

Abstract
Gastrointestinal stromal tumor has received a lot of attention over the last 10 years due to its unique biologic behavior, clinicopathological features, molecular mechanisms, and treatment implications. GIST is the most common mesenchymal neoplasm in the gastrointestinal tract and has emerged from a poorly understood and treatment resistant neoplasm to a well-defined tumor entity since the discovery of particular molecular abnormalities, KIT and PDGFRA gene mutations. The understanding of GIST biology at the molecular level promised the development of novel treatment modalities. Diagnosis of GIST depends on the integrity of histology, immunohistochemistry and molecular analysis. The risk assessment of the tumor behavior relies heavily on pathological evaluation and significantly impacts clinical management. In this review, historic review, epidemiology, pathogenesis and genetics, diagnosis, role of molecular analysis, prognostic factor and treatment strategies have been discussed.
Key Words: Gastrointestinal stromal tumor, GIST, KIT mutation, imatinib
Introduction
Gastrointestinal stromal tumor (GIST) is the most common (80%) mesenchymal tumor of the alimentary cannel (1-3). It accounts for less than 1% of all gastrointestinal tumors and about 5% all sarcomas (2-4). It represents a wide clinical spectrum of tumors with different clinical presentations, locations, histology and prognosis. GIST can occur throughout the entire gastrointestinal (GI) tract and may have extragastrointestinal involvement as well. The clinical relevance of this tumor was generated by the discovery of its molecular biology and, consequently, of a drug effective in treating the tumor. The following review will discuss the GISTs in all aspects including history, epidemiology, clinical presentation, diagnosis, prognosis and treatment and emphasize on those relevant to diagnosis.
Historic overview
Stromal tumors arising from the GI tract were initially classified as smooth muscle neoplasms including leiomyomas (5), leiomyoblastomas or sarcomas (6), following description by Stout and colleagues in 1940 (7). These descriptions were widely used until the 1970s when electron microscope found little evidence of the smooth muscle origin of these tumors (8,9). With the advent of immunohistochemistry during the 1980’s it was soon appreciated that a large number of these tumors did not have immunophenotypic features of smooth muscle, and conversely, expressed antigens related to neural crest cells (10).
The term of “stromal tumors” was first described as a separate entity by Mazur and Clark (11) in 1983 and Schaldenbrand and Appleman in 1984 (12). However, this term was not widely accepted. In 1989, a distinctive subset of these stromal tumors revealing autonomic neural features was recognized and named “plexosarcoma” (13) and subsequently as gastrointestinal autonomic nerve tumor (GANT) (14). There was considerable confusion regarding the origin, differentiation and even clinical behavior of these tumors. In 1994, it was discovered that a significant proportion of GANTs were immunopositive for CD34 (15,16), which was the first relatively specific marker of GISTs during the mid-1990s. Based on the CD34 immunopositivity the possibility that GIST might be related to the interstitial cells of Cajal was raised by investigators (17). Interstitial cells of Cajal, also known as pacemaker cells for peristaltic contraction, are a group of cells found in the muscularis propria and around the myenteric plexus along the GI tract and have the immunophenotypic and ultrastructural characteristics of both the neural and smooth muscle elements. Meantime, additional studies found that interstitial cells of Cajal express KIT and are developmentally dependent on stem cell factor which is regulated through the KIT kinase (17,18). However, the following critical issues were not resolved: the exact origin of GIST, the best way to diagnose GIST, and differentiation of benign from malignant GIST. As the developments in studies of GISTs, describing gain-of-function mutations and consequently, constitutive activation of KIT receptors in several human tumor cell lines was reported in the mid-1990s (19,20).
Finally in 1998, Hirota and colleagues (21) discovered a specific mutation in the intracellular domain of the c-KIT protooncogene in GISTs as well as a near-universal expression of KIT protein in GISTs by immunohistochemistry. In the same year, Kindblom and colleagues (22) corroborated findings from Hirota and colleagues by showing the immunoreactivity for KIT in 78 of 78 GISTs studied and GISTs shared striking ultrastructural and immunophenotypic similarities with interstitial cells of Cajal. Both studies supported the hypothesis that GIST may indeed derive from stem cells that differentiated toward interstitial Cajal phenotype and confirmed KIT as a diagnostic tool for GIST (23). The KIT mutation implied a gain-of function linked to the activation of the kinase even in the absence of the binding of the ligand. The identification of the KIT mutation was a major breakthrough in the biology of GIST and overall, in cancer biology.
The identification of the biologic driver, activating mutations in KIT provided a therapeutic target for the treatment of GIST. One patient with metastatic GIST refractory to multiple types of therapies was treated with STI-571 (Imatinib mesylate- Gleevec; Novartis, Basel, Switzerland), which is a small molecule tryosine kinase inhibitor (TKI) with potent activity against the transmembrane receptor KIT, ABL kinase and chimeric BCR-ABL fusion oncoprotein product of chronic myeloid leukemia. The treatment yielded an early, rapid, and sustained response (24) with supportive preclinical data (25,26). This case provided proof of principle that inhibition of KIT by drug therapy was associated with improvement in the disease and brought phenomenal growth in the understanding of GIST biology and therapeutics. Imatinib occupies the ATP binding pocket of KIT, thereby preventing substrate phosphorylation, downstream signaling, and thereby inhibiting cell proliferation and survival (23). The remarkable therapeutic efficacy of imatinib in patients with GIST along with accurate diagnoses using CD117 expression (a marker of KIT receptor tryosine kinase) resulted in subsequent approval of imatinib in this indication by the US Food and Drug Administration in February 2002 (27). In 2003, Heinrich and colleagues (28) and Hirota and colleagues (29) all found platelet-derived growth factor receptor alpha (PDGFRA) gene mutations as an alternative pathogenesis in GISTs without KIT gene mutation. In January 26, 2006, Sunitinib, a multitargeted TKI with activity against KIT, PDGFR, vascular endothelial growth factor (VEGF) receptor (VEGFR), and FLT-1/KDR, also received FDA approval for the management of patients who are refractory or intolerant to imatinib (30).
Overall, about 85% of GISTs are reported to have activating mutation in KIT or PDGFRA (28,31,32). CD117 (c-Kit) immunohistochemistry has proven to be a reliable and sensitive diagnostic tool (22,33,34). With the TKI therapies against KIT and PDGFRA (imatinib and sunitinib), inoperable or metastatic GISTs are now treatable, and a number of additional alternative drugs are in clinical trials.
Epidemiology
Although the exact incidence of GISTs in the world is hard to determine since the entity was not uniformly defined until the late 1990s, a few estimates and studies indicate the incidences of approximately 14.5 cases/million/year in Sweden (35), 14.2 in Northern Italy (36), 13.7 in Taiwan (37), 12.7 in Holland (38), 11 in Iceland (39) and 6.5 in Norway (40). In a recent report, about 5,000 new cases of GISTs were diagnosed annually (41) and a incidence of 6.8/million from 1992 to 2000 (38) in the United States. The overall incidence rates of GIST, therefore, ranges between 6.5 and 14.5 per million per year. In general, little information on the prevalence of GIST was available. It is believed that the prevalence of GIST is higher, as many patients live with the disease for many years or develop small GISTs only detected at autopsy or if a gastrectomy is performed for other causes (42). A study performed in Germany on consecutive autopsies revealed small (<10 mm) GISTs in 22.5% of individuals who were older than 50 years (43). Rubin and colleagues used the SEER (surveillance, epidemiology, and end results) cancer registry in US for patients with GIST from 1993-2002 to determine incidence, prevalence, and 3-year survival and found the overall incidence, prevalence, and 3-year-servival rate were 3.2/million, 16.2/million, and 73%, respectively (44).
GIST mainly affects middle aged to elderly adults, typically in their 60s (35,45) with no clear gender predilection (46) although some studies demonstrated a slight male predominance (39,47). GISTs are uncommonly seen in patients younger than 40, however, cases in children and young adults have been reported (46). The true incidence of GIST in children is unknown. An incidence rate of 0.06/million/year was reported among young adults (20-29 years of age) (37). Other large series studies showed the percentage of patients with GIST below the age of 21 years ranged from 0.5% to 2.7% (45,46,48). Data from the UK National Registry revealed an annual incidence of 0.02 per million children below the age of 14 years, which appears to be the most accurate epidemiological data to date on pediatric GIST (49). Pediatric GISTs are considered a rare entity that can be quite different from its adult counterpart and seen predominantly in the second decade (46,50,51) with a predilection for female patients (46).
Sporadic GISTs are most common and familial GISTs with germline mutation of the KIT gene are rare, but have been well described (52-55). These patients usually have multiple GISTs and cutaneous hyperpigmentation (53). In addition, GIST rarely occurs in association with other syndromes such as neurofibromatosis type I (56-59) or Carney’s triad, a nonfamilial condition with gastric GIST, paraganglioma, and pulmonary chondroma (60,61). The latter should be distinguished from Carney-Stratakis syndrome, an inherited tumor syndrome comprising gastric GIST and paragangliomas (62).
GIST co-existing with other tumors has been reported mainly as case report (63) and mostly with colorectal carcinomas or adenomas, followed by gastric carcinomas (64,65). p53, one of the most common involved genes in colorectal carcinogenesis, has also been found to have a prognostic significance in GISTs, and mutations in this tumor suppressor gene are more often observed in the high-risk GISTs (66). GIST colliding with other tumors, mostly gastric adenocarcinomas, is rarely seen in literature (67-69). Only one case of gastric GIST colliding with angiosarcoma was reported (70).
