

Abstract
Colorectal carcinoma is one of the most common cancers and one of the leading causes of cancer-related death in the United States. Pathologic examination of biopsy, polypectomy and resection specimens is crucial to appropriate patient managemnt, prognosis assessment and family counseling. Molecular testing plays an increasingly important role in the era of personalized medicine. This review article focuses on the histopathology and molecular pathology of colorectal carcinoma and its precursor lesions, with an emphasis on their clinical relevance.
Key Words: Colorectal carcinoma, pathology, adenoma, molecular, MSI, KRAS, BRAF
Introduction
Colorectal carcinoma is the third most common cancer in the United States after prostate and lung/bronchus cancers in men and after breast and lung/bronchus cancers in women. It is also the third leading cause of cancer-related death in the United States after lung/bronchus and prostate cancers in men and after lung/bronchus and breast cancers in women (1). In 2011, an estimated 141,210 new cases of colorectal carcinoma were diagnosed in United States, with an estimated 49,380 deaths, representing approximately 9% of all newly diagnosed cancers and all cancer-related deaths (excluding basal and squamous cell skin cancers).
With the rapid therapeutic advancement in the era of personalized medicine, the role of pathologists in the management of patients with colorectal carcinoma has greatly expanded from traditional morphologists to clinical consultants for gastroenterologists, colorectal surgeons, oncologists and medical geneticists. In addition to providing accurate histopathologic diagnosis, pathologists are responsible for accurately assessing pathologic staging, analyzing surgical margins, searching for prognistic parameters that are not included in the staging such as lymphovascular and perineural invasion, and assessing therapeutic effect in patients who have received neoadjavant therapy. Pathologists also play a central role in analyzing histologic features of the tumors that are suggestive of microsatellite instability (MSI), selecting appropriate tissue sections for MSI testing and mutation analysis for KRAS and BRAF, and interpreting the results of these important therapeutic and prognostic tests (2).
This review article focuses on the histolopathology of colorectal carcinoma and its precursor lesions. Recent advances in molecular pathology and molecular tests are discussed. Their clinical relevance is emphasized.
Histopathologic diagnosis of colorectal carcinoma
More than 90% of colorectal carcinomas are adenocarcinomas originating from epithelial cells of the colorectal mucosa (3). Other rare types of colorectal carcinomas include neuroendocrine, squamous cell, adenosquamous, spindle cell and undifferentiated carcinomas. Conventional adenocarcinoma is characterized by glandular formation, which is the basis for histologic tumor grading. In well differentiated adenocarcinoma >95% of the tumor is gland forming. Moderately differentiated adenocarcinoma shows 50-95% gland formation. Poorly differentiated adenocarcinoma is mostly solid with <50% gland formation. In practice, most colorectal adenocarcinomas (~70%) are diagnosed as moderately differentiated (Figure 1). Well and poorly differentiated carcinomas account for 10% and 20%, respectively.
Figure 1.
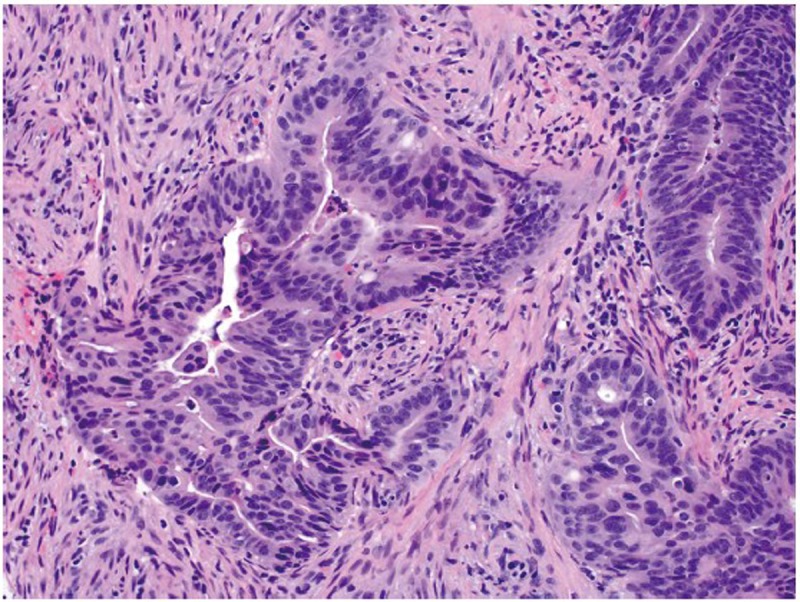
An example of moderately differentiated adenocarcinoma showing complicated glandular structures in a desmoplastic stroma (original magnification ×200)
It is apparent that the determination of tumor grade is a subjective exercise. Many studies have demonstrated that a 2-tiered grading system, which combines well and moderately differentiated to low grade (50% gland formation) and defines poorly differentiated as high grade (<50% gland formation), reduces interobserver variation and improves prognostic significance (4,5). Though controversial, tumor grade is generally considered as a stage-independent prognostic variable, and high grade or poorly differentiated histology is associated with poor patient survival (6-8). It should be emphasized, however, that histologic grading should apply only to conventional adenocarcinoma. Some of the histologic variants, which will be discussed later, may show high grade morphology but behave as low grade tumors because of their MSI status.
The vast majority of colorectal carcinomas are initially diagnosed by endoscopic biopsy or polypectomy. The key aspect of microscopic examination is to look for evidence of invasion. However, this can be difficult when the biopsy is superficial or poorly oriented. If the muscularis mucosae can be identified, it is important to determine whether it is disrupted by neoplastic cells. Invasive carcinoma typically invades through the muscularis mucosae into the submucosa, and is sometimes seen in close proximity to submucosal blood vessels. Another important feature of invasion is the presence of desmoplasia or desmoplastic reaction (Figure 2), a type of fibrous proliferation surrounding tumor cells secondary to invasive tumor growth. Invasive colorectal carcinoma also frequently shows characteristic necrotic debris in glandular lumina, so-called “dirty necrosis” (Figure 3). This unique feature can be quite useful to suggest a colorectal primary when a metastasis of unknown origin is encountered.
Figure 2.
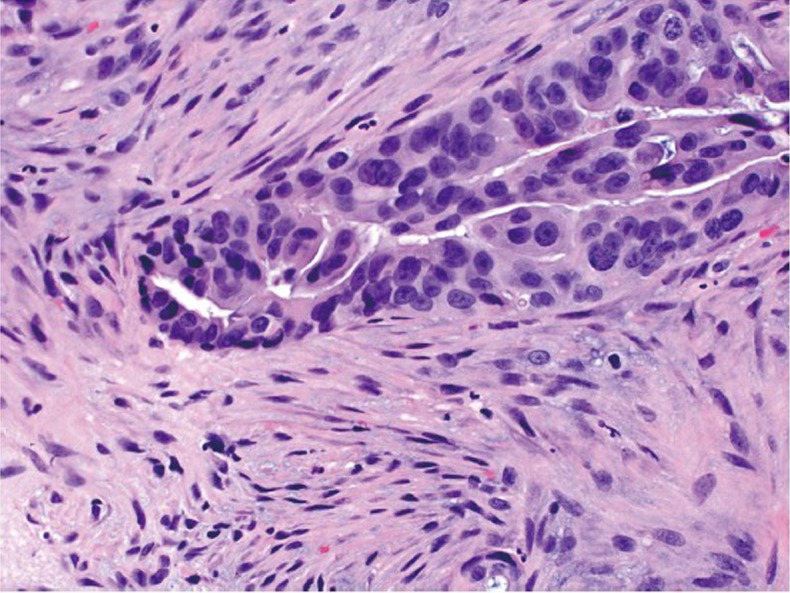
Desmoplastic reaction characterized by proliferation of spindle cells surrounding an adenocarcinomatous gland (original magnification ×400)
Figure 3.
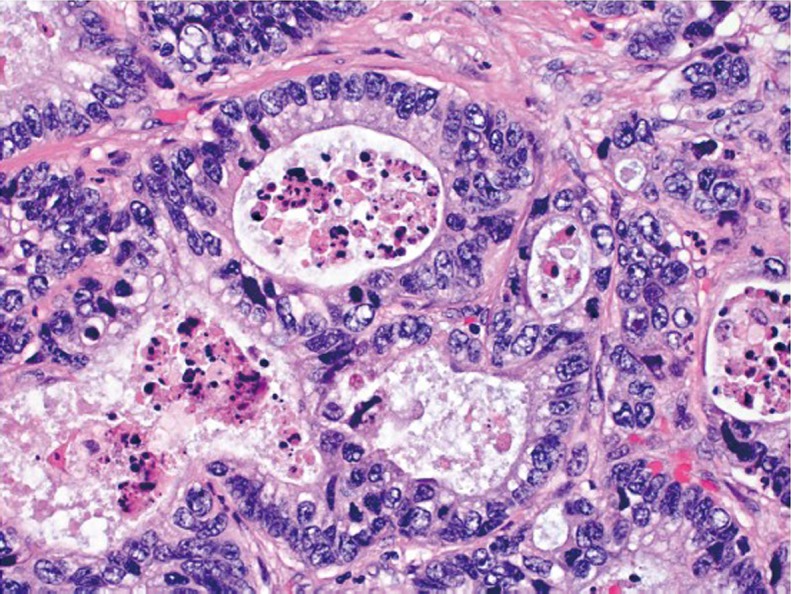
Necrotic debris (“dirty necrosis”) within the lumina of adenocarcinomatous glands (original magnification ×400)
It should be noted that when a diagnosis of invasive carcinoma is rendered, it means that carcinoma has at least invaded into the submucosa of the colorectum. This differs from the concept of invasion in other parts of the gastrointestinal tract (esophagus, stomach and small intestine), where the presence of mucosal invasion is sufficient for the diagnosis of invasive carcinoma (pT1). In the colorectum, submucosal invasion is required for the diagnosis of a pT1 tumor. For reasons that are not entirely clear but generally thought to be due to the relative paucity of lymphatics, invasion confined to the lamina propria and muscularis mucosae has no risk of nodal or distant metastasis. Thus, intramucosal carcinoma is preferably called high grade dysplasia (discussed later) by pathologists in order to avoid unnecessary surgical intervention. In the American Joint Committee on Cancer (AJCC) Cancer Staging Manual (9), mucosal invasion is classified as carcinoma in situ (Tis). Nevertheless, the term of intramucosal carcinoma may still be used by some pathologists. No matter what term is used by pathologists, the identification of high grade dysplasia or intramucosal carcinoma in a biopsy or polypectomy specimen should not affect the decision-making for patient management. The decision to perform surgical resection should be ultimately determined by the gross appearance of the lesion, endoscopic ultrasound findings, and endoscopic resectability.
