

Abstract
Retinal bipolar cells are interneurons that transmit visual signals from photoreceptors to ganglion cells. Although the visual pathways mediated by bipolar cells have been well characterized, the genes that regulate their development and function are largely unknown. To determine the role in bipolar cell development of the homeobox gene Vsx1, whose retinal expression is restricted to a major subset of differentiating and mature cone bipolar (CB) cells, we targeted the gene in mice. Bipolar cell fate was not altered in the absence of Vsx1 function, because the pan-bipolar markers Chx10 and Ret-B1 continued to be expressed in inner nuclear layer neurons labeled by the Vsx1-targeting reporter gene, τLacZ. The specification, number, and gross morphology of the subset of on-center and off-center (OFF)-CB cells defined by τLacZ expression from the Vsx1 locus were also normal in Vsx1τLacZ/Vsx1τLacZ mice. However, the terminal differentiation of OFF-CB cells in the retina of Vsx1τLacZ/Vsx1τLacZ mice was incomplete, as demonstrated by a substantial reduction in the expression of at least four markers (recoverin, NK3R, Neto1, and CaB5) for these interneurons. These molecular abnormalities were associated with defects in retinal function and documented by electroretinography and in vitro ganglion cell recordings specific to cone visual signaling. In particular, there was a general reduction in the light-mediated activity of OFF, but not on-center, ganglion cells. Thus, Vsx1 is required for the late differentiation and function of OFF-CB cells and is associated with a heritable OFF visual pathway-specific retinal defect.
Bipolar cells are classified as either on-center (ON) or off-center (OFF), according to their signaling response to increments or decrements of light intensity (1–4). Bipolar cells are also designated as either rod or cone, on the basis of their synaptic input from rod or cone photoreceptors, respectively, although a subset of mouse OFF–cone bipolar (CB) cells has been recently shown to contact rod photoreceptors directly (5–7). Mammalian CB cells have either ON or OFF physiology and synapse with corresponding ON and OFF ganglion cells in the inner plexiform layer (IPL) (3, 4). In contrast, all rod bipolar (RB) cells are thought to display ON physiology and, for the most part, do not contact ganglion cells. Rather, RB signals are relayed to ganglion cells through AII amacrine cell-mediated contacts between RB cells and both ON- and OFF-CB cells (4). This organization allows a single cohort of ganglion cells to subserve both rod and cone vision. Despite their name, therefore, CB cells are essential for the transmission of both rod- and cone-generated visual signals in mammals.
We have previously shown that the retinal expression of the homeobox gene, Vsx1, is restricted to differentiating and mature CB cells (8). Vsx1 is a paired-like homeo- and CVC-domain transcription factor (8–12) whose homologues, including ceh-10 in nematodes (13) and Chx10 in mammals (14, 15), are essential for sensory interneuron specification or maintenance. Because the retinal expression of Vsx1 is first detected in the mouse at postnatal day 5 in presumptive CB cells, we have suggested that this gene may be required for the differentiation as well as the maintenance of these interneurons (8). The molecules that regulate bipolar cell differentiation are largely unknown; the few molecules whose function in bipolar cell development has been determined (14, 16) or examined (17), appear to mediate bipolar cell specification and/or maintenance.
A requirement for the human VSX1 gene in visual signaling has been suggested by the presence of mild subclinical electroretinography (ERG) b/a-wave ratio defects in subjects carrying dominant VSX1 missense mutations (18), but the cellular and physiological basis of these defects is unknown. These ERG abnormalities may result from inner retinal defects, consistent with the expression of the human and mouse VSX1/Vsx1 gene in CB cells (8, 10). To determine the role of Vsx1 in CB development and activity, we generated mice carrying Vsx1 null alleles and examined the biochemical, cellular, and physiological consequences of the loss of Vsx1 function.
Materials and Methods
Immunostaining. Immunostaining of mouse retinal sections was performed as described (8). A table of antibodies and modifications of the immunostaining protocol is presented in Supporting Text, which is published as supporting information on the PNAS web site.
Retinal Electrophysiology. Full-field ERGs were obtained in a custom Ganzfeld dome by using techniques described previously (19). A flattened intact retinal–scleral preparation was used for ganglion cell recording experiments following previously described detailed descriptions (20) (descriptions for both techniques are presented in Supporting Text).
Results
We used two gene-targeting strategies to generate mice with Vsx1-null alleles, Δ1–4 and τLacZ (see Fig. 7, which is published as supporting information on the PNAS web site, and Methods in Supporting Text). Importantly, both homozygous mutants were complete Vsx1 protein nulls (see Fig. 7). The heterozygotes and homozygotes were viable and had no overt behavioral or neurological defects (data not shown). In particular, in contrast to human patients with missense mutations in VSX1 (18), no corneal abnormalities were observed either grossly or at the histological level (see Fig. 8, which is published as supporting information on the PNAS web site). Because no corneal expression of Vsx1/VSX1 has been observed (refs. 8 and 18 and data not shown), the pathogenesis of the VSX1-associated human corneal dystrophies is unclear.