Pathogenesis and genetics
In 1995 Huizinga and colleagues reported a knockout mice model of KIT failed to express in interstitial cells of Cajal cells (17). This finding led to the hypothesis that KIT was essential for the development of interstitial cells of Cajal cells. In 1998, Hirota and colleagues published a groundbreaking discovery of KIT mutations in GISTs (21) and 95% GISTs are immunohistochemically positive for the receptor tyrosine kinase KIT (also known as CD117) (21,22). It is now established that KIT mutations, which cause the constitutive activation of the kinase, are found in 70-80% of GISTs. CD117 becomes a crucial diagnostic marker for GIST, and mutant KIT provides an important therapeutic target clinically in GIST treatment.
Initially, GISTs lacking any evidence of KIT mutation were classified as “wild type” (WT). In 2003, novel mutations in PDGFRA were found in WT GIST by Heinrich and colleagues (28). Currently PDGFRA mutations account for 5-10% of known mutations in GIST. About 9-15% of all GISTs do not exhibit mutations in either KIT or PDGFRA and are now termed “wild type” (WT) (71).
KIT is a member of the type III transmembrane receptor tyrosine kinase (RTK) family that includes PDGFRA and PDGFRB, as well as macrophage colony-stimulating-factor receptor (CSF1R) and Fl cytokine receptor (FLT1) (72). Normally, binding of the KIT ligand, stem cell factor (SCF) to KIT results in receptor dimerization and kinase activation (73). In contrast, the presence of KIT receptor-activating mutations will bypass the ligand binding requirement for activation and therefore become oncogenic, which has been implicated in the pathogenesis of several human tumors in addition to GIST and chronic myelogenous leukemia (CML), including seminomas (74), mastocytosis (19), acute myelogenous leukemia (75) and, more recently, in melanomas (76).
KIT oncogenetic activation is the dominant pathogenetic mechanism in GIST (77). Although familial GIST with germline mutations have been reported (52,55), the majority of KIT mutations in GIST are somatic. The most common mutations in KIT are found in the juxtamembrane domain that is encoded by the 5' end of exon 11 of the KIT receptor (Figure 1). Mutations in exon 11 change the normal juxtamembrane secondary structure and cause the active conformation of the normal kinase activation loop (78). The mutations vary from in-frame deletions of variable sizes, point mutations to deletions preceded by substitutions (79). The deletions are associated with a more aggressive behavior in comparison to other exon 11 mutations (80-83). Particularly, deletions involving codon 557 and/or codon 558 are associated with malignant behavior (84,85). A less common mutant spot is located at the 3' end of exon 11, which includes mainly internal tandem duplications mutations (ITDs) (86). These ITD-type mutations are considered to have a more indolent clinical course and a predilection in GISTs located in the stomach (86). The second most common KIT mutation, between 10% and 15% of GISTs, is a mutation in an extracellular domain encoded by exon 9 (87). GISTs with KIT exon 9 mutations are characterized by small bowel location and aggressive clinical behavior (86).
Figure 1.

Schematic distribution of KIT or PDGFRA receptor mutations, frequency of mutations and TKI (Abbreviations: Ex, Exon; S, sensitive; R, resistant)
A minority of GISTs that lack KIT gene mutations have high levels of phosphorylation of PDGFRA resulted from an activation by mutations or small deletions (28). PDGFRA is a close homologue of KIT (28). Mutations in PDGFRA and KIT in GIST are mutually exclusive and about one-third of GISTs without KIT mutations harbor a mutation of PDGFRA, within exons 12, 14 or 18 (28,88,89). In GIST, mutant forms of PDGFRA have constitutive kinase activity in the absence of their ligand-PDGFRA similar to those for KIT mutations, and the activated downstream pathways (28,29) are identical to those in KIT-mutant GISTs (28,90). In spite of the similarities in molecular aspect, most GISTs with mutated PDGFRA have distinct pathologic features, including gastric location, epithelioid morphology, variable/absent CD117 by immunohistochemistry and an indolent clinical course (88,91,92).
Recent studies indicate that a small portion of GIST wild-type for both KIT and PDGFRA genes may harbor mutations of the BRAF gene (93) and KRAS and BRAF mutations predict primary resistance to imatinib in GISTs (94).
Furthermore, GISTs demonstrate typical patterns of chromosomal gains and losses, including losses at 1p, 14q, 15q, and 22q. Tumor site appears to be associated with distinct chromosomal imbalances; for example, gastric GISTs show predominantly losses 14q, whereas intestinal GISTs more frequently exhibit losses of 15q (95).
Clinical presentation
Most GISTs remain ‘silent’ until reaching a large size. Symptoms vary according to location and size. Symptomatic GIST patients generally present with nonspecific symptoms including abdominal pain, fatigue, dyspepsia, nausea, anorexia, weight loss, fever and obstruction. Patients may present with chronic GI or overt bleeding due to mucosal ulceration or tumor rupture with life-threatening intraperitoneal hemorrhage. Some patients with large GISTs may have externally palpable masses (96,97). Aggressive GISTs have a defined pattern of metastasis to the liver and throughout the abdomen or both (45). Lymph node metastasis is not common. Spreading to the lung and bone in advanced cases has been reported (98). Metastasis often occurs 10-15 years after initial surgery (45).
More than 80% of GISTs are primarily located in GI tract and may occur throughout the GI tract with extra-GI tract GISTs reported in omentum, mesentery, retroperitoneum, gallbladder and urinary bladder (99-101). The majority of GISTs (60%) are seen in the stomach, usually in the fundus (35,39). The percentages of GISTs found in other portions of GI tract are reported as 30% in jejunum and ileum, 5% in duodenum, 4% in colorectum, and rarely in the esophagus and appendix (45,46,48,65). Reported tumor size in the stomach varies from a few millimeters to >40 cm with a mean size of 6 cm in the largest reported series (65). Apparently, the tumor size is one of the factors contributing to the clinical symptoms. A population-based study showed that the tumor size is 8.9 cm in patients with clinical symptoms, which is about 70% of GISTs studied, 2.7 cm in patients without clinical symptoms, 20%, and 3.4 cm in patients with GISTs detected at autopsy, 10% (35). Many smaller GISTs are detected incidentally during endoscopy, surgery, or computed tomography (CT) scans (35).
Diagnosis
The diagnostic evaluation of GISTs is based on imaging techniques (Figure 2), with a special role of endoscopic examination because it is usually accessible when tumors are in the stomach, esophagus and large intestine. In addition, endoscopic ultrasonography (EUS) also plays an important role in the diagnostic work-up of GISTs and is accurate and efficient in the diagnosis of GISTs (102). In general, externally bilging tumors are more common than intraluminal masses (103). Punch-out ulcer is the classical appearance of a submucosal tumor (104).
Figure 2.
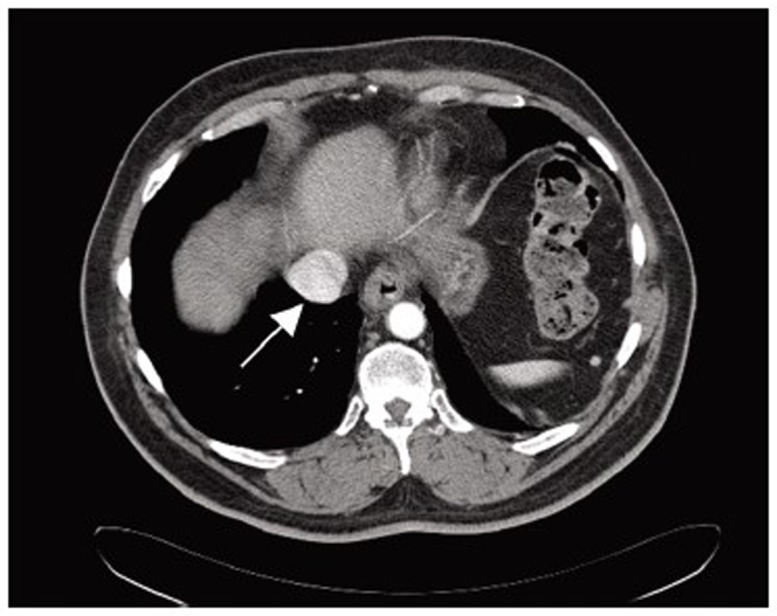
Computed tomography scan revealed a partially exophytic, dumbbell shaped solid mass (arrow) arising from the posterior aspect of the gastric fundus along the greater curvature, measuring approximately 6.7 cm × 4.5 cm
Macroscopy
Gastric GISTs are greyish-white sub-mucosal tumors with smooth contours and usually well-circumscribed and highly vascular tumors. They typically have a tan-white or fleshy pink cut surface often with hemorrhagic foci, central cystic degeneration, or necrosis (Figure 3). The overlying mucosa of large tumors is typically ulcerated (46).
Figure 3.

A gastric GIST with a nodulular surface and thin capusle. The cut surface reveals coarse granular and solid white tan suface with hemarrhage and cavities
Histopathology
Microscopically, GISTs have a broad morphological spectrum. Three main histological subtypes have been best widely accepted and they are spindle cell type (most common, 70%), epithelioid type (20-25%), and mixed spindle cell and epithelioid type (99,105,106) (Figure 4). In general, GISTs have a wide variation ranging from hypocellular to highly cellular with higher mitotic rates. Nuclear pleomorphism is relatively uncommon, and occurs more frequently in epithelioid type.
Figure 4.