Histologic variants
In World Health Organization (WHO) classification, a number of histologic variants of colorectal carcinomas are listed, such as mucinous, signet ring cell, medullary, micropapillary, serrated, cribriform comedo-type, adenosquamous, spindle cell, and undifferentiated. Only the first 3 variants are discussed here.
Mucinous adenocarcinoma
This special type of colorectal carcinoma is defined by >50% of the tumor volume composed of extracellular mucin (3). Tumors with a significant mucinous component (>10%) but <50% are usually termed adenocarcinoma with mucinous features or mucinous differentiation. Mucinous adenocarcinoma typically shows large glandular structures with pools of extracellular mucin (Figure 4). A variable number of individual tumor cells, including signet ring cells, may be seen. The prognosis of mucinous adenocarcinoma in comparison with conventional adenocarcinoma has been controversial among different studies (10,11). Many mucinous adenocarcinomas occur in patients with hereditary nonpolyposis colorectal cancer (HNPCC or Lynch syndrome) and thus represent high-level MSI (MSI-H) tumors (12). These tumors are expected to behave in a low grade fashion. In contrast, mucinous adenocarcinomas that are microsatellite stable (MSS) are expected to behave more aggressively, particularly when detected at an advanced stage.
Figure 4.
Mucinous adenocarcinoma showing abundant extracellular mucin (original magnification ×200)
Signet ring cell adenocarcinoma
In contrast to that in the stomach, signet ring cell adenocarcinoma is rare in the colorectum, representing <1% of all colorectal carcinomas. Similar to mucinous carcinoma, signet ring cell carcinoma is defined by the presence of >50% of tumor cells showing signet ring cell features characterized by a prominent intracytoplasmic mucin vacuole that pushes the nucleus to the periphery (Figure 5). Signet ring cells may show an infiltrative growth pattern or are present within the pools of extracellular mucin. By definition, signet ring cell carcinoma is poorly differentiated (high grade) and carries a worse outcome than conventional adenocarcinoma (11,13,14). However, some signet ring cell carcinomas may be MSI-H tumors and thus may behave as low grade tumors biologically (3).
Figure 5.
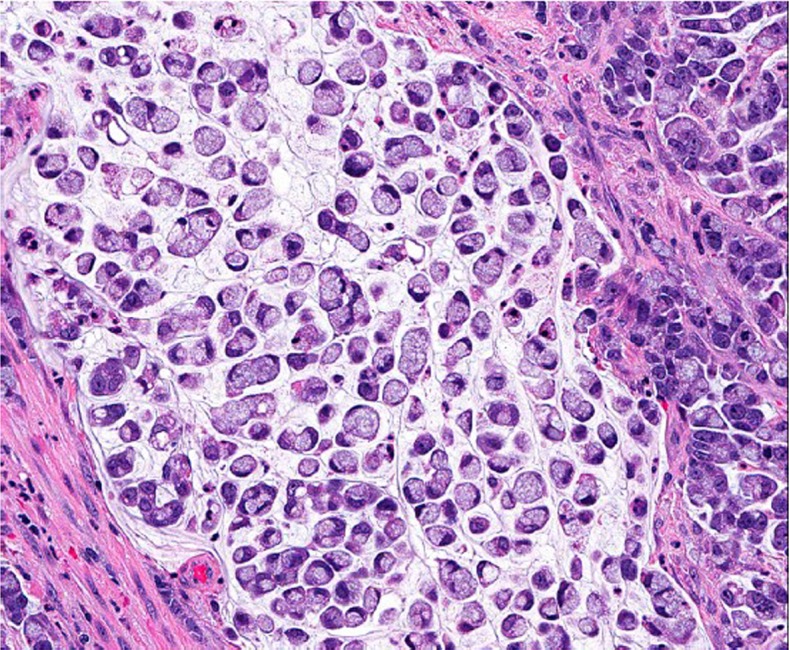
Signet ring cell carcinoma (original magnification ×400)
Medullary carcinoma
Medullary carcinoma is extremely rare, constituting approximately 5-8 cases for every 10,000 colorectal cancers diagnosed, with a mean annual incidence of 3.47 (±0.75) per 10 million population (15). This tumor is characterized by sheets of epithelioid neoplastic cells with large vesicular nuclei, prominent nucleoli, and abundant cytoplasm. It typically has a pushing border on resection specimens (Figure 6), and is characteristically associated with marked tumor-infiltrating lymphocytes (Figure 7). Medullary carcinoma is a distinctive histologic subtype that is strongly associated with MSI-H (16,17). It usually has a favorable prognosis despite its poorly differentiated or undifferentiated histology.
Figure 6.
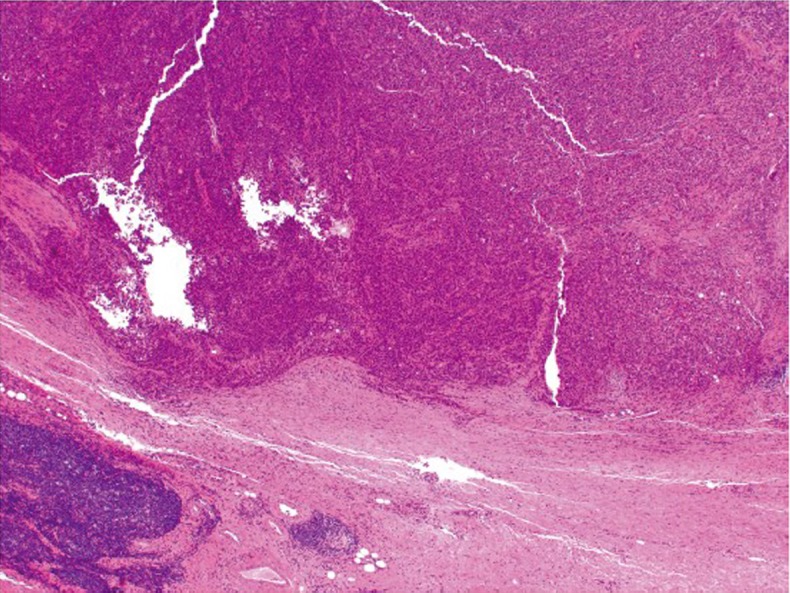
Medullary carcinoma showing a pushing border at the tumor edge (original magnification ×40)
Figure 7.
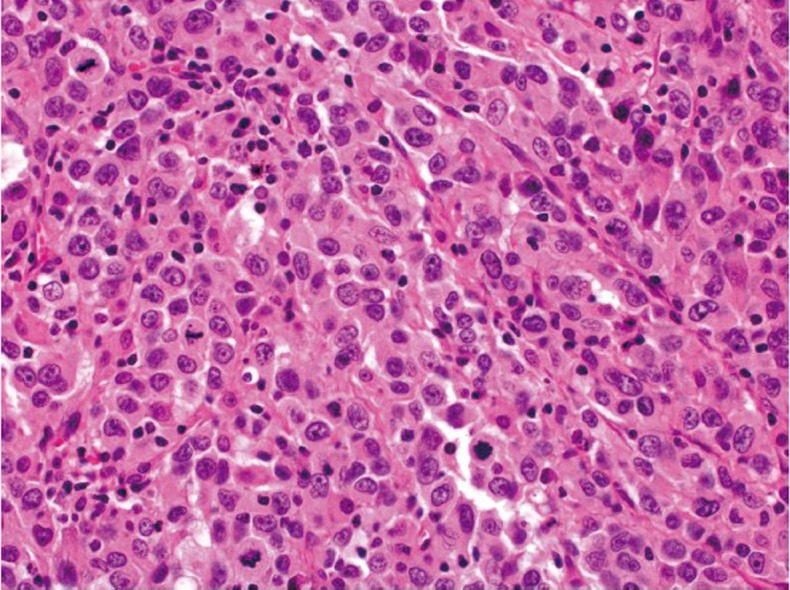
Medullary carcinoma showing poorly differentiated histology and tumor-infiltrating lymphocytes (original magnification ×400)
Immunohistochemical phenotype
The most widely used immunohistochemical markers for colorectal adenocarcinoma are cytokeratin (CK) 20, CK7 and CDX2. The most common immunophenotype of colorectal adenocarcinoma is positivity for CK20 and negativity for CK7, which is a relatively specific staining pattern for colorectal origin (18). However, up to 20% of the tumors may exhibit a CK7-positive/CK20 negative or CK7-negative/CK20-negative staining pattern. It has been suggested that reduced or absent CK20 expression in colorectal carcinoma is associated with MSI-H (19). CDX2 is a marker of enteric differentiation and is positive in >90% of colorectal adenocarcinomas (20,21). However, CDX2 can be positive in any carcinoma that shows enteric differentiation, and thus is not entirely colorectal-specific. Interestingly, medullary carcinomas of the colorectum are frequently CK20-negative and CDX2-negative, in line with the concept of MSI (16,19).