To determine whether the formation of CB cells was disrupted by the loss of Vsx1 function, β-galactosidase (β-gal) expression was examined in Vsx1-τLacZ mice. β-gal immunofluorescence in the retina of adult mice heterozygous (Fig. 1 C–G, I) or homozygous (Fig. 1B) for the τLacZ allele was restricted to a subset of cells in the outer tier of the inner nuclear layer, where CB cells normally reside. This expression is consistent with the immunohistochemical localization of the Vsx1 protein (8), which we estimate to be present in 60–70% of all CB cells (R.L.C. and R.R.M., unpublished data). In both heterozygotes and homozygotes, axonal labeling revealed cells with projections terminating in the IPL, in locations that have been shown to be characteristic of OFF- and ON-CB cells (1–3) [Fig. 1B (arrow and arrowhead, respectively), and D–G]. Moreover, in the Vsx1τLacZ/+ retina, OFF-CB β-gal expression was detected in at least two morphologically distinct cell types similar to those recently described in the mouse (6) (Fig. 1 D–G).
Fig. 1.

CB-like cells are specified in the Vsx1-null retina. Representative confocal micrographs of β-gal staining on frozen retinal sections of 1-yr-old littermates: Vsx1+/+ (A), Vsx1τLacZ/Vsx1τLacZ (B), and Vsx1τLacZ/+ (C). PR, photoreceptors; INL, inner nuclear layer; GC, ganglion cells. β-Gal staining in the IPL of the Vsx1τLacZ/Vsx1τLacZ retina was typical of the pattern normally seen for OFF (B, arrow) and ON (B, arrowhead) CB cells. In 5-mo-old Vsx1τLacZ/+ mice, β-gal-positive cells with both ON-CB morphology (D, arrowhead) and OFF-CB morphology (D–G, arrow) were present (in D, a and b indicate sublaminar regions bounded by dashed lines). Two types of OFF-CB types were observed (D–G): one type with a slightly longer axon (solid lines indicate axon length in E vs. F) and a less elaborate arbor (D and E, arrow) similar to the B2 OFF-CB cell (6), and a second type (F and G, arrow) with a shorter axon and a more elaborate arbor, similar to the B1 OFF-CB cell previously described in the mouse retina (6). β-Gal and Vsx1 staining colocalized in the Vsx1τLacZ/+ retina (H–J, arrows), although not all Vsx1-positive cells were colabeled with β-gal (H–J, arrowheads). The number of Chx10-positive cells (expressed as a percentage of 4′,6-diamidino-2-phenylindole-stained nuclei in the INL) was not altered in the Vsx1 heterozygotes (+/-)or Vsx1 homozygotes (-/-) (L); three sets of littermates (5–12 mo old) were examined (two with the τLacZ allele and one with the Δ1–4 allele). (M) The number of β-gal-positive cells in the mature (5–12 mo old) Vsx1τLacZ/+ retina (+/-, yellow bar) was significantly different (indicated by*, Student's t test, P < 0.05) from the number of Vsx1-positive cells (green bars) in the Vsx1 wild-type (+/+) and Vsx1τLacZ/+ (+/-) retina and from the number of β-gal-positive cells in the Vsx-τLacZ null (-/-) retina (yellow bar); the number above the bars is the total of Chx10-positive cells in the INL examined. β-Gal- and Vsx1-positive cells in M are expressed as a percentage of Chx10-positive cells. Each bar in M represents data from three mice except for the last bar (-/-), which is derived from four Vsx1τLacZ/Vsx1τLacZ mice and one Vsx1Δ1–4/Vsx1τLacZ mouse (the Vsx1Δ1–4/Vsx1τLacZ value is indicated on the bar by an arrowhead). [Bar = 85 μm(A–C); 70 μm(D–G); 61 μm(H–J); 75 μm(K).]
To confirm that the β-gal-positive CB-like cells in the Vsx1τLacZ/Vsx1τLacZ retina were bipolar cells, we examined the expression of the pan-bipolar markers Chx10 (14) and Ret-B1 (21). All β-gal-positive cells in the Vsx1τLacZ/Vsx1τLacZ retina expressed both pan-bipolar markers (Fig. 2 A and B), indicating that the β-gal-positive cells were indeed bipolar interneurons. Notably, the β-gal-positive cells in the Vsx1-null retina did not express the RB marker PKCα (22), indicating that they had not adopted a RB cell fate (Fig. 2C). The overall number of bipolar cells was also unchanged in the Vsx1-null mutants (Fig. 1L; see Methods in Supporting Text), and the number of putative CB cells labeled by β-gal in the Vsx1τLacZ/Vsx1τLacZ retina was comparable to the number of Vsx1-immunostained cells in the Vsx1+/+ and Vsx1τLacZ/+ retina (Fig. 1M). Altogether, the above findings indicate that the specification, gross morphology, and viability of at least the great majority of ON- and OFF-CB cells do not require Vsx1 function.
Fig. 2.