Common histologic al features of GISTs. A. Spindle cell GIST with short fascicles and whorls (×100); B. Spindle cell GIST with longer fascicles in bundles (×100); C. Spindle cell GIST with extensive perinuclear vacuolization (×100); D. Spindle cell GIST with prominent nuclear palisading (×100); E. Epithelioid cells GIST with pleomorphic nuclei and vacuolated cytoplasm (×400); F. Epithelioid cell GIST with rhabdoid features (×400)
Spindle cell type of GIST is composed of cells in short fascicles and whorls. They have pale eosinophilic fibrillary cytoplasm, ovoid nuclei, and ill-defined cell borders. Gastric spindle cell GISTs often reveal extensive perinuclear vacuolization, a diagnostic feature formerly used for tumors of smooth muscle origin. The stroma sometimes demonstrates myxoid change or, rarely osseous metaplasia. Distinctive histological patterns among spindle cell GISTs including sclerosing type and palisading-vacuolated type (65). The sclerosing spindle cell GISTs have slender spindle cells with no nuclear atypia and low mitotic activity and are usually paucicellular with extensive extracellular collagen. They are often small and contain calcifications. The palisading-vacuolated type is one of the most common gastric GISTs and usually cellular with plump and uniformed spindle cells. Nuclear palisading with perinuclear vacuolization is characteristic. There is usually limited atypia with mitotic activity rarely more than 10/50 high power fields (HPFs). However, some examples show diffuse hypercellular pattern, and others sarcomatoid features with significant nuclear atypia and mitotic activity (65,99,106).
Epithelioid cell GISTs are characterized by round cells arranged in nests or sheets and with eosinophilic to clear cytoplasm. They also have spectrums from sclerosing and paucicellular to sarcomatous and mitotically inactive to mitotically highly active. However, the epithelioid GISTs with atypia, even with pleomorphism are sometimes benign (65,99,106).
Immunohistochemically, the vast majority of GISTs (95%) are strongly and diffusely positive for KIT (CD117), which makes the KIT to be a very specific and sensitive marker in the differentiating GIST from other mesenchyma tumors in the GI tract (21,22,34,107). The stain appears as cytoplasmic, membrane-associated or sometimes as perinuclear dots (34). Although KIT positivity appears to have significant therapeutic implications, the intensity, extent and patters of KIT staining neither correlates with the type of KIT mutation nor have therapeutic significance (34). It is important to note that negative KIT does not exclude the patient from being treated with TKI (imatinib or sunitinib) since some wild-type GISTs for both KIT and PDGFRA genes respond to treatment with TKI (42). In addition, CD34 is another common marker for GISTs but it is not as sensitive or specific. It is positive in about 80% of gastric GISTs, 50% of small intestine GISTs, and in 95% of esophageal and colorectal GISTs (48,108) (Figure 5). Other markers which can be expressed by GISTs include h-caldesmon, SMA, S100, desmin, Vimentin, and cytokeratins 8 and 18 (100). Recently other CD markers for GISTs are reported including CD10 (109), CD133, and CD44 (110).
Figure 5.

Immunohistochemical features of GIST. A. Spindle cell GIST with strong and diffuse cytoplasmic staining of CD117 (c-kit) (×400); B. Spindel cell GIST with strong and diffuse membrane staining of CD34 (×400); C. Epithelioid cell GIST with strong cytoplasmic staining of CD117 (×100); D. Epithelioid cell GIST with patchy and heterogeneous staining of CD34 (×400); E. Epithelioid cell GIST with punctate staining of h-Caldesmon (×100); F. Epithelioid cell GIST with patchy mambrane staining of h-Caldesmon (×400)
A small minority of GIST (<5%) are negative for KIT, or minimally, if any, positive for KIT by immunohistochemistry. These tumors appear to be either KIT wild-type or with mutant PDGFRA, have a predilection to stomach or omentum/peritoneum, and be usually epithelioid or mixed subtype (91,111). For the special interest in this subgroup of KIT-negative GISTs, several new antibodies for the diagnosis of GIST have been discovered based on the molecular studies. DOG1 (discovered on GIST1), known also as TMEM16A and ANO1, a transmembrane protein, has been found specifically in GISTs and has emerged as a promising biomarker for GISTs (112,113). Recent studies have shown that antibodies against DOG1 have even higher sensitivity and specificity than KIT (CD117) and CD34 with 75% to 100% overall sensitivity (113-116). DOG1 is highly expressed in KIT mutant GISTs and also can detect up to one-third of KIT-negative GISTs, which mostly have PDGFRA mutation (113,116). In addition to GISTs, DOG1 is also positive in normal gastric epithelium, some carcinomas, germ cell tumors, melanomas, and some mesenchymal tumors (113,114), such as recently reported chondroblastoma (117). Like KIT, DOG1 is also expressed in interstitial cells of Cajal serving as an internal positive control. However, DOG1 does not stain mast cells which are usually positive for KIT (112,114).
Non-gastric gists and gists in specific populations
Non-gastric GISTs may vary in clinical presentation, histopathology, molecular profile, prognostic significance and management strategy compared with gastric GISTs. Small intestinal GISTs including the duodenal GISTs are more homogeneous histologically and have a significant tumor-related mortality if the tumor is >5 cm (48). They typically harbor KIT exon 11 mutations as seen in gastric GISTs and a small portion of small intestinal GISTs contain duplication of two codons in KIT exon 9 (86,118). Usually, small intestinal GISTs do not harbor PDGFRA mutations. The sigmoid colon is the most common segment involved by GISTs (39) in the colon. Histopathologic profile of colonic GISTs is similar to that of small intestinal GISTs.
Pediatric GISTs account for about 1-2% of GISTs. They are often misdiagnosed as having another acute or chronic abdominal condition and they are usually symptomatic and mostly located in the stomach with mainly epithelioid pattern (35,46,50,51). GIST occurs in children and young adults as a component of two distinct syndromes: Carney triad and Carney-Stratakis syndrome. Carney triad is composed of co-occurrence of GIST, pulmonary chondroma, and paraganglioma. Carney triad can be diagnosed when any of the two tumors are present in a patient. However, if only GIST and paraganglioma are present, it is considered to be Carney-Stratakis syndrome. GIST in patients with Carney triad tends to be multifocal and have high local recurrence rate and/or metastatic rate. However, the clinical course of GIST in Carney triad is usually indolent (61). Although pediatric GISTs express KIT protein, the majorities lack KIT or PDGFRA mutations (46,50,51). In 2002, a germline-inactivating mutation in the hereditary paraganglioma gene was found to be unique for Carbey-Stratakis (119,120). This germline mutation results in a cancer predisposition syndrome including GIST.
Patients with neurofibromatosis type 1 (NF1) have a high risk for GIST. Some autopsy studies have demonstrated as many as one of three NF1 patients to have GISTs (121). NF-associated GIST typically occur in duodenum or small intestine and often multifocal and small. They commonly have low risk parameters and are clinically indolent (57,121). In contrast to sporadic adults GISTs, NF1-associated GISTs lack KIT and PDGFRA mutations (57,121,122).
Familial GISTs were reported and account for a very small portion of GISTs (<0.1%). They have typically activated germline KIT or PDGFRA mutations with an autosomal dominant inheritance and high penetrance (52,55,123,124). They occur usually in middle age of life and typical multifocal or diffuse in the GI tract. Most of these GISTs have a benign course.
Differential diagnosis
Although GISTs are the most common mesenchymal tumor of the GI tract, a variety of other tumors should be included in the differential diagnosis. Accurate recognition of GIST is obviously important as the treatment differs according to the tumor type. The main differential diagnoses include smooth muscle tumors, schwannoma, desmoid fibromatosis, inflammatory myofibroblastic tumor, inflammatory fibroid polyp, solitary fibrous tumor, synovial sarcoma, follicular dendritic cell sarcoma, glomus tumor, and melanoma. Kirsch and colleagues have published extensive review of diagnostic challenges and practical approach to differential diagnosis of GISTs (125).
Anatomic location may be helpful in differential diagnosis. Intramural leiomyomas most commonly locate in the esophagus and are rare in the stomach and small intestine (126). Morphologically, leiomyomas have brightly eosinophilic cytoplasm with distinct cell borders whereas GISTs usually reveal syncytial cell morphology. Immunohistochemically, GISTs and leiomyomas share some markers, such as SMA and h-caldesmon, but spindle cell GISTs are rarely positive for desmin which is more specific for leiomyomas. Rare epithelioid GISTs that lack KIT expression do stain positive for desmin (116). Leiomyomas are negative for CD117.
Although gastric schwannomas are not commonly seen, they can be morphologically very similar to certain spindle cell GISTs. Distinct peripheral cuffing of lymphocytes and strong reactivity with S-100 and GFAP readily differentiate them from GIST in addition to the negativities of CD117 and CD34 (127).
Mesenteric fibrous lesions can be very challenging in terms of diagnosis of itself and confusion with GIST due to the location and gross appearance. Microscopically, intraabdominal desmoid fibromatosis usually display long sweeping fascicles of spindle cells embedded within a collagen matrix with an infiltrating patter at peripheral of the tumor. Immunohistochemical stain of beta-catenin is positive in about 75% of cases (128-130). Inflammatory myofibroblastic tumors are commonly seen in pediatric or young adult patients and recognized as a mesenteric mass. Microscopically, this tumor has cellular fascicular fibroblastic/myofibroblastic proliferation with a prominent mixed inflammatory components including significant number of plasma cells. About 50% of tumors express ALK-1 (131), which is essentially negative in GIST. Inflammatory fibroid polyp is a polypoid lesion of mucosa with collagenous or myxoid stroma admixed with fibroblasts. It can be CD34 positive but should be negative for CD117 and DOG1 (113,114,132). Interestingly, same PDGFRA mutations as seen in GISTs are also discovered in inflammatory fibroid polyps (133).
Histologically, epithelioid GISTs need to be distinguished from other epithelial or epithelioid tumors including carcinoma, melanoma, glomus tumor, germ cell tumor and clear cell sarcoma. Immunohistochemical studies play a major rule on the differential diagnosis and the evaluation of appropriate immunophenotypic markers in context with morphology in most cases allows an accurate classification (Table 1).