Pathologic staging
Tumor staging is by far the most important prognostic predictor of clinical outcome for patients with colorectal carcinoma. Histologic examination of surgically resected specimens serves an irreplaceable role in determining the depth of tumor invasion (T) and the extent of nodal metastasis (N). The histologic determination of T1 (tumor invades submucosa), T2 (tumor invades muscularis propria) and T3 (tumor invades through the muscularis propria into pericolorectal tissues) is usually straightforward when using the AJCC TNM staging system (9). However, determination of T4a (tumor penetrates to the surface of the visceral peritoneum) and T4b (tumor directly invades or is adherent to other organs or structures) can sometimes be problematic. First, serosal surface (visceral peritoneum) involvement can be missed if the specimen is not adequately sampled for histologic examination. Second, the serosal surface may be confused with the circumferential (radial) or mesenteric margin, which is a nonperitonealized surface created surgically by blunt or sharp dissection. A T3 tumor may involve the radial margin and a T4 tumor may have a negative radial margin. Third, a surgically induced perforation at the tumor site may be confused with true tumor perforation, which requires clarification from surgeons. Fourth, adherence of other organs or structures at the tumor site does not necessarily qualify for T4b. Histologically, the adherent site may show only inflammatory changes, abscess formation and/or fibrosis, but without direct tumor involvement. Finally, there is some confusion about the definition of visceral peritoneum involvement. Clearly, the interpretation of T4a can be unequivocal if, (I) tumor cells are present at the serosal surface with inflammatory reaction, mesothelial hyperplasia, and/or erosion; or (II) free tumor cells are seen on the serosal surface with underlying ulceration of the visceral peritoneum. However, identification of tumor cells close to, but not at, the serosal surface would be considered T4a by some investigators if there are associated mesothelial inflammatory and/or hyperplastic reactions (Figure 8) (22). Apparently, the application of this third criterion is prone to subjective judgment and lacks reproducibility. It is noted that in the updated cancer protocols and checklists by College of American Pathologists (CAP), only the first two criteria are listed as the diagnostic features of T4a, and the third criterion is deleted (23).
Figure 8.
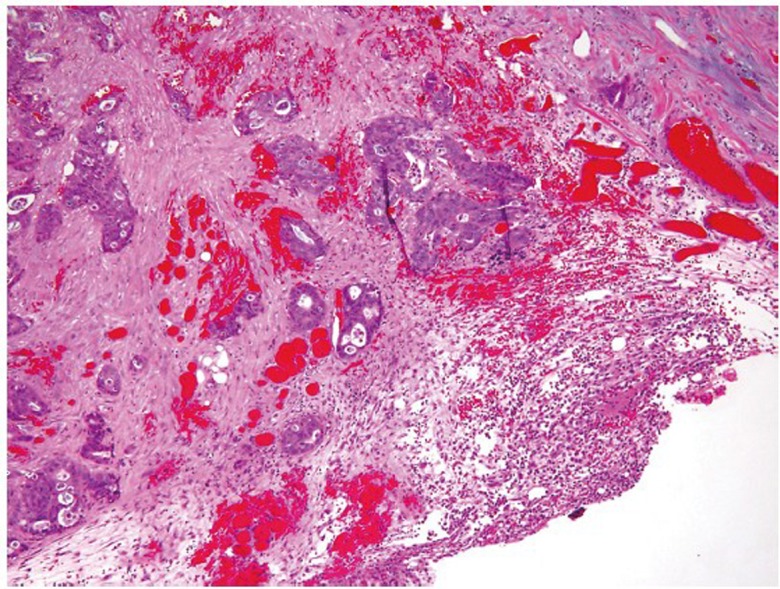
Tumor cells close to, but not at, the serosal surface, with mesothelial inflammatory and hyperplastic reactions, which may be considered T4a by some investigators (original magnification ×40)
It is pathologists’ obligation to retrieve as many lymph nodes as possible from surgical specimens. The vast majority of pathologists follow the guidelines of a minimum of 12 nodes (24). Extra efforts will be made if <12 nodes are retrieved, although this will increase the turnaround time for pathology reports. The extra efforts may include repeated manual searches, submitting more sections, utilizing fat clearance techniques (25,26), or ex vivo injection of methylene blue (27,28). The application of fat clearance techniques has several potential disadvantages, such as further delay in signout of the pathology reports, cost, toxicity and disposal of clearing solutions, and unknown effect on immunohistochemistry. As a result, fat clearance has not become a standard practice in pathology laboratories. Methylene blue injection is a relatively new method for colorectal cancer. There have been only a few publications in this area, mostly from the same study group (27,28). Its clinical application needs further investigation.
It should be realized that the total number of nodes retrieved is not only dissector-dependent, but also influenced by a number of specimen and patient variables. Studies have shown a positive correlation with the specimen length, pericolorectal fat width, female gender and tumor size; and a negative correlation with the age of patient and the rectosigmoid location of tumors (29,30). Not surprisingly, fewer than 12 nodes may be expected if patients have received preoperative neoadjuvant therapies (31,32). It is recommended that pathologists document the degree of diligence of their efforts to find lymph nodes in a specimen in pathology reports, if <12 nodes are retrieved.
One of the interesting issues in nodal staging is the interpretation of discrete tumor deposits in pericolorectal fat away from the main tumor but without identifiable residual lymph node tissue. In AJCC Cancer Staging Manual 5th edition, a tumor nodule >3 mm was counted as a positive node, whereas a nodule ≤3 mm was classified in the category of discontinuous extension (T3). In the 6th edition, tumor deposits were considered as positive nodes if they are round and have a smooth contour irrespective of size, but classified in the T category as well as venous invasion if they are irregular in shape. The current edition (7th edition) recognizes the fact that tumor deposits may represent discontinuous extension, venous invasion with extravascular spread, or truly totally replaced lymph nodes. Given their association with reduced disease-free and overall survival (33,34), these tumor deposits are now considered nodal metastasis, irrespective of size or contour, and are designated N1c in the absence of regional lymph node metastasis to favor additional postoperative treatment. However, if a single positive lymph node is also identified, the N stage will be changed from N1c to N1a. The presence of discontinuous tumor deposits does not change the T stage in the 7th edition (9).
The prognostic significance of isolated tumor cells (ITCs), defined as single tumor cells or small clusters of tumor cells ≤0.2 mm, detected by either immunohistochemical staining or standard hematoxylin and eosin staining in regional lymph nodes remains unclear at present. In the absence of overt nodal metastasis, ITCs are classified as N0 but annotated as N0 (i+) with “i” standing for “isolated tumor cells”. On the other hand, micrometastasis (>0.2 mm but ≤2.0 mm) is reported as N1(mic). The number of lymph nodes involved by ITCs or micrometastasis should be stated (9,23).
Pathology reporting
Most pathologists use standardized synoptic report for colorectal carcinoma following the checklist recommended by CAP (23). The details that should be included in the report are specimen type, tumor site, tumor size, macroscopic tumor perforation, histologic type, histologic grade, microscopic tumor extension, margins (proximal, distal and radial), treatment effect (for tumors treated with neoadjuvant therapy), lymphovascular invasion, perineural invasion, tumor deposits (discontinuous extramural extension), TNM staging (including the total number of lymph nodes examined and the total number of nodes involved). Some pathology reports may also include leading edge of the tumor (infiltrative or expansile), presence or absence of tumor budding, and assessment of histologic features that are suggestive of MSI such as tumor-infiltrating lymphocytes, peritumoral Crohn-like lymphoid response and the percentage of mucinous component.
Specimen handling and sampling
In pathology laboratories, surgically resected specimens are processed in a systematic manner to ensure completeness and accuracy of pathology report. The external surface of the specimen is inspected before opening for possible serosal involvement, radial margin involvement, tumor perforation, and distant tumor implants. For rectal resections, the intactness of the mesorectum is examined. Once the specimen is oriented and the specimen is measured, the radial margin around tumor is inked. The specimen is then opened, usually along the antimesenteric border with an attempt to avoid cutting through the tumor. The location and size of the tumor and its distance from the closest margin are recorded. Small portions of fresh tumor and nonneoplastic tissues may be procured for tissue bank, but this should not compromise the quantity of tumor for diagnosis.
The opened and cleaned specimen is pinned down on a wax board and immersed in an adequate volume of formalin for fixation overnight. The tumor is then sliced at 3-4 mm intervals to assess the depth of invasion. The rest of the specimen is also examined for additional lesions. Adequate sections of the tumor (usually 5 sections depending on the size of the tumor) should be submitted for microscopic examination to include the area of deepest invasion and to maximize the chance to find lymphovascular and perineural invasion. Including both tumor and adjacent uninvolved colorectum in the same sections is desirable because there is always a possibility that the case may be used for research in the future. Additional sections should include proximal and distal margins, radial margin (if not included in tumor sections), any additional polyps or lesions, and random uninvolved colorectum.
