Repair. 2010;9:1209–1213. doi: 10.1016/j.dnarep.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hargreaves VV, Shell SS, Mazur DJ, Hess MT, Kolodner RD. Interaction between the Msh2 and Msh6 nucleotide-binding sites in the Saccharomyces cerevisiae Msh2-Msh6 complex. J Biol Chem. 2010;285:9301–9310. doi: 10.1074/jbc.M109.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran PT, Simon JA, Liskay RM. Interactions of Exo1p with components of MutLalpha in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2001;98:9760–9765. doi: 10.1073/pnas.161175998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SD, Alani E. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. J Mol Biol. 2006;355:175–184. doi: 10.1016/j.jmb.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 42.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 43.Rose MD, Winston F, Hieter P. Methods in yeast genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1990. [Google Scholar]
- 44.Heck JA, Argueso JL, Gemici Z, Reeves RG, Bernard A, Aquadro CF, Alani E. Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair. Proc Natl Acad Sci U S A. 2006;103:3256–3261. doi: 10.1073/pnas.0510998103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall MC, Kunkel TA. Purification of eukaryotic MutL homologs from Saccharomyces cerevisiae using self-affinity technology. Protein Expr Purif. 2001;21:333–342. doi: 10.1006/prep.2000.1379. [DOI] [PubMed] [Google Scholar]
- 46.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 47.Gietz RD, Schiestl RH. Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:35–37. doi: 10.1038/nprot.2007.14. [DOI] [PubMed] [Google Scholar]
- 48.Demogines A, Wong A, Aquadro C, Alani E. Incompatibilities involving yeast mismatch repair genes: a role for genetic modifiers and implications for disease penetrance and variation in genomic mutation rates. PLoS Genet. 2008;4:e1000103. doi: 10.1371/journal.pgen.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 50.Kijas AW, Studamire B, Alani E. Msh2 separation of function mutations confer defects in the initiation steps of mismatch repair. J Mol Biol. 2003;331:123–138. doi: 10.1016/s0022-2836(03)00694-6. [DOI] [PubMed] [Google Scholar]
- 51.Surtees JA, Alani E. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J Mol Biol. 2006;360:523–536. doi: 10.1016/j.jmb.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 52.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Two repetitions (a, b) of EMSA assays are shown for the indicated Mlh complexes (see Materials and Methods and Fig. 5).