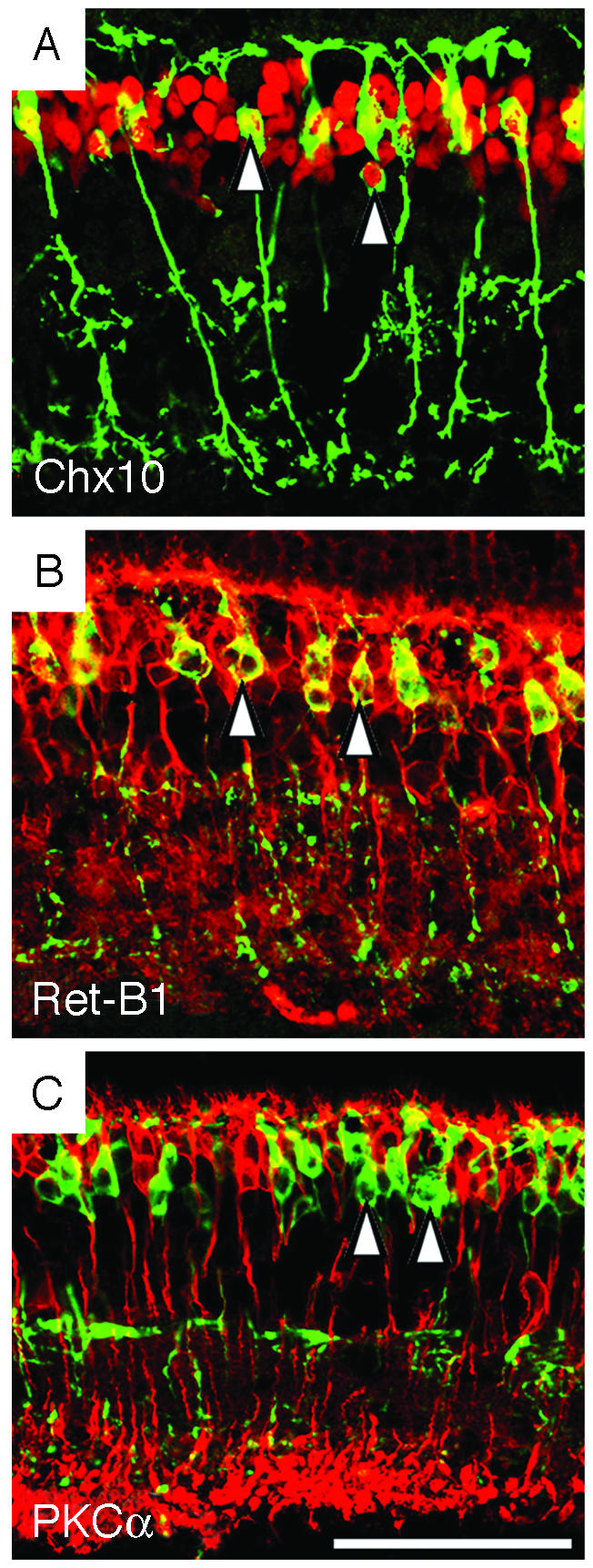
Cells lacking Vsx1 function express pan-bipolar cell markers. In the Vsx1τLacZ/Vsx1τLacZ retina of 5-mo-old mice, β-gal-labeled cells (green) colocalized with a subset of cells (red) expressing two pan-bipolar markers, Chx10 (A) and Ret-B1 (B) (Chx10 and Ret-B1 colocalized with β-gal in the cell body region indicated by arrowheads in A and B). β-Gal staining did not colocalize with the RB marker PKCα in the Vsx1τLacZ/Vsx1τLacZ retina (C, arrowheads indicate two of the β-gal-positive cells). (Bar = 50 μm.)
Although the number of Vsx1-immunostained nuclei in the heterozygous Vsx1τLacZ/+ retina was comparable to that of wild-type littermates (Fig. 1M), we observed, unexpectedly, that the number of cells that expressed β-gal in the heterozygous Vsx1τLacZ/+ retina was substantially lower than in the homozygous mutant (Fig. 1, compare C to B; M; see Fig. 9, which is published as supporting information on the PNAS web site). This reduction in the number of β-gal-staining cells in Vsx1τLacZ/+ heterozygotes was not due to the possibility that the expression of Vsx1 in mammalian retina is monoallelic (i.e., from either the wild-type Vsx1 allele alone or from the Vsx1τLacZ allele alone, but not both), because single cells expressing both Vsx1 and β-gal were evident (Fig. 1 H–J, arrows). Furthermore, the reduced number of β-gal staining cells was not due to the Vsx1τLacZ/+ heterozygotes having only a single copy of the τLacZ allele vs. two in the homozygous mutants, because the number of β-gal-staining cells in the retinas of Vsx1Δ1–4/Vsx1τLacZ compound null mice was comparable to the homozygous Vsx1τLacZ/Vsx1τLacZ retina (Fig. 1 K and M). The reduced β-gal staining in the heterozygous Vsx1τLacZ/+ retina therefore suggests that the retinal expression of Vsx1 is regulated in a Vsx1-dependent manner.