Table 1. Immunophenotypic features of gastrointestinal mesenchymal tumors.
| Diagnosis | KIT | DOG1 | Desmin | SMA | h-Cal | S100 | CD34 | HMB45 | EMA | β-Cat | Clusterin | Keratin | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GIST |
+++ |
+++ |
- |
++ (40)* |
++ |
- |
+++ |
- |
- |
- |
- |
- |
|
| Leiomyoma |
- |
- |
+++ |
+++ |
+++ |
- |
- |
- |
- |
- |
- |
- |
|
| Leiomyosarcoma |
- |
- |
+ to ++ |
+++ |
++ |
- |
+ (10) |
- |
- |
- |
- |
- |
|
| Schwannoma |
- |
- |
- |
- |
- |
+++ |
- |
- |
- |
- |
- |
- |
GFAP |
| Fibromatosis |
- |
- |
- |
++ |
- |
± |
- |
- |
- |
++ |
- |
- |
|
| Synovial sarcoma |
- |
- |
- |
- |
- |
++ (30) |
- |
- |
++ |
- |
- |
± |
|
| PEComa |
- |
- |
++ |
++ |
- |
- |
- |
+++ |
- |
- |
- |
- |
Melan-A |
| FDCS |
- |
- |
- |
- |
- |
± |
- |
- |
± |
- |
+++ |
- |
CD21/23/35 |
| Dermatofibroma |
- |
- |
- |
+++ |
- |
- |
+++ |
- |
- |
++ |
- |
- |
|
| IFP |
- |
- |
- |
± |
- |
- |
++ |
- |
- |
- |
- |
- |
|
| IMT |
- |
- |
- |
++ |
- |
- |
± |
- |
- |
- |
- |
- |
ALK-1 |
| SFT | - | - | - | + | - | - | +++ | - | - | - | - | - |
*Parenthetical numbers represent approximate percentage of cases that are positive. Abbreviations: SMA, smooth muscle actin; h-Cal, h-Caldesmon; -Cat, -Catenin; PEComa, Perivascular epithelioid cell tumour; FDCS, Follicular dendritic cell sarcoma; IFP, Inflammatory fibroid polyp; IMT, Inflammatory myofibroblastic tumor; SFT, Solitary fibrous tumor; -, negative stain; ±, sometimes positive and sometimes negative stain; +, <25% of cases positive; ++, 25-50% of cases positive; +++, >50% of cases positive
Role of molecular analysis
Mutational analysis of the KIT gene including exons 11, 9, 13, and 17, and PDGFRA gene including exons 12, 14, and 18 can be helpful in confirming the diagnosis of GISTs if immunohistochemical studies fail to support the diagnosis (particularly in CD117/DOG1-negative spindle cell suspect cases). Corless and colleagues (134) summarized the mutations of GISTs and classified GISTs based on the molecular findings (Table 2). Furthermore, mutational analysis probably has more clinical significance in therapeutic aspect as it has predictive value for sensitivity to molecular-targeted therapy (including dosage) and prognostic value. It is strongly recommended that it should be included in the diagnostic work-up of all GISTs (135). The correlation between KIT and PDGFRA mutational status and the response to tyrosine kinase inhibitors and their role in primary and secondary resistance has been widely investigated (31,136). Tumors harboring KIT exon 11 mutations have a better outcome under imatinib treatment than tumors harboring different mutation, whereas tumors with PDGFRA exon 18 mutations (D842V) have primary resistance to imatinib both in vivo and in vitro (27,71,137). Therefore, GIST mutational analysis is strongly recommended in current NCCN (National Comprehensive Cancer Network) clinical practice guidelines (Figure 6) and in ESMO (European Society for Medical Oncology) clinical recommendations (138,139).
Table 2. Molecular classification of GISTs (134)*.
| Genetic type | Relative frequency | Anatomic distribution |
|---|---|---|
|
KIT mutation (relative frequency 75-80%) |
||
| Exon 8 |
Rare |
Small intestine |
| Exon 9 insertion AY502-503 |
10% |
Small intestine and colon |
| Exon 11 (deletion, single nucleotide substitution and insertions |
67% |
All sites |
| Exon 13 K642E |
1% |
All sites |
| Exon 17 D820Y, N822K and Y823D |
1% |
All sites |
|
PDGFRA mutation (relative frequency 5-8%) |
||
| Exon 12 (such as V561D) |
1% |
All sites |
| Exon 14 N659K |
<1% |
Stomach |
| Exon 18 D842V |
5% |
Stomach, mesentery and momentum |
| Exon 18 (such as deletion of amino acids IMHD 842-846 |
1% |
All sites |
|
KIT and PDGFRA wild-type (relative frequency 12-15% |
||
|
BRAF V600E |
~7-15% |
|
|
SDHA, SDHB, SDHC and SDHD mutations |
~2% |
Stomach and small intestine |
|
HRAS and NRAS mutation |
<1% |
|
| Sporadic pediatric GISTs |
~1% |
Stomach |
| GISTs as part of the Carney triad |
~1% |
Stomach |
| NF1-related | Rare | Small intestine |
Figure 6.

NCCN Guidelines Version 1.2012, Gastrointestinal Stromal Tumors (GIST) (Abbreviations: H&P, history & physical examination; Mets, metastatic disease; IM, imatinib; Preop, preoperative; DX, diagnosis; SU; sunitinib; mo, month; y, year)
Prognostic factors, grade and stage
The risk of relapse of GISTs is estimated based on mitotic rate, tumor size, tumor site, surgical margins and the status of tumor rupture. Tumor size and mitotic count are considered to be the most useful and best studied prognostic factors by the 2002 Consensus risk classification (Table 3) (99). It is believed that indicating a risk level of GIST (low, intermediate, or high) is more appropriate than definitively labeling the tumor as benign or malignant. This risk classification was based on the cumulative experience of the authors in the committee. The most important cut-offs as indicators of aggressive clinical behavior were tumor size of 5 cm and 5 mitoses/50 HPF. This consensus guideline indicated that all GISTs may have malignant potential (99). Based on long-term follow-up of more than 1,600 GISTs (1,055 gastric, 629 small intestinal, 144 duodenal, and 111 rectal), Miettinen and colleagues proposed risk classification incorporates primary tumor site in addition to the mitotic count and tumor size (Table 4) (140). It demonstrates the fact that gastric GISTs have a better prognosis than small intestine or rectal GISTs. The more recently updated consensus NCCN guidelines from 2007 (141) includes anatomic site as an additional parameter in risk assessment for GIST. Based on those guidelines, GISTs that are smaller than 2 cm are considered to be essentially benign. Recently, Gold and colleagues proposed a nomogram for estimating the risk of tumor progression (142), in which each GIST was assigned points on a scale based on tumor site, size, and mitotic index. The total points of a tumor should determine the 2- and 5-year recurrence free survival probabilities. From a clinical point of view, additional prognostic factors including non-radical resection and tumor rupture, whether spontaneous or at the time of surgical resection, are both associated with adverse outcome independent of any other prognostic factors (143). Furthermore, Takahashi and colleagues suggested the inclusion of a “clinically malignancy group” to include patients with peritoneal dissemination, metastasis, and invasion into adjacent organs or tumor rupture (144). In 2008, a proposal by Joensuu based on the NIH system included the presence of tumor rupture as a high risk factor irrespective of size and mitotic count (145). The Joensuu’s revised NIH risk system is shown in Table 5.
Table 3. Risk assessment of GIST, 2002 by NIH.
| Risk category | Size (cm) | Mitotic count (50 HPF) |
|---|---|---|
| Very low risk |
<2 |
<5 |
| Low risk |
2-5 |
5 |
| Intermediate risk |
5 |
6-10 |
| 5-10 |
5 |
|
| High risk |
>5 |
>5 |
| >10 |
Any mitotic rate |
|
| Any size | >10 |
Table 4. Risk assessment of GIST, 2006 by miettinen and lasota (ref 140).
| Mitotic rate (50 HPF) | Tumor size (cm) | Stomach | Duodenum | Jejunum or ileum | Rectum |
|---|---|---|---|---|---|
| 5 |
2 |
None |
None |
None |
None |
| >25 |
Very low |
Low |
Low |
Low |
|
| >510 |
Low |
Moderate |
Insufficient data |
Insufficient data |
|
| >10 |
Moderate |
High |
High |
High |
|
| >5 |
2 |
None* |
High* |
Insufficient data |
High |
| >25 |
Moderate |
High |
High |
High |
|
| >510 |
High |
High |
Insufficient data |
Insufficient data |
|
| >10 | High | High | High | High |
Adopted from Miettinen and Lasota (140). Abbreviation: HPF, high-power field; *Very small number of cases
Table 5. Risk Assessment of GIST, 2008 by Joensuu (145).
| Risk category | Tumor size (cm) | Mitotic rate | Duodenum |
|---|---|---|---|
| (50 HPF) |
Primary tumor site |
None |
None |
| Very low risk |
<2 |
5 |
Any |
| Low risk |
2.1-5.0 |
5 |
Any |
| Intermediate risk |
2.1-5.0 |
>5 |
Gastric |
| <5.0 |
6-10 |
Any |
|
| 5.1-10.0 |
5 |
Gastric |
|
| High risk |
Any |
Any |
Tumor rupture |
| >10.0 |
Any |
Any |
|
| Any |
>10 |
Any |
|
| >5.0 |
>5 |
Any |
|
| 2.1-5.0 |
>5 |
Nongastric |
|
| 5.1-10.0 | 5 | Nongastric |
In the TNM staging (AJCC, 7th edition, 2010) (146), grading of GISTs is based on mitotic rate. Mitotic rate less than 5/50 HPFs is considered to be low (grade 1) and greater than 5/50 HPFs is considered to be high (grade 2). Please note that the staging criteria are different for gastric GISTs and small intestinal GISTs to emphasize the more aggressive clinical course of small intestinal GISTs even with similar tumor parameters (147). The seventh edition of the international union against cancer (UICC) published at the beginning of 2010 included for the first time a classification and staging system for GIST (148). This represents a significant step towards a more standardized surgical and oncological treatment for patients with GIST and, more importantly, may facilitate the establishment of a uniformed follow-up system based on tumor stage (Table 6) (149).