After taking the above sections, the mesenteric fat or pericolorectal soft tissue is stripped off and dissected for lymph nodes. All grossly negative lymph nodes are entirely submitted for microscopic examination. Grossly positive lymph nodes may be submitted in part or entirely depending on their size.
A polypectomy specimen is inked at the cauterized base, but the stalk may retract and thus be difficult to identify. The specimen is either bisected or serially sectioned depending on its size, and entirely submitted. Sectioning should follow the vertical plane of the stalk to maximize the histologic evaluation of polypectomy margin and submucosal involvement. If the specimen is received in multiple pieces, however, margin evaluation may become impossible.
Precursor lesions
It has been well established that the vast majority of colorectal adenocarcinomas derive from precursor lesions such as adenomas and dysplasia. Residual adenoma is a common finding in colorectal adenocarcinomas. Endoscopic polypectomy decreases the incidence of colorectal cancers in treated population and prevents death from colorectal cancer (35,36). Some of the common precursor lesions are discussed here.
Adenomas
At least half of adults in Western countries will have an adenomatous polyp in their lifetime and one-tenth of these lesions will progress to adenocarcinoma (37). The risk increases after the age of 50. Endoscopically, adenomas can be pedunculated or sessile. By definition, adenomas are clonal lesions that show at least low grade dysplasia characterized by enlarged, hyperchromatic and elongated (pencillate) nuclei arranged in a stratified configuration along the basement membrane. The adenomatous cells may show mucin depletion and increased apoptotic activity. Interestingly, adenomatous polyps appear to develop through a “top-down” mechanism (38). As such, small lesions will often only have adenomatous epithelium in their superficial portions.
Conventional adenomas are subclassified as tubular, tubulovillous and villous based on their architectural features (Figure 9). Tubular adenomas are composed of simple crypt-like dysplastic glands and contain <25% villous component. Villous adenomas consist of >75% villous component that resemble finger-like projections. Tubulovillous adenomas are intermediate lesions with 25-75% villous component. Adenomas that are large in size (>1 cm) or predominantly villous, or contain high grade dysplasia (discussed below) are considered “advanced adenomas” (39), which require more aggressive endoscopic surveillance.
Figure 9.

Examples of tubular adenoma (A. original magnification ×200), and tubulovillous adenoma (B. original magnification ×100)
Serrated polyps
Serrated polyp is a general term for any polyp that shows a serrated (sawtooth or stellate) architecture of the epithelial compartment. It is a heterogeneous group of lesions that mainly include hyperplastic polyp, sessile serrated adenoma/polyp, and traditional serrated adenoma (40).
Hyperplastic polyps (HP) are the most common serrated lesions that are more likely to be found in the distal colon and generally small in size (<5 mm). Only rare HPs are >1 cm. Endoscopically, HPs can be difficult to distinguish from adenomas (41). Histologically, HPs are characterized by a simple tubular architecture with elongated and straight crypts and by luminal serration that is more pronounced in the upper portions of the crypts with an appearance of surface maturation (Figure 10). The proliferation zone is limited to the basal portion of the crypts, which remains narrow and is not serrated (42). HPs can be further divided into microvesicular, goblet cell and mucin-poor subtypes (43), but this histologic subclassification does not appear to have any clinical relevance.
Figure 10.
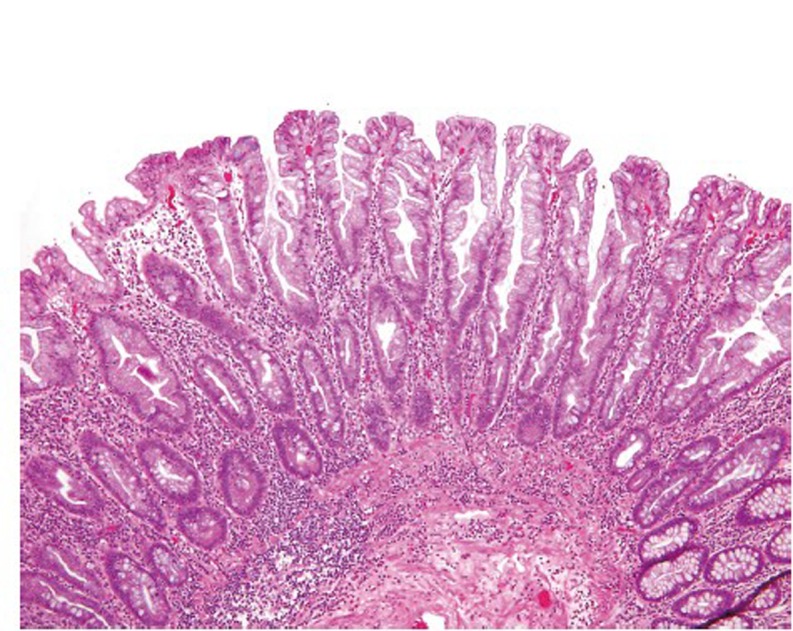
Hyperplastic polyp (original magnification ×100)
Sessile serrated adenoma (SSA) and sessile serrated polyp (SSP) refer to the same serrated lesion and are currently used interchangeably. SSA/Ps are more commonly seen in the proximal colon and are usually larger than HPs (44). Histologic diagnosis of SSA/Ps are entirely based on architectural features characterized by exaggerated crypt serration, serration throughout the crypt length, hypermucinous epithelium, crypt dilatation, crypt branching, horizontal crypt extensions at the crypt base, and aberrant proliferation (45). Despite the name, SSA/Ps lack the dysplastic nuclear changes that characterize conventional adenomas. It should be remembered that SSA/P is a relatively new entity that used to be classified as HP in the past. Thus, pathologists may have difficulty to separate between SSA/P and HP on histologic ground (46-48). In cases where the separation is not easy, a descriptive diagnosis of “serrated polyp” with a comment may be rendered.
Nevertheless, the separation of SSA/P from HP appears important because SSA/P is now thought to be the precursor lesion for colorectal carcinomas with MSI and probably also for CpG island methylated MSS carcinomas (40), whereas HP is generally believed to be innocuous. The most reliable features for SSA/P to distinguish from HP are dilation of the crypts at the base, often assuming a L, inverted T, or anchor-shaped configuration (Figure 11). These unusual shapes (“architectural dysplasia”) are often observed in two or more contiguous crypts and are thought to result from abnormal proliferation and/or decreased apoptosis (42-44). Given the presumed premalignant potential, it is probably warranted for patients with SSA/Ps to undergo endoscopic surveillance similar to those with conventional adenomas. In addition, a subset of these lesions may potentially progress to carcinoma more rapidly than conventional adenomas. Patients with these lesions may thus need an even more aggressive endoscopic surveillance (49,50).
Figure 11.

Low power (A. original magnification ×40) and high power (B. original magnification ×200) views of sessile serrated polyp. Note the presence of basal serration
Traditional serrated adenoma (TSA) is a unique and uncommon type of true adenoma that exhibits low grade nuclear dysplasia similar to that seen for conventional adenoma, and also shows a serrated architecture similar to that seen for HP and SSA/P. Prominent cytoplasmic eosinophilia and a villous growth pattern are characteristic (Figure 12).
Figure 12.
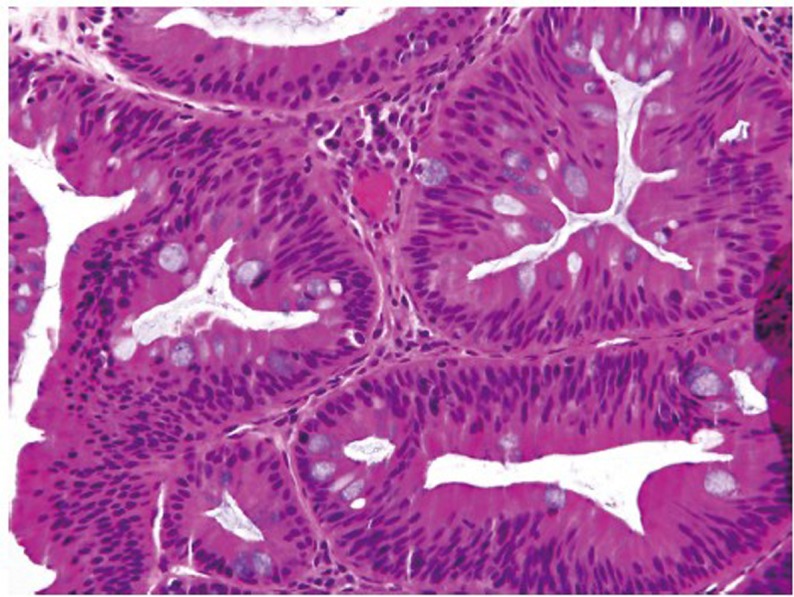
An example of traditional serrated adenoma (original magnification ×400). Note the presence of luminal serration, low grade cytologic dysplasia and cytologic eosinophilia
Dysplasia in inflammatory bowel disease
Inflammatory bowel disease (IBD) is a well-known risk factor for the development of dysplasia and carcinoma. Dysplastic lesions in the setting of IBD can be flat (endoscopically invisible) or raised (51,52), which are both graded as indefinite for dysplasia, low grade dysplasia or high grade dysplasia. Raised lesions are commonly termed dysplasia-associated lesions or masses (DALMs) and can be difficult or impossible to distinguish from sporadic adenomas. However, several studies have shown that adenoma-like lesions in IBD patients, regardless of whether it represents an IBD-associated DALM lesion or a sporadic adenoma, can be adequately managed by polypectomy and continued endoscopic surveillance if there is no coexisting flat dysplasia (53-55).