To establish whether Vsx1 regulates, directly or indirectly, the expression of proteins characteristic of CB cells, we examined the expression of five CB cell markers in the Vsx1-null retina. In the mutant retina, the expression of three OFF-CB markers was greatly reduced: recoverin (Fig. 3 A and B) (23, 24), NK3R (Fig. 3 C and D) (24, 25), and Neto1 (Fig. 3 E and F and Fig. 10, which is published as supporting information on the PNAS web site) (ref. 26; D.N., R.K.S., and R.R.M., unpublished data; see Methods in Supporting Text). Although the expression of these markers was completely extinguished in the majority of cells in the Vsx1-null retina, their expression persisted, to varying degrees, in a very small subset of β-gal-expressing cells (Fig. 11, which is published as supporting information on the PNAS web site). These findings establish that in the great majority of OFF-CB cells, the expression of recoverin, NK3R, and Neto1 is entirely regulated, directly or indirectly, by Vsx1: in a small minority of OFF-CB cells, the expression of these three proteins is only partially regulated by Vsx1. Additional evidence of the molecular heterogeneity of OFF-CB cells (24) was apparent from the observation that a variable number of NK3R- and CaB5-positive cells did not express β-gal (Fig. 11 B and D).
Fig. 3.

Vsx1 is essential for late CB differentiation. Confocal micrographs of retinal expression of CB markers in 1-yr-(A–D) and 5-mo-old (E–L) littermates. Recoverin expression in OFF-CB cells is absent from the Vsx1τLacZ/Vsx1τLacZ retina (B) compared to the wild-type Vsx1+/+ retina (A), whereas photoreceptor recoverin staining (A and B, staining at top) is unchanged. Arrow in B indicates a single weakly labeled recoverin-positive cell. NK3R CB staining is greatly reduced in the Vsx1τLacZ/Vsx1τLacZ retina (D) compared to the Vsx1+/+ retina (C). In the Vsx1+/+ retina, punctate Neto1 staining is observed in both the outer plexiform layer (E, arrowheads) and IPL (E, between dotted lines) of the Vsx1+/+ retina. In contrast to the Vsx1+/+ retina, Neto1 immunofluorescence is greatly reduced in both the outer plexiform layer of the Vsx1τLacZ/Vsx1τLacZ retina (F, arrow indicates a single patch of Neto1-positive staining in this layer) and the IPL of the Vsx1τLacZ/Vsx1τLacZ retina (F, the bracketed region indicates an isolated region of faint Neto1 staining). CaB5 staining (G–J) was moderately diminished in the Vsx1Δ1–4/Vsx1Δ1–4 retina in the OFF-CB axon terminal region (G–J, bracketed region) but was normal in the ON-CB axon terminal region (H and J, arrowhead) and RB axonal terminal regions (H and J, arrow). The area between the ON-CB axonal terminal region and the RB axonal terminal region that is normally free of CaB5 staining was absent in the Vsx1Δ1–4/Vsx1Δ1–4 retina (H and J, compare region between dashed lines on left). PMCA1 staining (K and L) was not diminished in the Vsx1Δ1–4/Vsx1Δ1–4 retina in the OFF-CB axon terminal region (K and L, arrow) or in the ON-CB axon terminal region (K and L, arrowhead). Frozen sections, A–D; paraffin sections, E–L. [Bar = 50 μm(A–D, K, and L); 40 μm(E and F); 95 μm(G and I); 34 μm(H and J).
We also observed that the expression of a fourth marker, the ON- and OFF-CB protein CaB5 (Fig. 3 G–J) (24), was moderately reduced in the OFF-CB cell population of Vsx1-null mice. However, the reduction in expression was apparent only in retinal sublamina a (Fig. 3 G–J, bracketed regions), a region of the IPL occupied by the axonal termini of OFF-CB cells (1–3). This reduction in CaB5 staining was associated with a severe reduction of the well-defined unstained region that normally separates the CaB5-immunostained axonal termini of RB and ON-CB cells (24) (Fig. 3 H and J; compare region between dashed lines on the left).
To ascertain whether the abnormalities in CaB5-staining reflect the absence of the axonal termini of OFF-CB cells or gross disruptions in their structure or organization, we also examined the structure of these axons by staining for PMCA1, an OFF-CB marker that normally colocalizes with CaB5 (24). The overall expression of PMCA1 and distribution of the axonal termini expressing PMCA1 were both normal (Fig. 3 K and L).
To determine whether the molecular abnormalities in the CB cells of Vsx1-mutant mice were accompanied by changes in retinal function, ERG experiments were performed (Fig. 4); the brief (20-ms) flashes used in these experiments merge the ON and OFF components of the ERG into a single b-wave (27). All of the major rod responses (Fig. 4) were comparable between wild-type and the Vsx1-mutant animals. The only alteration observed in Vsx1-mutant mice was a selective loss of b-wave amplitude, in response to white flashes eliciting a mixed rodand-cone response (Fig. 4 B and D). Furthermore, and consistent with a Vsx1-gene dosage effect, b-wave amplitudes from heterozygous mice were observed to lie between wild-type and Vsx1-null values (Fig. 4D). Although the cone b-wave amplitude from Vsx1-null mice was reduced below that of wild-type mice (Fig. 4C), this reduction did not reach statistical significance due to small and variable cone a-wave amplitudes among mice, which precluded use of the same type of b/a-wave normalization used with mixed rod–cone responses. These ERG studies demonstrate a selective effect of Vsx1 loss-of-function on the ERG b-wave and suggest that the mutant mice have a cone-dependent abnormality in bipolar interneuron function.