Table 6. UICC TNM classification for GIST, 7th Edition, 2010.
| Mitotic rate (50HPFs) | Tumor size (cm) | T |
N | M | UICC stage |
||
|---|---|---|---|---|---|---|---|
| Gastric | Non-gastric | Gastric GIST | Non-gastric GIST | ||||
| 5 |
2 |
T1 |
T1 |
N0 |
M0 |
IA |
I |
| 2-5 |
T2 |
T2 |
N0 |
M0 |
IA |
I |
|
| 5-10 |
T3 |
T3 |
N0 |
M0 |
IB |
II |
|
| >10 |
T4 |
T4 |
N0 |
M0 |
II |
IIIA |
|
| >5 |
2 |
T1 |
T1 |
N0 |
M0 |
II |
IIIA |
| 2-5 |
T2 |
T2 |
N0 |
M0 |
II |
IIIB |
|
| 5-10 |
T3 |
T3 |
N0 |
M0 |
IIIA |
IIIB |
|
| >10 |
T4 |
T4 |
N0 |
M0 |
IIIB |
IIIB |
|
| Any | Any | Any |
Any |
N1 |
M0 |
IV |
IV |
| Any | Any | Any | M1 | IV | IV | ||
Abbreviation: UICC, the international union against cancer; GIST, gastrointestinal stromal tumor; HPF, high-power field
Treatment
Treatment of localized disease
Surgery
The only potentially curative treatment of GISTs, still, is complete surgical resection if it is a locally resectable or marginally resectable tumor (141,150). GISTs rarely metastasize to lymph node (142,151) and therefore regional lymph node dissection is generally not needed. In addition, organ-sparing resection (segmental resection) is also appropriate oncologically. However, about 40-90% of surgically treated patients experience disease recurrence (152). A recent study of 127 patients with localized GISTs who underwent complete resection demonstrated a 5-year recurrence-free survival (RFS) rate of 63% (153). This study concludes tumor size 10 cm, mitotic rate 5/50HPFs, and tumor location in the small intestine were all independently associated with an increased risk of recurrence. In addition, intraperitoneal rupture or bleeding is also associated with a high risk of postoperative recurrence of nearly 100% (143,154,155).
Adjuvant therapy
Understanding the molecular changes of GISTs along with target treatments resulted in a considerable transformation in the management of GISTs. The remarkable efficacy of imatinib in treating metastatic GISTs has prompted interest in developing an adjuvant after complete resection of GISTs. Resent phase III randomized trial involved 778 patients with localized GISTs who underwent complete surgical resection followed by 1 year of imatinib (400 mg/day) and revealed that adjuvant imatinib significantly improved the 1-year RFS rate (98%) compared with the placebo (83%) (P<0.0001) (156). Based on the results of this trial, FDA approved imatinib as adjuvant therapy for GISTs (157). The most recent management guidelines in US (NCCN) (138) and Europe (ESMO) (139) recommended adjuvant imatinib for at least 1 year following complete surgical resection in patients with intermediate- to high-risk GIST. However, the optimal duration of adjuvant therapy has not been established yet.
Treatment of localized unresectable or metastatic gists
Although surgical intervention was applied to patients with metastases prior to the imatinib era, it was unlikely to completely resect the tumor and consequently with earlier recurrence than localized disease (45). Nunoby and colleagues (158) in Japan studied the outcome of surgical resection in 18 patients with liver metastases of GISTs and showed 83% complete resection of liver metastases with 64% 3-year postoperative overall survival (OS) rate and 34% 5-year postoperative OS rate. However, the recurrence rate in the remnant liver and in other organs reached 94% in this study. Surgical treatment alone for metastatic GISTs, therefore, is only palliative (158).
The application of imatinib for patients with advanced and non-resectable GISTs was first evaluated in the palliative setting in 2000 (24). A recent large clinical study of imatinib for unresectable or metastatic GISTs revealed up to 57 months of median OS rate (159), which is almost a threefold increase in OS from about 20 months (45) prior to the application of imatinib. Based on the clinical practice guidelines (NCCN & ESMO), treatment with imatinib (400 mg/day) now is the standard of care for patients with locally advanced, recurrent, or metastatic disease (138,139). Multiple phase III clinical trials have confirmed the effectiveness of imatinib with standard-dose (400 mg/day) or high-dose (800 mg/day) (159,160). Furthermore, the efficacy of imatinib certainly also depends on the mutant profile of GISTs. KIT exon 11 mutations show the greatest benefit from imatinib treatment (400 mg/day) (Figure 1) (135,161). KIT exon 11 codon 557/558 deletion/insertion mutations have a more aggressive clinical behavior (162). KIT exon 9 mutant GIST requires a higher imatinib dosage to reach a better response (135,163). In addition, sunitinib, another TKI, is beneficial for exon 9 mutated-GIST (30). Although wild-type patients are not likely to benefit from imatinib (161), some in vivo and in vivo studies on sunitinib (164), nilotinib, and dasatinib (165) are promising. Regarding PDGFRA-mutated GISTs, PDGFRA exon 18 mutations have better response to imatinib therapy but not with PDFGRA exon 18 D842V-mutation (71).
According to the NCCN guidelines, patients with progressive disease after imatinib treatment are allowed to be re-assessed for surgery. Surgical resection has been achieved in those cases (166-168). However, the timing of the surgical intervention is very important and was recommended as the time at which patients reached maximum benefit from imatinib but before tumor progression occurs (139,169). In addition, neoadjuvant therapy with TKI should be considered to facilitate complete resection and allow for a less morbid operation, especially in duodenal GIST which can be sometimes hardly resected completely (170,171). With a short neoadjuvant imatinib therapy, tumor blood flow was decreased and apoptosis was increased within 3-7 days of starting therapy compared with pre-imatinib tumor tissue, although minimal size reduction was observed (171).
Assessment of treatment response
According to the NCCN guidelines, imaging study of contrast-enhanced CT scan is the technique of choice to detect recurrence or progression of GISTs (138,139,172). In rectal GIST, MRI should be used or additional PET or PET-CT/MRI may be useful for early detection of tumor response to neoadjuvant therapy (172). Choi and colleagues (173) proposed modified response evaluation criteria which is considered to predict response more accurately than previously proposed Response Evaluation Criteria in Solid Tumor (RECIST) (174) and has a better correlation with time to progression (175).
Resistant disease and alterative treatments
Although TKIs, especially imatinib, have resulted in disease-free survival for patients following surgical resection of their primary tumors and increased response rates and survival for patients with metastatic disease, some patients will eventually develop resistance to imatinib (176). Several potential mechanisms of resistance were proposed and include specific types of mutations (KIT exon 9, KIT wild-type or PDGFRA exon 18) (31,135), acquisition of secondary mutations within the KIT gene, KIT gene amplification, loss of the wild-type allele, or inadequate imatinib plasma levels (176-179). Sunitinib is the only second-line TKI approved for use after imatinib failure due to its inhibitory function on multi-kinases receptors (136). It has also been shown to be effective against secondary mutations in vitro and in vivo studies (136,161). However, as with imatinib, resistance has recently been documented in patients with prolonged exposure to sunitinib (180,181). In addition, it has been shown that sunitinib can cause serious, life-threatening adverse effects, including hypertension, cardiotoxicity, and hypothyroidism (30,182,183). According to the NCCN and ESMO guidelines, sunitinib is recommended as a second-line therapy in patients who experience disease progression after high-dose imatinib or who have life-threatening side effects. If further progression occurs with sunitinib, patients should be considered for clinical trials of new agents or new combinations or discontinuation of anti-cancer therapy.
The role of newer generation KIT and PDGFRA kinase inhibitors, e.g., nilotinib, remains to be determined in GIST patients with multiple resistants after imatinib and sunitinib therapies. Nilotinib has demonstrated activity against imatinib- and sunitinib resistant GISTs (184) and displays, by an ongoing pilot study (185), substantial clinical benefit and is safe in the first-line treatment of advanced GIST. Other agents, such as dasitinib (186), sorafenib (187), and masitinib (188), target multiple oncogenic receptor tyrosine kinases that have been implicated in the development and growth of GIST. These newer agents and a wide number of others (189) are currently under clinical trials for the management of advanced and resistant GISTs and likely to change the treatment of this disease soon.
Conclusions
GISTs have received much attention for many reasons. The rapid expansion of molecular and clinicopathological knowledge of GIST has given this disease a promising future. The molecular targets for therapeutic interventions are not only of importance for the treatment of GIST patients, but also in the development of novel drugs and new strategies in basic cancer therapy. Pathologists need to know their role as the diagnostic information they provided impacts on the choice of treatment as well as on estimation of its efficacy. Molecular testing of GISTs should be performed for treatment selection and assessment of disease progression. The cause of GIST is still unknown; therefore, little has been done preventively. However, with gradual understanding the molecular mechanisms of GIST, the etiology will be elucidated eventually.
Acknowledgments
Disclosure: The authors declare no confict of interest.