Given the treatment implications, it is recommended that the diagnosis of dysplasia in the setting of IBD be confirmed by an experienced pathologist (56). The diagnosis of indefinite for dysplasia should not become a waste basket, and should be reserved for cases showing worrisome cytologic and architectural changes but also showing surface maturation or abundant inflammation. The diagnosis is also appropriate if the mucosal surface cannot be evaluated due to tangential sectioning of the tissue, the presence of marked cautery effect, or the presence of other processing artifacts.
Lynch syndrome
Lynch syndrome is the most common inherited colorectal cancer syndrome (57). It is characterized by increased lifetime cancer risks primarily in the gastrointestinal and gynecologic tracts, with colorectal and endometrial carcinomas being most common. The cumulative lifetime risk for colorectal cancer is estimated to be 66% for men and 43% for women (58). Patients with Lynch syndrome tend to develop mucinous, poorly differentiated, undifferentiated, or medullary carcinomas in the right colon at a relatively young age. Tumor-infiltrating lymphocytes and Crohn-like peritumoral lymphoid reaction may be prominent.
Lynch syndrome results from germline mutation in one of the four DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2), and is inherited in an autosomal dominant mood. Almost 90% of patients have mutations in either MLH1 or MSH2 gene (57,59). Mutations in MSH6 and PMS2 genes are much less frequent. The diagnosis is established by following Amsterdam Criteria II (Table 1) (60) and MSI testing following the revised Bethesda guidelines (Table 2) (61). Patients with a MSI tumor but without an identifiable germline defect in a MMR gene may still have Lynch syndrome if other causes of MSI, such as methylation of the MLH1 promoter, are excluded.
Table 1. Amsterdam criteria II for Lynch syndrome (60).
| There should be at least three relatives with a Lynch syndrome-associated cancer (colorectal cancer, cancer of the endometrium, small bowel, ureter or renal pelvis) |
| All of the following criteria should be present |
| 1. One should be a first-degree relative of the other two |
| 2. At least two successive generations should be affected |
| 3. At least one should be diagnosed before the age of 50 years |
| 4. Familial adenomatous polyposis should be excluded in colorectal cancer case(s), if any |
| 5. Tumors should be verified by pathological examination |
Table 2. Revised Bethesda guidelines for MSI testing (61).
| 1. Colorectal cancer diagnosed in a patient who is less than 50 years of age |
| 2. Presence of synchronous, metachronous colorectal, or other Lynch syndrome-related tumors,*regardless of age |
| 3. Colorectal cancer with the MSI-H histology,**diagnosed in a patient who is less than 60 years of age |
| 4. Colorectal cancer diagnosed in one or more first-degree relatives with a Lynch syndrome-related tumor, with one of the cancers being diagnosed under age 50 years |
| 5. Colorectal cancer diagnosed in two or more first- or second-degree relatives with Lynch syndrome-related tumors, regardless of age |
*Lynch syndrome-related tumors include colorectal, endometrial, stomach, ovarian, pancreas, ureter, renal pelvis, biliary tract, and brain (usually glioblastoma) tumors, sebaceous gland adenomas, keratoacanthomas, and carcinoma of the small bowel; **Presence of tumor-infiltrating lymphocytes, Crohn-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern
Familial adenomatous polyposis
Familial adenomatous polyposis (FAP) is a rare autosomal dominant inherited colorectal cancer syndrome (62,63), characterized by early development of hundreds to thousands of adenomatous polyps in the colorectum (Figure 13). If left untreated, there is an almost inevitable progression to colorectal cancer at an average age of 35-40 years (63,64). These patients are also at risk of developing adenomatous polyps in the small bowel (65) and fundic gland polyps in the stomach (66). Although syndromic fundic gland polyps more frequently show low grade dysplasia than sporadic counterparts (67-69), the likelihood to progress to high grade dysplasia or invasive carcinoma is exceedingly low.
Figure 13.

A case of familial adenomatous polyposis. Note the presence of innumerable polyps in the colon
The diagnostic criteria for FAP include: (I) 100 colorectal adenomatous polyps; (II) germline mutation of the adenomatous polyposis coli (APC) gene; or (III) family history of FAP and any number of adenomas at a young age (70). Patients with attenuated FAP have <100 colorectal adenomatous polyps, usually averaging approximately 30. Their lifetime risk to develop colorectal cancers drops to roughly 70% and most patients tend to develop cancers later in life (63,71). Gardner syndrome is a variant of FAP. Patients with this syndrome also have epidermoid cysts, osteomas, dental anomalies and desmoid tumors. Turcot syndrome is another variant which includes brain tumors, typically medulloblastoma (70).
The APC tumor suppressor gene is a large gene that contains 21 exons spanning a region of 120 kb and encoding a 2,843 amino-acid protein. Most of the germline mutations are nonsense and frameshift mutations and cluster within a “hot spot” in the largest exon 15 (72,73), leading to the synthesis of a truncated protein, which, in turn, leads to aberrant nuclear accumulation of β-catenin and subsequent activation of the β-catenin/Tcf transcription factor complex to promote uncontrolled activation of the Wnt signaling pathway of tumorigenesis (74).
Peutz-Jeghers syndrome
This is an autosomal dominant inherited cancer syndrome characterized by hamartomatous polyps in the gastrointestinal tract, pigmented mucocutaneous lesions, and an increased risk of gastrointestinal and extragastrointestinal malignancies (75). The cumulative lifetime risk for colorectal cancer approaches 40% (76). It remains questionable, however, whether the malignancies occurring in the intestines derive from direct transformation of hamartomatous polyps because dysplasia is exceedingly rare in these polyps. Patients with Peutz-Jeghers syndrome have germline mutations in the LKB1/STK11 gene (77-79). The hamartomatous polyps in Peutz-Jeghers syndrome are most commonly seen in the small intestine, but can also occur in the colon. They are composed of proliferative epithelium, stroma and smooth muscle arranged in an arborizing pattern (Figure 14).
Figure 14.
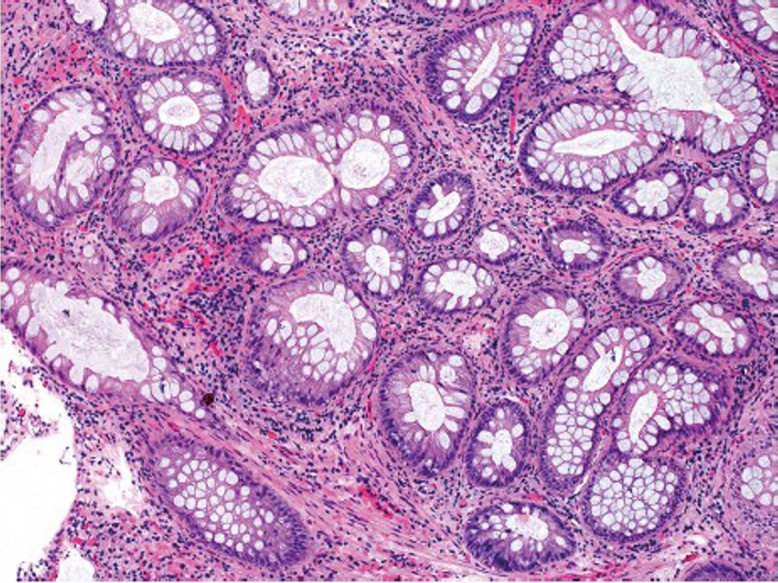
Peutz-Jeghers polyp in the colon. Note the lobular pattern of colonic crypts divided by smooth muscle bundles (original magnification ×100)
Juvenile polyposis syndrome
This is also an autosomal dominant inherited cancer syndrome diagnosed if, (I) 5 juvenile polyps in the colorectum; (II) juvenile polyps throughout the gastrointestinal tract; or (III) any number of juvenile polyps and a family history of juvenile polyposis (80). Similar to Peutz-Jeghers syndrome, the cumulative lifetime risk to develop colorectal cancer in patients with juvenile polyposis syndrome also approaches 40% (80,81). In contrast to Peutz-Jeghers syndrome, however, colorectal cancers in patients with juvenile polyposis syndrome are believed to develop directly from neoplastic transformation within a juvenile polyp because dysplasia is a frequent finding in these polyps. Approximately 50-60% of the patients have germline mutations in the SMAD4 or BMPR1A genes (82). Histologically, juvenile polyps feature cystically dilated crypts with edematous and inflamed stroma (Figure 15). The surface of the polyp may be eroded, with granulation tissue and epithelial regenerative changes.
Figure 15.