Fig. 4.

b-wave defects in the electroretinograms of Vsx1 mutants. (A) Representative rod full-field ERGs from a Vsx1-null mouse (solid lines). Shown are responses from a Vsx1-null mouse to a series of retinal illuminances spanning over 7 log units at ≈0.6 log unit intervals. Responses to high-energy flashes used for a-wave modeling are shown as broken curves. (Inset) Expanded view of a-waves to high-energy flashes. Solid lines show Vsx1-null responses, and dashed lines show best fit of the Lamb and Pugh model (37). (B) Relative loss of b-wave response in mixed rod-and-cone-mediated response in Vsx1-null mice. Each trace was derived from six mice, except for Vsx1+/+, which was based on five. Responses to 20-ms white flashes were normalized so that a-wave amplitudes are comparable. The double-headed arrow between the lines indicates the amplitude difference between Vsx1+/+ (upper line) and Vsx1-/- mice (lower line). (C) Rod a/b-wave amplitudes in Vsx1+/- and Vsx1-/- mice were comparable to wild-type mice. Cone a-waves are too small to measure in mice, but cone b-wave amplitudes were not significantly different among Vsx1+/+, Vsx1+/-, and Vsx1-/- animals. (D)b/a-wave amplitude ratios were comparable for the rod response in Vsx1+/+, Vsx1+/-, and Vsx1-/- mice. The b/a-wave amplitude ratio of the mixed rod-and-cone-mediated response was significantly smaller in Vsx1-/- mice. Consistent with a Vsx1-gene dosage effect, b/a-wave amplitude ratios from Vsx1+/- mice were observed to lie between Vsx1+/+ and Vsx-/- values. The differences among Vsx1+/+ (n = 6 mice), Vsx1+/- (n = 6; combined results from three Vsx1τLacZ/Vsx1τLacZ and three Vsx1Δ1–4/Vsx1Δ1–4 mice), and Vsx1-/- groups (n = 6; combined results from three Vsx1τLacZ/Vsx1τLacZ and three Vsx1Δ1–4/Vsx1Δ1–4 mice) were significant [ANOVA, F (2, 15) = 5.07; 0 = 0.02].
Because the reduced b-wave in Vsx1 mutant mice suggested a disruption of bipolar interneuron function, we asked whether the output of visual signals from the retina was affected by the loss of Vsx1 activity. Retinal output was assessed by performing single-unit recordings from ganglion cells in an in vitro intact retina-eyecup preparation (28) (Fig. 5). We were able to categorize ganglion cells of the Vsx1-null retina into the same physiological groups previously reported (20) for wild-type mouse retina, suggesting that the full cohort of ganglion cell types (B.V. and S.A.B., unpublished work) remained responsive in the absence of Vsx1 activity (data not shown). In addition, ganglion cell threshold responses in the Vsx1-null retina revealed no changes in the thresholds of rod-and-cone photoreceptors (Fig. 5A). Whereas ON ganglion cells in the Vsx1-null retina displayed spike activity indistinguishable from that in wild-type retinas (t test, P > 0.1 for all intensity levels), there was a clear abnormality in OFF ganglion cells (Fig. 5 C and D). The spike activity of OFF ganglion cells in Vsx1-null and wild-type retinas were identical at scotopic intensity levels (i.e., with rod signaling only), but the same cells in the Vsx1-null retina showed a significant reduction in spike activity at intensity levels above cone threshold. These results demonstrate a specific requirement for Vsx1 activity in OFF visual signaling and are consistent with the ERG defects in the Vsx1 mutants, which suggested an impairment in the transmission of cone-generated signals.
Fig. 5.

OFF ganglion cell response defects in the Vsx1-null retina. (A) Histogram showing response thresholds of high- and low-sensitivity ganglion cells in the Vsx1-null retina, which provide an index of rod-and-cone photoreceptor thresholds, respectively. The thresholds in the Vsx1-nulls (asterisks) matched those reported previously in the wild-type mouse (20). (B) Representative extracellular recordings from ON and OFF ganglion cells in the Vsx1-/- and Vsx1+/+ mouse retinas to three stimulus intensities. Because cross referencing of different ganglion cell types was not performed in this study, a direct comparison of the responses between Vsx-/- and Vsx1+/+ ganglion cells cannot be made. The OFF ganglion cell response to higher light intensities showed reduced spike frequency in the Vsx1-null mutant compared to wild-type, whereas the responses of ON ganglion cells in the wild-type and Vsx1-/- retinas were similar. Bars at bottom of trace indicate onset and offset of the light stimulus. (Bars = 100 μV/500 ms.) (C) Scatter plot showing the average light-evoked spike count to full-field light stimulation over a 6-log unit range. There was no difference between the responses of ON cells in the Vsx1-/- and Vsx1+/+ mice; t test, P > 0.1 for all light intensities. (D) Scatter plot comparing the light-evoked spike count of OFF ganglion cells in Vsx1-/- and Vsx1+/+ mice. There was no difference between wild-type and Vsx1-null mice in the responses of the ganglion cells to low (scotopic) stimulus intensities, but the responses of ganglion cells in Vsx1-null mice were markedly reduced to stimulus intensities above the cone threshold; t test, P < 0.05 for intensities above 100 Rh*/rod per second (Rh*/rod, time-average rate of photoisomerizations per rod). Data points indicate averages and standard errors of 48 ON/37 OFF ganglion cell recordings from Vsx1-null retinas and 48 ON/24 OFF ganglion cell recordings from wild-type retinas.