References
- 1.Joensuu H, Fletcher C, Dimitrijevic S, et al. Management of malignant gastrointestinal stromal tumours. Lancet Oncol 2002;3:655-64 [DOI] [PubMed] [Google Scholar]
- 2.Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet 2007;369:1731-41 [DOI] [PubMed] [Google Scholar]
- 3.Steigen SE, Eide TJ. Gastrointestinal stromal tumors (GISTs): a review. APMIS 2009;117:73-86 [DOI] [PubMed] [Google Scholar]
- 4.Katz SC, DeMatteo RP. Gastrointestinal stromal tumors and leiomyosarcomas. J Surg Oncol 2008;97:350-9 [DOI] [PubMed] [Google Scholar]
- 5.Appleman HD, Helwig EB. Gastric epithelioid leiomyoma and leiomyosarcoma (leiomyoblastoma). Cancer 1976;38:708-28 [DOI] [PubMed] [Google Scholar]
- 6.Akwari OE, Dozois RR, Weiland LH, et al. Leiomyosarcoma of the small and large bowel. Cancer 1978;42:1375-84 [DOI] [PubMed] [Google Scholar]
- 7.Stout AP. Bizarre smooth muscle tumors of the stomach. Cancer 1962;15:400-9 [DOI] [PubMed] [Google Scholar]
- 8.Welsh RA, Meyer AT. Ultrastructure of gastric leiomyoma. Arch Pathol 1969;87:71-81 [PubMed] [Google Scholar]
- 9.Weiss RA, Mackay B. Malignant smooth muscle tumors of the gastrointestinal tract: an ultrastructural study of 20 cases. Ultrastruct Pathol 1981;2:231-40 [DOI] [PubMed] [Google Scholar]
- 10.Yagihashi S, Kimura M, Kurotaki H, et al. Gastric submucosal tumours of neurogenic origin with neuroaxonal and Schwann cell elements. J Pathol 1987;153:41-50 [DOI] [PubMed] [Google Scholar]
- 11.Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol 1983;7:507-19 [DOI] [PubMed] [Google Scholar]
- 12.Schaldenbrand JD, Appelman HD. Solitary solid stromal gastrointestinal tumors in von Recklinghausen’s disease with minimal smooth muscle differentiation. Hum Pathol 1984;15:229-32 [DOI] [PubMed] [Google Scholar]
- 13.Herrera GA, Cerezo L, Jones JE, et al. Gastrointestinal autonomic nerve tumors. ‘Plexosarcomas’. Arch Pathol Lab Med 1989;113:846-53 [PubMed] [Google Scholar]
- 14.Lauwers GY, Erlandson RA, Casper ES, et al. Gastrointestinal autonomic nerve tumors. A clinicopathological, immunohistochemical, and ultrastructural study of 12 cases. Am J Surg Pathol 1993;17:887-97 [DOI] [PubMed] [Google Scholar]
- 15.Miettinen M, Virolainen M, Maarit Sarlomo R.Gastrointestinal stromal tumors--value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol 1995;19:207-16 [DOI] [PubMed] [Google Scholar]
- 16.Mikhael AT, Bacchi CE, Zarbo RJ, et al. CD34-expression in stromal tumors of the gastrointestinal tract. Appl Immunohistochem 1994;2:89-93 [Google Scholar]
- 17.Huizinga JD, Thuneberg L, Kluppel M, et al. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995;373:347-9 [DOI] [PubMed] [Google Scholar]
- 18.Isozaki K, Hirota S, Nakama A, et al. Disturbed intestinal movement, bile reflux to the stomach, and deficiency of c-kit-expressing cells in Ws/Ws mutant rats. Gastroenterology 1995;109:456-64 [DOI] [PubMed] [Google Scholar]
- 19.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995;92:10560-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longley BJ, Tyrrell L, Lu SZ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet 1996;12:312-4 [DOI] [PubMed] [Google Scholar]
- 21.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577-80 [DOI] [PubMed] [Google Scholar]
- 22.Kindblom LG, Remotti HE, Aldenborg F, et al. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;152:1259-69 [PMC free article] [PubMed] [Google Scholar]
- 23.Quek R, George S.Gastrointestinal stromal tumor: a clinical overview. Hematol Oncol Clin North Am 2009;23:69-78 viii. [DOI] [PubMed] [Google Scholar]
- 24.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344:1052-6 [DOI] [PubMed] [Google Scholar]
- 25.Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther 2000;295:139-45 [PubMed] [Google Scholar]
- 26.Heinrich MC, Griffith DJ, Druker BJ, et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000;96:925-32 [PubMed] [Google Scholar]
- 27.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472-80 [DOI] [PubMed] [Google Scholar]
- 28.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708-10 [DOI] [PubMed] [Google Scholar]
- 29.Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660-7 [DOI] [PubMed] [Google Scholar]
- 30.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329-38 [DOI] [PubMed] [Google Scholar]
- 31.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342-9 [DOI] [PubMed] [Google Scholar]
- 32.Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813-25 [DOI] [PubMed] [Google Scholar]
- 33.Hornick JL, Fletcher CD. Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am J Clin Pathol 2002;117:188-193 [DOI] [PubMed] [Google Scholar]
- 34.Hornick JL, Fletcher CD. The role of KIT in the management of patients with gastrointestinal stromal tumors. Hum Pathol 2007;38:679-87 [DOI] [PubMed] [Google Scholar]
- 35.Nilsson B, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era--a population-based study in western Sweden. Cancer 2005;103:821-9 [DOI] [PubMed] [Google Scholar]
- 36.Mucciarini C, Rossi G, Bertolini F, et al. Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. BMC Cancer 2007;7:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran T, Davila JA, El-Serag HB. The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol 2005;100:162-8 [DOI] [PubMed] [Google Scholar]
- 38.Goettsch WG, Bos SD, Breekveldt-Postma N, et al. Incidence of gastrointestinal stromal tumours is underestimated: results of a nation-wide study. Eur J Cancer 2005;41:2868-72 [DOI] [PubMed] [Google Scholar]
- 39.Tryggvason G, Gislason HG, Magnusson MK, et al. Gastrointestinal stromal tumors in Iceland, 1990-2003: the icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 2005;117:289-93 [DOI] [PubMed] [Google Scholar]
- 40.Sandvik OM, Soreide K, Kvaloy JT, et al. Epidemiology of gastrointestinal stromal tumours: single-institution experience and clinical presentation over three decades. Cancer Epidemiol 2011;35:515-20 [DOI] [PubMed] [Google Scholar]
- 41.Pisters PW, Blanke CD, von Mehren M, et al. A USA registry of gastrointestinal stromal tumor patients: changes in practice over time and differences between community and academic practices. Ann Oncol 2011;22:2523-9 [DOI] [PubMed] [Google Scholar]
- 42.Liegl-Atzwanger B, Fletcher JA, Fletcher CD. Gastrointestinal stromal tumors. Virchows Arch 2010;456:111-27 [DOI] [PubMed] [Google Scholar]
- 43.Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol 2007;31:113-20 [DOI] [PubMed] [Google Scholar]
- 44.Rubin JL, Sanon M, Taylor DC, et al. Epidemiology, survival, and costs of localized gastrointestinal stromal tumors. Int J Gen Med 2011;4:121-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000;231:51-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005;29:1373-81 [DOI] [PubMed] [Google Scholar]
- 47.Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol 2006;37:1527-35 [DOI] [PubMed] [Google Scholar]
- 48.Miettinen M, Makhlouf H, Sobin LH, et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006;30:477-89 [DOI] [PubMed] [Google Scholar]
- 49.Stiller C. Childhood cancer in Britain: incidence, survival, mortality. Oxford University Press Oxford. 2007. [Google Scholar]
- 50.Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 2007;67:9084-8 [DOI] [PubMed] [Google Scholar]
- 51.Prakash S, Sarran L, Socci N, et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 2005;27:179-87 [DOI] [PubMed] [Google Scholar]
- 52.Nishida T, Hirota S, Taniguchi M, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998;19:323-4 [DOI] [PubMed] [Google Scholar]
- 53.Maeyama H, Hidaka E, Ota H, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology 2001;120:210-5 [DOI] [PubMed] [Google Scholar]
- 54.Beghini A, Tibiletti MG, Roversi G, et al. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001;92:657-62 [DOI] [PubMed] [Google Scholar]
- 55.Li FP, Fletcher JA, Heinrich MC, et al. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J Clin Oncol 2005;23:2735-43 [DOI] [PubMed] [Google Scholar]
- 56.Takazawa Y, Sakurai S, Sakuma Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type I (von Recklinghausen’s disease). Am J Surg Pathol 2005;29:755-63 [DOI] [PubMed] [Google Scholar]
- 57.Miettinen M, Fetsch JF, Sobin LH, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006;30:90-6 [DOI] [PubMed] [Google Scholar]
- 58.Kang DY, Park CK, Choi JS, et al. Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007;31:224-32 [DOI] [PubMed] [Google Scholar]
- 59.Maertens O, Prenen H, Debiec-Rychter M, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006;15:1015-23 [DOI] [PubMed] [Google Scholar]
- 60.Carney JA, Sheps SG, Go VL, et al. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med 1977;296:1517-8 [DOI] [PubMed] [Google Scholar]
- 61.Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clinic proceedings Mayo Clinic 1999;74:543-52. [DOI] [PubMed]
- 62.Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002;108:132-9 [DOI] [PubMed] [Google Scholar]
- 63.Melis M, Choi EA, Anders R, et al. Synchronous colorectal adenocarcinoma and gastrointestinal stromal tumor (GIST). Int J Colorectal Dis 2007;22:109-14 [DOI] [PubMed] [Google Scholar]
- 64.Liszka Ł, Zielińska-Pajak E, Pajak J, et al. Coexistence of gastrointestinal stromal tumors with other neoplasms. J Gastroenterol 2007;42:641-9 [DOI] [PubMed] [Google Scholar]
- 65.Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005;29:52-68 [DOI] [PubMed] [Google Scholar]
- 66.Ryu MH, Kang YK, Jang SJ, et al. Prognostic significance of p53 gene mutations and protein overexpression in localized gastrointestinal stromal tumours. Histopathology 2007;51:379-89 [DOI] [PubMed] [Google Scholar]
- 67.Liu SW, Chen GH, Hsieh PP. Collision tumor of the stomach: a case report of mixed gastrointestinal stromal tumor and adenocarcinoma. J Clin Gastroenterol 2002;35:332-4 [DOI] [PubMed] [Google Scholar]
- 68.Katsoulis IE, Bossi M, Richman PI, et al. Collision of adenocarcinoma and gastrointestinal stromal tumour (GIST) in the stomach: report of a case. Int Semin Surg Oncol 2007;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toyoda A, Komaba A, Yoshizumi H, et al. Collision of advanced gastric adenocarcinoma and gastrointestinal stromal tumour: a case report. BMJ case reports 2009;2009. [DOI] [PMC free article] [PubMed]
- 70.Adhikari M, Wu ML, Zhao X. Gastrointestinal stromal tumor colliding with angiosarcoma. Int J Surg Pathol 2006;14:252-6 [DOI] [PubMed] [Google Scholar]
- 71.Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357-64 [DOI] [PubMed] [Google Scholar]
- 72.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 1988;241:42-52 [DOI] [PubMed] [Google Scholar]
- 73.Lev S, Yarden Y, Givol D.Dimerization and activation of the kit receptor by monovalent and bivalent binding of the stem cell factor. J Biol Chem 1992;267:15970-7 [PubMed] [Google Scholar]