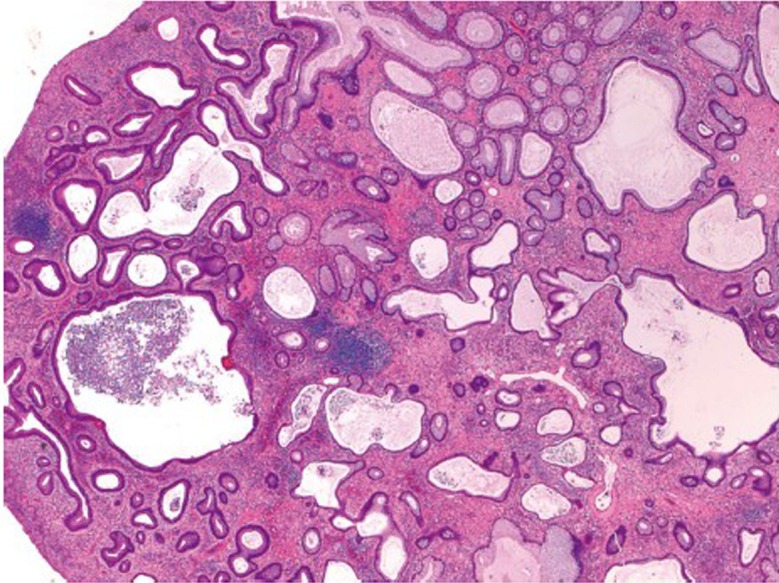
Juvenile polyp showing dilated crypts and inflamed stroma (original magnification ×40)
It should be pointed out that syndromic juvenile polyps cannot be distinguished from sporadic counterparts, and can be confused with inflammatory polyps on histologic ground. Despite the name, juvenile polyps can occur in adults or even elderly. Patients with sporadic juvenile polyps do not have an increased risk for malignancy (83).
MUTYH-associated polyposis
MUTYH-associated polyposis (MAP) is an autosomal recessive polyposis syndrome that carries an increased risk for colorectal cancers (84,85). It is caused by biallelic germline mutations in the MUTYH gene (also known as MYH gene) that encodes a base excision repair (BER) enzyme responsible for preventing mutations following oxidative DNA damage. The most common mutations are missense variants Y165C and G382D, accounting for >70% of all mutant alleles (86,87). MAP patients usually have >10 synchronous colorectal adenomas and can have several hundreds or even up to 1,000 polyps. Most patients have <100 polyps at the time of diagnosis, however (88). Therefore, MAP is often phenotypically indistinguishable from attenuated FAP. In contrast to FAP, however, there is a lack of APC gene mutations in MAP patients. In addition, serrated polyps (hyperplastic and sessile serrated polyps) are a common finding in MAP patients (89), which can be confused with serrated polyposis (described below). Furthermore, due to its recessive mode of inheritance, MAP has a tendency to skip generations, which makes identification of MAP patients more difficult since many patients seemingly present as sporadic cases.
Serrated polyposis
Serrated polyposis is a new term used by WHO, which was historically called hyperplastic polyposis (40). It is defined by: (I) at least 5 serrated polyps proximal to the sigmoid colon with 2 or more polyps >1 cm; (II) any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with serrated polyposis; or (III) >20 serrated polyps of any size throughout the colon. The polyps can be either SSA/Ps or HPs.
High grade dysplasia
Pathologic evaluation of an adenomatous polyp and dysplasia includes the determination of the presence or absence of high grade dysplasia, which represents the immediate precursor to invasive colorectal adenocarcinoma. High grade dysplasia manifests as a constellation of architectural complexity and cytologic atypia that are more malignant-appearing than those seen in a conventional adenoma (Figure 16). Architecturally, high grade areas typically show increased glandular density with crowded glands that have a cribriform or back-to-back growth pattern. Cytologically, cells with high grade dysplasia exhibit rounded nuclei, coarse chromatin, prominent nucleoli, and loss of nuclear polarity with nuclei no longer being oriented perpendicular to the basement membrane. Necrotic debris within the lumina of dysplastic glands may be seen.
Figure 16.
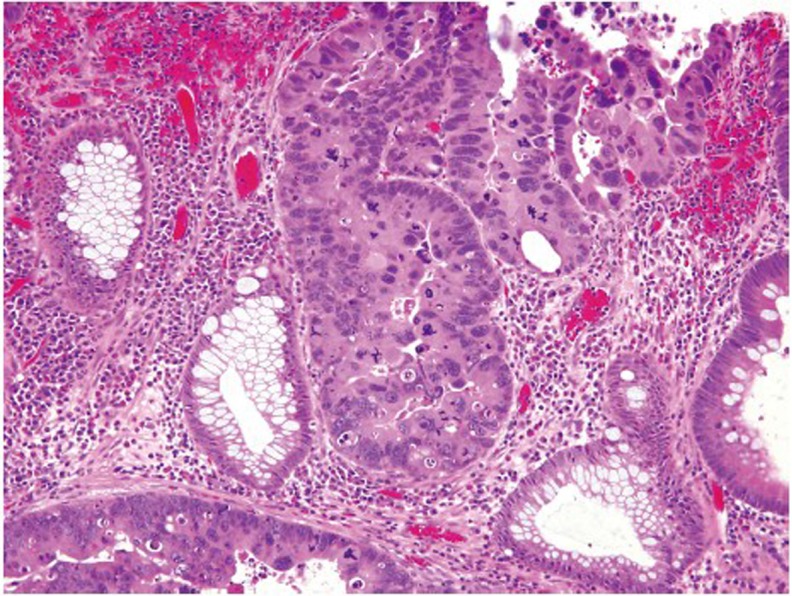
High grade dysplasia showing complex architecture and marked nuclear atypia (original magnification ×400)
High grade dysplasia is usually focal and situated on the superficial portion of the polyp, and thus requires no additional treatment beyond polypectomy if the polyp is completely removed endoscopically. As discussed earlier, high grade dysplasia in the colorectum is synonymous with carcinoma in situ or intraepithelial carcinoma. Intramucosal adenocarcinoma, defined by lamina propria invasion including invasion into (but not through) the muscularis mucosae, still belongs to the category of high grade dysplasia because of its negligible potential of metastasis and can still be successfully managed by polypectomy alone (90).
Malignant polyp
The term malignant polyp is used to describe a polyp that contains invasive adenocarcinoma in the submucosa. Prior studies have suggested a prevalence of 2-5% in endoscopically removed adenomas (91). When a malignant polyp is encountered, several critical histologic features need to be assessed, which include the status of the resection margin, histologic grade, and the presence or absence of lymphovascular invasion. These factors are related to the risk of adverse outcomes such as nodal metastasis and/or local recurrence following polypectomy. Polyps with a negative polypectomy margin, low grade histology, and no lymphovascular invasion can be safely treated with endoscopic polypectomy. An increased risk of adverse outcomes has been shown to be associated with positive margin (defined as <2 mm from deep cauterized margin) (Figure 17), high grade (poorly differentiated) histology, and lymphovascular invasion. If any of these features is present, surgical resection is indicated (91-93). Therefore, it is important that polypectomy specimens be received in one intact piece in order for margins to be accurately evaluated by pathologists. Inability to assess margin status because of piecemeal resection should also be considered as a risk factor (91,93), and surgical resection may be recommended in clinically fit patients.
Figure 17.
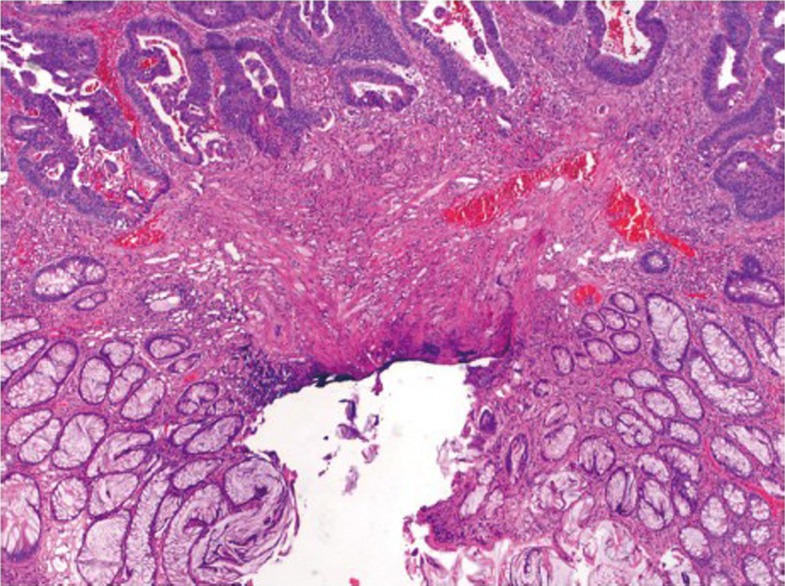
A malignant polyp showing adenocarcinomatous glands present within 1 mm of polypectomy margin (original magnification ×100)
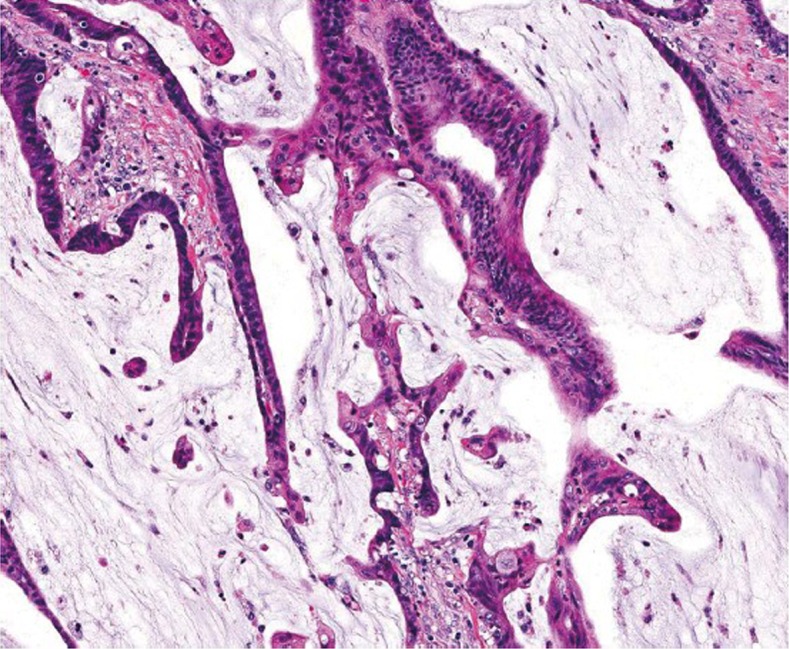
A pitfall in the assessment of an adenomatous polyp is pseudoinvasion where adenomatous elements are misplaced or herniated into the submucosa, usually secondary to traumatization such as twisting and torsion of the stalk (Figure 18). Histologic features that help distinguish from true invasion include a lobular configuration of herniated elements, lack of overt high grade architectural and cytologic atypia, presence of a rim of lamina propria inflammatory cells around entrapped elements, lack of desmoplastic reaction, lack of direct contact with submucosal muscular vessels, and presence of hemosiderin or hemorrhage. Occasionally, herniated adenomatous glands exhibit high grade histology, which can be even more difficult to distinguish from invasive adenocarcinoma. However, other histologic features that favor pseudoinvasion may still be present. For rare cases in which a definitive distinction cannot be made, complete polypectomy or surgical resection may be considered based on the clinical and endoscopic circumstances of the patient.