Discussion
We have identified essential roles for Vsx1 in late stages of CB cell differentiation and visual signaling. In mice lacking Vsx1 function, a selective impairment of late differentiation was evident from substantial reductions in the expression of at least four OFF-CB cell proteins (recoverin, NK3R, Neto1, and CaB5). The normalcy of PMCA1 immunostaining, however, demonstrates that Vsx1 is not essential for the expression of all OFF-CB proteins. Furthermore, the absence of Vsx1 function had no noticeable effect on ON or OFF rod signaling or on cone signaling in the ON pathway. Rather, Vsx1 was critical only for the function of the OFF visual pathway and specifically for its ability to transmit cone visual signals. A model of the role of Vsx1 function in retinal bipolar interneuron development is presented in Fig. 6.
Fig. 6.

A model of the role of Vsx1 function in retinal bipolar interneuron development. We have established an essential role for Vsx1 function in the late differentiation of OFF-CB cells. Analysis of reporter β-gal staining in the Vsx1τLacZ/Vsx1τLacZ retina indicated that the number, specification, and gross morphology of OFF-CB cells is unaffected by the absence of Vsx1 function. Early roles in pan-bipolar specification and differentiation have previously been established for the Vsx1 homologue Chx10 (14, 15) and the basic helix–loop–helix transcription factor genes Mash1 and Math3 (16). The dashed line and question mark indicate it is unclear whether bipolar cells are first specified as a homogeneous group, which is then further specified into the various bipolar types, or whether each bipolar type is specified independently and directly from a retinal progenitor. Asterisks indicate that multiple OFF- and ON-CB cell types are present: at least three OFF-CB types and one ON type have been identified in mouse (6, 24).
Although a number of genetic abnormalities of the ON visual pathway have been reported (20, 29–34), the effect of the loss of Vsx1 function reported here is the initial description of a heritable OFF visual pathway-specific abnormality. Our findings suggest that the mild subclinical reduction in the ERG b/a-wave ratio observed in humans with dominant VSX1 missense mutations (18) is the result of OFF-CB cell dysfunction that reflects the incomplete terminal differentiation of these cells. Further characterization of vision defects in Vsx1-/- mice, by behavioral and/or visual evoked potential studies, will allow firmer definition of the retinal phenotype likely to be present in humans with a complete loss of VSX1 function.
The molecular basis of the defect in OFF-CB signaling in the Vsx1-null retina remain to be identified, but our studies provide substantial insight into the role of Vsx1 in the retina. First, the photopic light-evoked activity in OFF ganglion cells suggests there is diminished excitatory drive from OFF-CB cells in the mutant mice. However, the scotopic OFF ganglion cell responses were normal. Because the scotopic OFF responses are mediated through the same OFF-CB cells as photopic light-evoked responses (1–3), the reduced OFF-CB signaling is not likely to be due to a defect in cell structure or in signal transmission, such as voltage-regulated transmitter release from the axonal termini of OFF-CB cells to ganglion cells. Furthermore, the gross structure and the axonal projections of OFF-CB cells appeared normal in Vsx1-null mice, as shown by both β-gal and PMCA1 immunostaining (although we cannot rule out the possibility that a small fraction of OFF-CB cells is missing in the Vsx1-null retina or that the loss of Vsx1 function results in subtle OFF-CB axonal arborization defects). Consequently, the OFF ganglion cell abnormality would appear to arise from Vsx1-dependent changes in OFF-CB cells at sites proximal to their axonal termini.
Second, although central roles for yet-unidentified Vsx1-dependent proteins are fully possible, the OFF-CB signaling defects may arise from the decreased expression of recoverin, NK3R, Neto1, or CaB5, because the biochemical functions of these proteins in bipolar cells are poorly defined. Whether the abnormalities in CaB5 staining reflect reduced CaB5 production in OFF-CB cells or result from abnormal localization of the CaB5 protein in the axonal termini of OFF-CB cells is unclear. Because the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptors GluR1 and -2 and the kainate glutamate receptor GluR5 are present in putative OFF-CB cells at the flat contacts of cone synaptic terminals (24, 35), we examined the expression of these proteins by immunostaining but observed no abnormalities (data not shown).
Third, in contrast to the abnormalities in photopic OFF responses observed in Vsx1-null mice, scotopic OFF responses were normal, indicating that Vsx1 activity and the expression of recoverin, NK3R, Neto1, and CaB5 are not critical for the major known rod OFF signaling pathways (4, 6, 7, 36). Because Vsx1 is not expressed in all CB cells, however, and because no markers that label all OFF-CB cells are currently known, it is not possible at present to determine whether Vsx1 expression identifies all OFF-CB cells, or only a subset. Consequently, the absence of any detectable rod-signaling defect in the OFF pathway of Vsx1-null mice may be due to compensation by a population of non-Vsx1-expressing OFF-CB cells.