- 74.Tian Q, Frierson HF, Jr, Krystal GW, et al. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol 1999;154:1643-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gari M, Goodeve A, Wilson G, et al. c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 1999;105:894-900 [DOI] [PubMed] [Google Scholar]
- 76.Curtin JA, Busam K, Pinkel D, et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340-6 [DOI] [PubMed] [Google Scholar]
- 77.Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA 2009;106:1542-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 2004;279:31655-63 [DOI] [PubMed] [Google Scholar]
- 79.Corless CL, McGreevey L, Town A, et al. KIT gene deletions at the intron 10-exon 11 boundary in GI stromal tumors. J Mol Diagn 2004;6:366-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ernst SI, Hubbs AE, Przygodzki RM, et al. KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998;78:1633-6 [PubMed] [Google Scholar]
- 81.Lasota J, Jasinski M, Sarlomo-Rikala M, et al. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999;154:53-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taniguchi M, Nishida T, Hirota S, et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999;59:4297-300 [PubMed] [Google Scholar]
- 83.Andersson J, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 2006;130:1573-81 [DOI] [PubMed] [Google Scholar]
- 84.Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887-95 [DOI] [PubMed] [Google Scholar]
- 85.Martín J, Poveda A, Llombart-Bosch A, et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol 2005;23:6190-8 [DOI] [PubMed] [Google Scholar]
- 86.Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329-37 [PubMed] [Google Scholar]
- 87.Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000;156:791-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lasota J, Dansonka-Mieszkowska A, Sobin LH, et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 2004;84:874-83 [DOI] [PubMed] [Google Scholar]
- 89.Pauls K, Merkelbach-Bruse S, Thal D, et al. PDGFRalpha- and c-kit-mutated gastrointestinal stromal tumours (GISTs) are characterized by distinctive histological and immunohistochemical features. Histopathology 2005;46:166-75 [DOI] [PubMed] [Google Scholar]
- 90.Kang HJ, Nam SW, Kim H, et al. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene 2005;24:1066-74 [DOI] [PubMed] [Google Scholar]
- 91.Medeiros F, Corless CL, Duensing A, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol 2004;28:889-94 [DOI] [PubMed] [Google Scholar]
- 92.Sakurai S, Hasegawa T, Sakuma Y, et al. Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: a subtype of GIST with mutations of platelet-derived growth factor receptor alpha gene. Hum Pathol 2004;35:1223-30 [DOI] [PubMed] [Google Scholar]
- 93.Hostein I, Faur N, Primois C, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010;133:141-8 [DOI] [PubMed] [Google Scholar]
- 94.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF Mutations Predict Primary Resistance to Imatinib in Gastrointestinal Stromal Tumors. Clin Cancer Res 2012;18:1769-76 [DOI] [PubMed] [Google Scholar]
- 95.Gunawan B, von Heydebreck A, Sander B, et al. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol 2007;211:463-70 [DOI] [PubMed] [Google Scholar]
- 96.Ueyama T, Guo KJ, Hashimoto H, et al. A clinicopathologic and immunohistochemical study of gastrointestinal stromal tumors. Cancer 1992;69:947-55 [DOI] [PubMed] [Google Scholar]
- 97.Miettinen M, Sarlomo-Rikala M, Sobin LH, et al. Gastrointestinal stromal tumors and leiomyosarcomas in the colon: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases. Am J Surg Pathol 2000;24:1339-52 [DOI] [PubMed] [Google Scholar]
- 98.Miettinen M, Furlong M, Sarlomo-Rikala M, et al. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the rectum and anus: a clinicopathologic, immunohistochemical, and molecular genetic study of 144 cases. Am J Surg Pathol 2001;25:1121-33 [DOI] [PubMed] [Google Scholar]
- 99.Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol 2002;33:459-65 [DOI] [PubMed] [Google Scholar]
- 100.Miettinen M, Monihan JM, Sarlomo-Rikala M, et al. Gastrointestinal stromal tumors/smooth muscle tumors (GISTs) primary in the omentum and mesentery: clinicopathologic and immunohistochemical study of 26 cases. Am J Surg Pathol 1999;23:1109-18 [DOI] [PubMed] [Google Scholar]
- 101.Reith JD, Goldblum JR, Lyles RH, et al. Extragastrointestinal (soft tissue) stromal tumors: an analysis of 48 cases with emphasis on histologic predictors of outcome. Mod Pathol 2000;13:577-85 [DOI] [PubMed] [Google Scholar]
- 102.Gu M, Ghafari S, Nguyen PT, et al. Cytologic diagnosis of gastrointestinal stromal tumors of the stomach by endoscopic ultrasound-guided fine-needle aspiration biopsy: cytomorphologic and immunohistochemical study of 12 cases. Diagn Cytopathol 2001;25:343-50 [DOI] [PubMed] [Google Scholar]
- 103.Levy AD, Remotti HE, Thompson WM, et al. Gastrointestinal stromal tumors: radiologic features with pathologic correlation. Radiographics 2003;23:283-304, 456; quiz 532. [DOI] [PubMed]
- 104.Saleem TB, Ahmed I. Gastrointestinal stromal tumour--evolving concepts. Surgeon 2009;7:36-41 [DOI] [PubMed] [Google Scholar]
- 105.Miettinen M, Lasota J.Gastrointestinal stromal tumors-- definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1-12 [DOI] [PubMed] [Google Scholar]
- 106.Miettinen M, Lasota J.Histopathology of gastrointestinal stromal tumor. J Surg Oncol 2011;104:865-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, et al. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998;11:728-34 [PubMed] [Google Scholar]
- 108.Miettinen M, Lasota J.KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol 2005;13:205-20 [DOI] [PubMed] [Google Scholar]
- 109.Wong NA, Melegh Z. Gastrointestinal stromal tumours can express CD10 and epithelial membrane antigen but not oestrogen receptor or HMB45. Histopathology 2011;59:781-5 [DOI] [PubMed] [Google Scholar]
- 110.Chen J, Guo T, Zhang L, et al. CD133 and CD44 are universally overexpressed in GIST and do not represent cancer stem cell markers. Genes Chromosomes Cancer 2012;51:186-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Debiec-Rychter M, Wasag B, Stul M, et al. Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol 2004;202:430-8 [DOI] [PubMed] [Google Scholar]
- 112.West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 2004;165:107-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Espinosa I, Lee CH, Kim MK, et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol 2008;32:210-8 [DOI] [PubMed] [Google Scholar]
- 114.Miettinen M, Wang ZF, Lasota J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol 2009;33:1401-8 [DOI] [PubMed] [Google Scholar]
- 115.Ardeleanu C, Arsene D, Hinescu M, et al. Pancreatic expression of DOG1: a novel gastrointestinal stromal tumor (GIST) biomarker. Appl Immunohistochem Mol Morphol 2009;17:413-8 [DOI] [PubMed] [Google Scholar]
- 116.Liegl B, Hornick JL, Corless CL, et al. Monoclonal antibody DOG1.1 shows higher sensitivity than KIT in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes. Am J Surg Pathol 2009;33:437-46 [DOI] [PubMed] [Google Scholar]
- 117.Akpalo H, Lange C, Zustin J.Discovered on gastrointestinal stromal tumour 1 (DOG1): a useful immunohistochemical marker for diagnosing chondroblastoma. Histopathology 2012;60:1099-106 [DOI] [PubMed] [Google Scholar]
- 118.Lasota J, Miettinen M.Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008;53:245-66 [DOI] [PubMed] [Google Scholar]
- 119.McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med 2007;357:1054-6 [DOI] [PubMed] [Google Scholar]
- 120.Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008;16:79-88 [DOI] [PubMed] [Google Scholar]
- 121.Andersson J, Sihto H, Meis-Kindblom JM, et al. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005;29:1170-6 [DOI] [PubMed] [Google Scholar]
- 122.Kinoshita K, Hirota S, Isozaki K, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol 2004;202:80-5 [DOI] [PubMed] [Google Scholar]
- 123.Agarwal R, Robson M.Inherited predisposition to gastrointestinal stromal tumor. Hematol Oncol Clin North Am 2009;23:1-13 vii. [DOI] [PubMed] [Google Scholar]
- 124.Chompret A, Kannengiesser C, Barrois M, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004;126:318-21 [DOI] [PubMed] [Google Scholar]
- 125.Kirsch R, Gao ZH, Riddell R. Gastrointestinal stromal tumors: diagnostic challenges and practical approach to differential diagnosis. Adv Anat Pathol 2007;14:261-85 [DOI] [PubMed] [Google Scholar]
- 126.Miettinen M, Sarlomo-Rikala M, Sobin LH, et al. Esophageal stromal tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 17 cases and comparison with esophageal leiomyomas and leiomyosarcomas. Am J Surg Pathol 2000;24:211-22 [DOI] [PubMed] [Google Scholar]
- 127.Sarlomo-Rikala M, Miettinen M.Gastric schwannoma-- a clinicopathological analysis of six cases. Histopathology 1995;27:355-60 [DOI] [PubMed] [Google Scholar]
- 128.Montgomery E, Torbenson MS, Kaushal M, et al. Beta-catenin immunohistochemistry separates mesenteric fibromatosis from gastrointestinal stromal tumor and sclerosing mesenteritis. Am J Surg Pathol 2002;26:1296-301 [DOI] [PubMed] [Google Scholar]
- 129.Bhattacharya B, Dilworth HP, Iacobuzio-Donahue C, et al. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol 2005;29:653-9 [DOI] [PubMed] [Google Scholar]
- 130.Carlson JW, Fletcher CD. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cell lesions: analysis of a series and review of the literature. Histopathology 2007;51:509-14 [DOI] [PubMed] [Google Scholar]
- 131.Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol 2002;15:931-8 [DOI] [PubMed] [Google Scholar]
- 132.Ozolek JA, Sasatomi E, Swalsky PA, et al. Inflammatory fibroid polyps of the gastrointestinal tract: clinical, pathologic, and molecular characteristics. Appl Immunohistochem Mol Morphol 2004;12:59-66 [DOI] [PubMed] [Google Scholar]
- 133.Lasota J, Wang ZF, Sobin LH, et al. Gain-of-function PDGFRA mutations, earlier reported in gastrointestinal stromal tumors, are common in small intestinal inflammatory fibroid polyps. A study of 60 cases. Mod Pathol 2009;22:1049-56 [DOI] [PubMed] [Google Scholar]
- 134.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 2011;11:865-78 [DOI] [PubMed] [Google Scholar]
- 135.Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093-103 [DOI] [PubMed] [Google Scholar]
- 136.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tamborini E, Pricl S, Negri T, et al. Functional analyses and molecular modeling of two c-Kit mutations responsible for imatinib secondary resistance in GIST patients. Oncogene 2006;25:6140-6 [DOI] [PubMed] [Google Scholar]
- 138.NCCN. Clinical Practice Guidelines in Oncology, Soft tissue sarcoma. Available online: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp Accessed 2012.