Figure 18.

Low power (A. original magnification ×40) and high power (B. original magnification ×200) views of pseudoinvasion in a tubular adenoma. Note the presence of hemorrhage and hemosiderin
Pathogenesis and molecular classification
Colorectal cancer is a heterogeneous group of diseases with distinctive genetic and epigenetic background (94). In order to improve clinical management and better predict patient outcome, attempts have been made to classify colorectal cancers based on location, histology, etiologic factors, and molecular mechanisms of tumorigenesis. As early as in the 1980’s, it has been recognized that cancers arising in the proximal colon and distal colon involve different genetic mechanisms (95,96). For instance, Lynch syndrome preferentially involves the proximal colon whereas FAP tends to show more polyps in the left colon. These familial forms of colorectal cancer have served as prototypes for understanding distinct molecular mechanisms of tumorigenesis. As discussed earlier, Lynch syndrome results from loss of function in one of the MMR genes and follows the MSI pathway (“mutator” pathway). In contrast, FAP arises in patient with inherited mutations in the APC gene, which has been the center of the original Fearon-Vogelstein model of colorectal tumorigenesis (97) that forms the basis of chromosomal instability (CIN) pathway (“suppressor” pathway).
Both MSI and CIN pathways describe colorectal cancer pathogenesis based on genetic abnormities that lead to loss of function of tumor suppressor genes and/or gain of function of oncogenes. In the last decade, epigenetic instability has gained considerable attention and is now believed to be implicated in the pathogenesis of almost one third of colorectal cancers (49). In addition to DNA sequence and structure, gene expression is controlled by a number of epigenetic modifications that include DNA methylation, histone alterations and chromatin remodeling (98). One of the best characterized epigenetic modifications associated with colorectal tumorigenesis is silencing of genes (tumor suppressor and/or MMR genes) through hypermethylation of their promoter regions. Although it was debated whether the phenomenon of epigenetic instability represents an adaptive cellular mechanism during carcinogenesis aimed to abort cellular proliferation, a secondary alteration to yet unidentified genetic mutations, a phenomenon expected to occur during tumor cell senescence, or simply an artifact (99-104), transcriptional silencing of certain genes by hypermethylation has undoubtedly shown to result in tumor development (105-110). In particular, promoter hypermethylation of the MLH1, one of the MMR genes, is demonstrated in the majority of sporadic colorectal cancers with a MSI phenotype (108,111,112). Since many genes are rich in cytosine and guanine dinucleotides (CpG islands) in their promoters, methylation of the cytosine residues in CpG islands is a common phenomenon, which leads to alterations of the chromosomal structure and suppression of gene expression. Colorectal cancers with CpG island methylator phenotype (CIMP) are characterized by epigenetic loss of function of tumor suppressor genes without mutations (49,113).
Figure 19 summaries the current understanding of the molecular pathways involved in colorectal tumorigenesis. CIN pathway is implicated in both sporadic and syndromic colorectal cancers. CIN tumors are characterized by karyotypic abnormalities and chromosomal gains and losses, which can be assessed by DNA ploidy or loss of heterozygosity (LOH) analyses. These tumors almost always harbor APC mutations, frequently show KRAS and p53 mutations, and often have 18q allelic loss (3,94). MSI pathway is also implicated in both sporadic and syndromic colorectal cancers and tends to be mutually exclusive with CIN. As discussed earlier, MSI tumors are characterized by loss of the DNA mismatch repair function. In sporadic colorectal cancers, the loss of function is primarily due to methylation of the MLH1 gene promoter that leads to epigenetic inhibition of protein expression of MLH1 and its binding partner PMS2. These tumors often show BRAF mutation but only rarely KRAS mutations. In Lynch syndrome, the loss of function usually results from germline mutations in one of the MMR genes. These tumors never harbor BRAF mutations. Finally, CIMP pathway represents a unique molecular mechanism in colorectal tumorigenesis, which can occur in either MSI-H or MSS tumors.
Figure 19.

Molecular pathways in colorectal tumorigenesis. CRC, colorectal cancer; MSS, microsatellite stable; MSI-H, high level microsatellite instability; FAP, familial adenomatous polyposis; AFAP, attenuated FAP; PJS, Peutz-Jeghers syndrome; JPS, juvenile polyposis syndrome; MAP, MUTYH-associated polyposis; CIN, chromosomal instability pathway; MSI, microsatellite instability; CIMP, CpG island methylator phenotype; ?, pathways yet undefined
Molecular genetic testing
With rapid advances in the understanding of colorectal tumorigenesis and pharmacogenetics, more and more molecular and genetic tests are demanded in order to optimally design personalized therapies for individual patients, to better predict patient prognosis, and to more accurately determine the necessity for family counseling. Currently, MSI, KRAS and BRAF are the most commonly performed tests in pathology laboratories.
MSI testing
As discussed earlier, MSI tumors account for ~15% of colorectal adenocarcinomas. These tumors tend to show unique clinicopathologic features, tend to have a better stage-adjusted prognosis when compared with MSS tumors, and appear to be resistant to treatment with 5-fluorouracil (114).
Microsatellites are repetitive DNA sequences that are prone to errors during DNA replication if the MMR system is defective. MSI is defined as alterations in the length of the microsatellite sequences. It is typically assessed by analyzing two mononucleotide repeats (BAT-25 and BAT-26) and three dinucleotide repeats (D2S123, D5S346, and D17S250), known as the Bethesda panel (61,115), by comparison between DNA samples extracted from normal and tumor tissues from the same patient. The test is polymerase chain reaction (PCR)-based, and can be performed on formalin-fixed paraffin-embedded tissues. A tumor is designated as MSI-H if two or more (>40%) of the five microsatellite markers show instability, MSI-L (low-level) if only one marker shows instability, or MSS if none of the markers show instability. The clinical significance of MSI-L remains unclear and controversial (116), and it may be helpful if additional microsatellite markers are tested in order to increase the accuracy of MSI classification.
An indirect analysis of MSI status can be achieved by immunohistochemical stains for MMR proteins. These proteins are ubiquitously present in normal cells but show loss of expression in MSI tumor cells. Several staining patterns may be observed based on the underlying genetic or epigenetic abnormalities (Table 3). It is interesting to note that loss of MLH1 protein expression is always accompanied by the loss of its binding partner PMS2 (Figure 20), but loss of PMS2 expression can occur by itself. The same holds true for MSH2 and its binding partner MSH6.
Table 3. Immunohistochemical staining patterns and interpretation for MMR proteins.
| MLH1 | MSH2 | MSH6 | PMS2 | Interpretation |
|---|---|---|---|---|
| + |
+ |
+ |
+ |
Intact MMR* |
| – |
+ |
+ |
– |
MLH1 germline mutation or hypermethylation |
| + |
– |
– |
+ |
MSH2 germline mutation |
| + |
+ |
– |
+ |
MSH6 germline mutation |
| + | + | + | – | PMS2 germline mutation |
+, positive nuclear staining (normal expression); –, negative staining (loss of expression); *There are rare examples of germline mutations in other genes that may produce detectable protein by immunohistochemistry but still cause MSI
Figure 20.

A MSI tumor showing loss of MLH1 (A) and PMS2 (D) protein expression, and normal expression of MSH2 (B) and MSH6 (C). Note the presence of positive staining in benign colonic crypts and inflammatory cells, which serve as good internal controls for the stains (original magnification ×200)
The sensitivity of PCR-based MSI test using the Bethesda panel ranges from 55% to 84% for the detection of mutations in different MMR gene. The sensitivity is increased if three or more mononucleotide repeat markers are used. The specificity of MSI test is 90%. Immunohistochemistry has been accepted as a reliable substitute for MSI with a concordance rate of >90%. It also provides additional information over PCR-based MSI test in that it allows gene-specific DNA sequence analysis based on the staining pattern. However, immunohistochemistry may miss rare MSI cases that are caused by germline mutations by other genes and does not discriminate germline mutation from epigenetic alteration when loss of MLH1 protein expression is detected. Thus, the most recent recommendation is to perform both PCR-based MSI test and immunohistochemistry in order to minimize the chance of missing the diagnosis of Lynch syndrome (117). It is also recently advocated to test all newly diagnosed colorectal cancers regardless of patient’s age and family history because ~25% of the patients with Lynch syndrome do not meet Amsterdam Criteria II or Bethesda guidelines (117). In that setting, only one test, either immunohistochemistry or MSI analysis, may be performed because the cost of the tests will become an issue.