Fourth, our findings suggest that ON-CB function is either not critically dependent on or is independent of Vsx1 function. Thus, ON ganglion cell responses were normal in Vsx1-null retinas stimulated in the scotopic, mesopic, or photopic light ranges. Moreover, we identified only subtle abnormalities in the putative ON-CB axonal termini region of the mutant mice by using CaB5 immunolabeling. The current lack of specific markers for ON-CB cells (24) prevented us from determining, at a molecular level, whether ON-CB cell differentiation was also disrupted in Vsx1 mutants. Alternatively, some ON-CB cells may have molecular defects, and the apparent normalcy of ON-CB signaling may arise from compensatory responses in a population of ON-CB cells that do not normally express Vsx1.
The presence of CB cell defects in Vsx1-null mice provides further evidence of the strong conserved role of CVC-domain transcription factors in sensory interneuron development that has been documented throughout higher eukaryotes (13, 14). In contrast to the importance of Chx10 in the development of all retinal bipolar cells (14, 15), however, Vsx1 appears to be required solely for the late differentiation of OFF-CB cells (Fig. 6). The normal expression of PMCA1 in OFF-CB cells of Vsx1-null mice demonstrates that Vsx1 is essential for the expression of only a subset of mature OFF-CB cell proteins. Furthermore, the continued expression of three OFF-CB cell markers in a very small subset of OFF-CB cells in Vsx1-null mice demonstrates that, at least in that subset, molecules other than Vsx1 regulate CB gene expression.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Danka Vidgen. We thank K. Locke, J. Panakos, M. Borden, Y. Tsukamoto, S. Wu, C. C. Hui, and J. Horsford for assistance; and A. Dizhoor (Pennsylvania College of Optometry, Elkins Park, PA), Y. Q. Ding (Washington University, St. Louis, MO), K. Palczewsk (University of Washington, Seattle), C. Barnstable (Yale University, New Haven, CT), and R. Bremner (Toronto Western Research Institute, Toronto) for antibodies. This work was supported by grants from the Canadian Institutes of Health Research and the Canadian Genetic Diseases Network (to R.R.M.), from the Canadian Institutes of Health Research and Genome Canada (to C.M.), from National Institutes of Health (Grants EY07360 and EY05235, to S.A.B. and D.G.B., respectively), and an unrestricted grant from Research to Prevent Blindness, Inc., to the Department of Ophthalmology, New York University School of Medicine (to S.A.B.). R.K.S. was supported by a Postdoctoral Fellowship from the Alberta Heritage Foundation for Medical Research, and R.L.C. was supported by a Senior Postdoctoral Fellowship from the Canadian Institutes of Health Research/Estate of Betty Irene West.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ON, on-center; OFF, off-center; CB, cone bipolar; IPL, inner plexiform layer; RB, rod bipolar; ERG, electroretinography; β-gal, β-galactosidase.
References
- 1.Rodieck, R. W. (1973) The Vertebrate Retina: Principles of Structure and Function (Freeman, San Francisco).
- 2.Famiglietti, E. V., Jr., Kaneko, A. & Tachibana, M. (1977) Science 198, 1267-1269. [DOI] [PubMed] [Google Scholar]
- 3.Wassle, H. & Boycott, B. B. (1991) Physiol. Rev. 71, 447-480. [DOI] [PubMed] [Google Scholar]
- 4.Sharpe, L. T. & Stockman, A. (1999) Trends Neurosci. 22, 497-504.10529817 [Google Scholar]
- 5.Soucy, E., Wang, Y., Nirenberg, S., Nathans, J. & Meister, M. (1998) Neuron 21, 481-493. [DOI] [PubMed] [Google Scholar]
- 6.Tsukamoto, Y., Morigiwa, K., Ueda, M. & Sterling, P. (2001) J. Neurosci. 21, 8616-8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hack, I., Peichl, L. & Brandstatter, J. H. (1999) Proc. Natl. Acad. Sci. USA 96, 14130-14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow, R. L., Snow, B., Novak, J., Looser, J., Freund, C., Vidgen, D., Ploder, L. & McInnes, R. R. (2001) Mech. Dev. 109, 315-322. [DOI] [PubMed] [Google Scholar]
- 9.Levine, E. M., Hitchcock, P. F., Glasgow, E. & Schechter, N. (1994) J. Comp. Neurol. 348, 596-606. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi, T., Huang, J. & Deeb, S. S. (2000) Genomics 67, 128-139. [DOI] [PubMed] [Google Scholar]