- 139.Casali PG, Blay JY. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21:v98-102 [DOI] [PubMed] [Google Scholar]
- 140.Miettinen M, Lasota J.Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70-83 [DOI] [PubMed] [Google Scholar]
- 141.Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)--update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 2007;5:S1-29; quiz S30. [PubMed]
- 142.Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol 2009;10:1045-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rutkowski P, Nowecki ZI, Michej W, et al. Risk criteria and prognostic factors for predicting recurrences after resection of primary gastrointestinal stromal tumor. Ann Surg Oncol 2007;14:2018-27 [DOI] [PubMed] [Google Scholar]
- 144.Takahashi T, Nakajima K, Nishitani A, et al. An enhanced risk-group stratification system for more practical prognostication of clinically malignant gastrointestinal stromal tumors. Int J Clin Oncol 2007;12:369-74 [DOI] [PubMed] [Google Scholar]
- 145.Joensuu H.Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008;39:1411-9 [DOI] [PubMed] [Google Scholar]
- 146.Edge SB, American Cancer Society, American College of Surgeons, et al. AJCC cancer staging handbook from the AJCC cancer staging manual 16. Gastrointestinal Stromal Tumor Springer New York 2010:16-1-16-2.
- 147.Miettinen M, Fletcher CDM, Kindblom LG, et al. WHO classification of tumours of the digestive system: Mesenchymal tumor of the stomach Bosman FT World Health Organization classification of tumours. International Agency for Research on Cancer Lyon 2010; 74-79. [Google Scholar]
- 148.(UICC). IUaC. TNM classification of malignant tumours Sobin LH, Gospodarowicz MK and Wittekind C Wiley-Blackwell Chichester, West Sussex, UK ; Hoboken, NJ 2010.
- 149.Agaimy A.Gastrointestinal stromal tumors (GIST) from risk stratification systems to the new TNM proposal: more questions than answers? A review emphasizing the need for a standardized GIST reporting. Int J Clin Exp Pathol 2010;3:461-71 [PMC free article] [PubMed] [Google Scholar]
- 150.Gervaz P, Huber O, Morel P.Surgical management of gastrointestinal stromal tumours. Br J Surg 2009;96:567-78 [DOI] [PubMed] [Google Scholar]
- 151.Pierie JP, Choudry U, Muzikansky A, et al. The effect of surgery and grade on outcome of gastrointestinal stromal tumors. Arch Surg 2001;136:383-9 [DOI] [PubMed] [Google Scholar]
- 152.Rossi CR, Mocellin S, Mencarelli R, et al. Gastrointestinal stromal tumors: from a surgical to a molecular approach. Int J Cancer 2003;107:171-6 [DOI] [PubMed] [Google Scholar]
- 153.Dematteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer 2008;112:608-15 [DOI] [PubMed] [Google Scholar]
- 154.Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumour. Br J Surg 2010;97:1854-9 [DOI] [PubMed] [Google Scholar]
- 155.Demetri GD, Antonia S, Benjamin RS, et al. Soft tissue sarcoma. J Natl Compr Canc Netw 2010;8:630-74 [DOI] [PubMed] [Google Scholar]
- 156.Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Administration: USFaD. FDA approves Gleevec to prevent recurrence of rare gastrointestinal cancer. Available online: http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm129210.htm 2008
- 158.Nunobe S, Sano T, Shimada K, et al. Surgery including liver resection for metastatic gastrointestinal stromal tumors or gastrointestinal leiomyosarcomas. Jpn J Clin Oncol 2005;35:338-41 [DOI] [PubMed] [Google Scholar]
- 159.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620-5 [DOI] [PubMed] [Google Scholar]
- 160.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364:1127-34 [DOI] [PubMed] [Google Scholar]
- 161.Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kontogianni-Katsarou K, Dimitriadis E, Lariou C, et al. KIT exon 11 codon 557/558 deletion/insertion mutations define a subset of gastrointestinal stromal tumors with malignant potential. World J Gastroenterol 2008;14:1891-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Patel S, Zalcberg JR. Optimizing the dose of imatinib for treatment of gastrointestinal stromal tumours: lessons from the phase 3 trials. Eur J Cancer 2008;44:501-9 [DOI] [PubMed] [Google Scholar]
- 164.Janeway KA, Albritton KH, van Den Abbeele AD, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 2009;52:767-71 [DOI] [PubMed] [Google Scholar]
- 165.Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 2008;14:3204-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Andtbacka RH, Ng CS, Scaife CL, et al. Surgical resection of gastrointestinal stromal tumors after treatment with imatinib. Ann Surg Oncol 2007;14:14-24 [DOI] [PubMed] [Google Scholar]
- 167.Bonvalot S, Eldweny H, Pechoux CL, et al. Impact of surgery on advanced gastrointestinal stromal tumors (GIST) in the imatinib era. Ann Surg Oncol 2006;13:1596-603 [DOI] [PubMed] [Google Scholar]
- 168.Rutkowski P, Nowecki Z, Nyckowski P, et al. Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J Surg Oncol 2006;93:304-11 [DOI] [PubMed] [Google Scholar]
- 169.Pisters PW, Patel SR. Gastrointestinal stromal tumors: current management. J Surg Oncol 2010;102:530-8 [DOI] [PubMed] [Google Scholar]
- 170.Lopes LF, Bacchi CE. Imatinib treatment for gastrointestinal stromal tumour (GIST). J Cell Mol Med 2010;14:42-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McAuliffe JC, Hunt KK, Lazar AJ, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol 2009;16:910-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reichardt P, Blay JY, Mehren M. Towards global consensus in the treatment of gastrointestinal stromal tumor. Expert Rev Anticancer Ther 2010;10:221-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 2007;25:1753-9 [DOI] [PubMed] [Google Scholar]
- 174.Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol 2007;25:1760-4 [DOI] [PubMed] [Google Scholar]
- 175.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47 [DOI] [PubMed] [Google Scholar]
- 176.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24:4764-74 [DOI] [PubMed] [Google Scholar]
- 177.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005;128:270-9 [DOI] [PubMed] [Google Scholar]
- 178.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bauer S, Yu LK, Demetri GD, et al. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 2006;66:9153-61 [DOI] [PubMed] [Google Scholar]
- 180.Guo T, Hajdu M, Agaram NP, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res 2009;15:6862-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nishida T, Takahashi T, Nishitani A, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol 2009;14:143-9 [DOI] [PubMed] [Google Scholar]
- 182.Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007;370:2011-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Desai J, Yassa L, Marqusee E, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med 2006;145:660-4 [DOI] [PubMed] [Google Scholar]
- 184.Montemurro M, Schoffski P, Reichardt P, et al. Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib. Eur J Cancer 2009;45:2293-7 [DOI] [PubMed] [Google Scholar]
- 185.Reichardt P, Montemurro M.Clinical experience to date with nilotinib in gastrointestinal stromal tumors. Semin Oncol 2011;38:S20-7 [DOI] [PubMed] [Google Scholar]
- 186.Dewaele B, Wasag B, Cools J, et al. Activity of dasatinib, a dual SRC/ABL kinase inhibitor, and IPI-504, a heat shock protein 90 inhibitor, against gastrointestinal stromal tumor-associated PDGFRAD842V mutation. Clin Cancer Res 2008;14:5749-58 [DOI] [PubMed] [Google Scholar]
- 187.Guo T, Agaram NP, Wong GC, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res 2007;13:4874-81 [DOI] [PubMed] [Google Scholar]
- 188.Soria JC, Massard C, Magne N, et al. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur J Cancer 2009;45:2333-41 [DOI] [PubMed] [Google Scholar]
- 189.NIH. Clinical Trial Database. Available online: http://clinicaltrials.gov/ 2012