- 11.Chen, C. M. & Cepko, C. L. (2000) Mech. Dev. 90, 293-297. [DOI] [PubMed] [Google Scholar]
- 12.Ohtoshi, A., Justice, M. J. & Behringer, R. R. (2001) Biochem. Biophys. Res. Commun. 286, 133-140. [DOI] [PubMed] [Google Scholar]
- 13.Altun-Gultekin, Z., Andachi, Y., Tsalik, E. L., Pilgrim, D., Kohara, Y. & Hobert, O. (2001) Development (Cambridge, U.K.) 128, 1951-1969. [DOI] [PubMed] [Google Scholar]
- 14.Burmeister, M., Novak, J., Liang, M. Y., Basu, S., Ploder, L., Hawes, N. L., Vidgen, D., Hoover, F., Goldman, D., Kalnins, V. I., et al. (1996) Nat. Genet. 12, 376-384. [DOI] [PubMed] [Google Scholar]
- 15.Green, E. S., Stubbs, J. L. & Levine, E. M. (2003) Development (Cambridge, U.K.) 130, 539-552. [DOI] [PubMed] [Google Scholar]
- 16.Hatakeyama, J., Tomita, K., Inoue, T. & Kageyama, R. (2001) Development (Cambridge, U.K.) 128, 1313-1322. [DOI] [PubMed] [Google Scholar]
- 17.Viczian, A. S., Vignali, R., Zuber, M. E., Barsacchi, G. & Harris, W. A. (2003) Development (Cambridge, U.K.) 130, 1281-1294. [DOI] [PubMed] [Google Scholar]
- 18.Heon, E., Greenberg, A., Kopp, K. K., Rootman, D., Vincent, A. L., Billingsley, G., Priston, M., Dorval, K. M., Chow, R. L., McInnes, R. R., et al. (2002) Hum. Mol. Genet. 11, 1029-1036. [DOI] [PubMed] [Google Scholar]
- 19.Clarke, G., Goldberg, A. F., Vidgen, D., Collins, L., Ploder, L., Schwarz, L., Molday, L. L., Rossant, J., Szel, A., Molday, R. S., et al. (2000) Nat. Genet. 25, 67-73. [DOI] [PubMed] [Google Scholar]
- 20.Deans, M. R., Volgyi, B., Goodenough, D. A., Bloomfield, S. A. & Paul, D. L. (2002) Neuron 36, 703-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnstable, C. J., Akagawa, K., Hofstein, R. & Horn, J. P. (1983) Cold Spring Harbor Symp. Quant. Biol. 2, 863-876. [DOI] [PubMed] [Google Scholar]
- 22.Negishi, K., Kato, S. & Teranishi, T. (1988) Neurosci. Lett. 94, 247-252. [DOI] [PubMed] [Google Scholar]
- 23.Milam, A. H., Dacey, D. M. & Dizhoor, A. M. (1993) Visual Neurosci. 10, 1-12. [DOI] [PubMed] [Google Scholar]
- 24.Haverkamp, S., Ghosh, K. K., Hirano, A. A. & Wassle, H. (2003) J. Comp. Neurol. 455, 463-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding, Y. Q., Shigemoto, R., Takada, M., Ohishi, H., Nakanishi, S. & Mizuno, N. (1996) J. Comp. Neurol. 364, 290-310. [DOI] [PubMed] [Google Scholar]
- 26.Stohr, H., Berger, C., Frohlich, S. & Weber, B. H. (2002) Gene 286, 223-231. [DOI] [PubMed] [Google Scholar]
- 27.Bush, R. A. & Sieving, P. A. (1996) J. Opt. Soc. Am. A 13, 557-565. [DOI] [PubMed] [Google Scholar]
- 28.Hu, E. H., Dacheux, R. F. & Bloomfield, S. A. (2000) J. Neurosci. Methods 103, 209-216. [DOI] [PubMed] [Google Scholar]
- 29.Alexander, K. R., Barnes, C. S., Fishman, G. A. & Milam, A. H. (2002) Invest. Ophthalmol. Visual Sci. 43, 1189-1197. [PubMed] [Google Scholar]
- 30.Masu, M., Iwakabe, H., Tagawa, Y., Miyoshi, T., Yamashita, M., Fukuda, Y., Sasaki, H., Hiroi, K., Nakamura, Y., Shigemoto, R., et al. (1995) Cell 80, 757-765. [DOI] [PubMed] [Google Scholar]
- 31.Dhingra, A., Lyubarsky, A., Jiang, M., Pugh, E. N., Jr., Birnbaumer, L., Sterling, P. & Vardi, N. (2000) J. Neurosci. 20, 9053-9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quigley, M., Roy, M. S., Barsoum-Homsy, M., Chevrette, L., Jacob, J. L. & Milot, J. (1996) Doc. Ophthalmol. 92, 159-165. [DOI] [PubMed] [Google Scholar]
- 33.Shinoda, K., Ohde, H., Mashima, Y., Inoue, R., Ishida, S., Inoue, M., Kawashima, S. & Oguchi, Y. (2001) Am. J. Ophthalmol. 131, 489-494. [DOI] [PubMed] [Google Scholar]
- 34.Barnes, C. S., Alexander, K. R. & Fishman, G. A. (2002) Ophthalmology 109, 575-583. [DOI] [PubMed] [Google Scholar]
- 35.Hack, I., Frech, M., Dick, O., Peichl, L. & Brandstatter, J. H. (2001) Eur. J. Neurosci. 13, 15-24. [PubMed] [Google Scholar]
- 36.DeVries, S. H. & Baylor, D. A. (1995) Proc. Natl. Acad. Sci. USA 92, 10658-10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamb, T. D. & Pugh, E. N., Jr. (1992) J. Physiol. 449, 719-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.