KRAS testing
Mutations in the KRAS (Kirsten rat sarcoma viral oncogene homolog) gene lead to expression of a constitutively activated KRAS protein, which are detected in ~40% of colorectal cancers (2,118). As a critical downstream molecule in the epidermal growth factor receptor (EGFR) signaling pathway, mutant KRAS renders tumors resistant to EGFR-targeted therapies (2,119-121). As a result, the American Society for Clinical Oncology (ASCO) and the National Comprehensive Cancer Network (NCCN) have recommended mutation analysis of the KRAS gene for candidate patients who will receive anti-EGFR therapies (122,123).
Greater than 95% of KRAS mutations occur in codons 12 and 13 in exon 2 (118,124,125), and thus PCR-based methodologies designed to detect KRAS mutations are primarily for these mutations. Mutations can also occur in other loci such as codons 61 and 146 (126), but they are generally not screened because of rarity. Clinically available real-time PCR-based methods include allele-specific amplification assay and post-PCR melting curve analysis. The allele-specific real-time PCR technique detects seven most common mutations in the codons 12 and 13 (127), whereas the melting-curve analysis detects all possible mutations in these two codons (128,129).
Another detection method is DNA sequencing, which can detect all possible mutations in the KRAS gene, not just limited to codons 12 and 13. In comparison to the traditional Sanger sequencing method, the pyrosequencing technology offers a higher analytical sensitivity and is more advantageous for the analysis of DNA samples extracted from paraffin-embedded tissue blocks (130,131).
BRAF testing
In addition to KRAS, mutations in other members of the EGFR signaling pathway can also cause resistance to anti-EGFR therapy. A good example is BRAF (v-raf murine sarcoma viral oncogene homolog B1) gene mutation, which has been reported in ~10% of colorectal cancers (132-134). There are several interesting facts about BRAF mutation in colorectal cancers. First, activating BRAF and KRAS mutations are almost always mutually exclusive (135,136), and thus mutation testing of the BRAF gene should be considered following a negative KRAS mutation analysis. In fact, many laboratories offer reflex BRAF test if no KRAS mutation is detected in a specimen. Second, almost all BRAF mutations are identical V600E point mutation (134), which can be readily detected by a number of commercially available PCR-based assays (137). Third, BRAF mutation is almost exclusively seen in sporadic MSI tumors that are presumed to develop through the serrated tumorigenic pathway, but has never been reported in Lynch syndrome (138). More specifically, activating mutation of the BRAF gene is associated with a high level of global DNA methylation and epigenetic silencing of the MLH1 gene, found in 70-90% of sporadic colorectal tumors with a microsatellite unstable phenotype (136,139). Therefore, further testing BRAF mutation in a MSI tumor will help clarify the sporadic or syndromic nature of the tumor (140). Fourth, the impact of BRAF mutation on prognosis appears MSI-dependent. As expected, BRAF wild-type MSI-H tumors have the best prognosis, whereas BRAF-mutated MSS tumors are associated with the worst outcome. BRAF-mutated MSI-H tumor and BRAF wild-type MSS tumor are intermediate in terms of prognosis (132,133). Therefore, testing for both MMR abnormalities and BRAF mutations offers additional prognostic information.
Conclusions
Colorectal adenocarcinoma is a heterogeneous disease that involves multiple tumorigenic pathways. Pathologic analysis provides histologic and molecular information critical to appropriate patient treatment, prognosis assessment, and family counseling. Further understanding the molecular mechanisms in tumorigenesis will certainly lead to the development of new targeted therapies and new molecular tests, which will ultimately benefit the patients and their families.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- 1.Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011;61:212-36 [DOI] [PubMed] [Google Scholar]
- 2.Wang HL, Lopategui J, Amin MB, et al. KRAS mutation testing in human cancers: the pathologist’s role in the era of personalized medicine. Adv Anat Pathol 2010;17:23-32 [DOI] [PubMed] [Google Scholar]
- 3.Hamilton SR, Bosman FT, Boffetta P, et al. Carcinoma of the colon and rectum. In: WHO Classification of Tumours of the Digestive System. Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. Lyon: IARC Press, 2010:134-46. [Google Scholar]
- 4.Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979-94 [DOI] [PubMed] [Google Scholar]
- 5.Compton CC. Updated protocol for the examination of specimens from patients with carcinomas of the colon and rectum, excluding carcinoid tumors, lymphomas, sarcomas, and tumors of the vermiform appendix: a basis for checklists. Cancer Committee. Arch Pathol Lab Med 2000;124:1016-25 [DOI] [PubMed] [Google Scholar]
- 6.Blenkinsopp WK, Stewart-Brown S, Blesovsky L, et al. Histopathology reporting in large bowel cancer. J Clin Pathol 1981;34:509-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jass JR, Atkin WS, Cuzick J, et al. The grading of rectal cancer: historical perspectives and a multivariate analysis of 447 cases. Histopathology 1986;10:437-59 [DOI] [PubMed] [Google Scholar]
- 8.Compton CC. Pathology report in colon cancer: what is prognostically important? Dig Dis 1999;17:67-79 [DOI] [PubMed] [Google Scholar]
- 9.Edge SB, Byrd DR, Compton CC, et al. AJCC Cancer Staging Handbook, 7th edition. New York: Springer, 2010:173-206. [Google Scholar]
- 10.Verhulst J, Ferdinande L, Demetter P, et al. Mucinous subtype as prognostic factor in colorectal cancer: a systematic review and meta-analysis. J Clin Pathol 2012;65:381-8 [DOI] [PubMed] [Google Scholar]
- 11.Kang H, O’Connell JB, Maggard MA, et al. A 10-year outcomes evaluation of mucinous and signet-ring cell carcinoma of the colon and rectum. Dis Colon Rectum 2005;48:1161-8 [DOI] [PubMed] [Google Scholar]
- 12.Leopoldo S, Lorena B, Cinzia A, et al. Two subtypes of mucinous adenocarcinoma of the colorectum: clinicopathological and genetic features. Ann Surg Oncol 2008;15:1429-39 [DOI] [PubMed] [Google Scholar]
- 13.Chen JS, Hsieh PS, Chiang JM, et al. Clinical outcome of signet ring cell carcinoma and mucinous adenocarcinoma of the colon. Chang Gung Med J 2010;33:51-7 [PubMed] [Google Scholar]
- 14.Makino T, Tsujinaka T, Mishima H, et al. Primary signet-ring cell carcinoma of the colon and rectum: report of eight cases and review of 154 Japanese cases. Hepatogastroenterology 2006;53:845-9 [PubMed] [Google Scholar]
- 15.Thirunavukarasu P, Sathaiah M, Singla S, et al. Medullary carcinoma of the large intestine: a population based analysis. Int J Oncol 2010;37:901-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinoi T, Tani M, Lucas PC, et al. Loss of CDX2 expression and microsatellite instability are prominent features of large cell minimally differentiated carcinomas of the colon. Am J Pathol 2001;159:2239-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander J, Watanabe T, Wu TT, et al. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol 2001;158:527-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology 2002;40:403-39 [DOI] [PubMed] [Google Scholar]
- 19.McGregor DK, Wu TT, Rashid A, et al. Reduced expression of cytokeratin 20 in colorectal carcinomas with high levels of microsatellite instability. Am J Surg Pathol 2004;28:712-8 [DOI] [PubMed] [Google Scholar]
- 20.Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol 2003;27:303-10 [DOI] [PubMed] [Google Scholar]
- 21.Kaimaktchiev V, Terracciano L, Tornillo L, et al. The homeobox intestinal differentiation factor CDX2 is selectively expressed in gastrointestinal adenocarcinomas. Mod Pathol 2004;17:1392-9 [DOI] [PubMed] [Google Scholar]
- 22.Shepherd NA, Baxter KJ, Love SB. The prognostic importance of peritoneal involvement in colonic cancer: a prospective evaluation. Gastroenterology 1997;112:1096-102 [DOI] [PubMed] [Google Scholar]
- 23.CAP Cancer Protocols and Checklists. Available online: http://www.cap.org/apps/cap.portal
- 24.Nelson H, Petrelli N, Carlin A, et al. Guidelines 2000 for colon and rectal cancer surgery. J Natl Cancer Inst 2001;93:583-96 [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Safar B, Wexner SD, et al. The clinical significance of fat clearance lymph node harvest for invasive rectal adenocarcinoma following neoadjuvant therapy. Dis Colon Rectum 2009;52:1767-73 [DOI] [PubMed] [Google Scholar]
- 26.Hernanz F, García-Somacarrera E, Fernández F.The assessment of lymph nodes missed in mesenteric tissue after standard dissection of colorectal cancer specimens. Colorectal Dis 2010;12:e57-e60 [DOI] [PubMed] [Google Scholar]
- 27.Märkl B, Kerwel TG, Jähnig HG, et al. Methylene blue-assisted lymph node dissection in colon specimens: a prospective, randomized study. Am J Clin Pathol 2008;130:913-9 [DOI] [PubMed] [Google Scholar]
- 28.Kerwel TG, Spatz J, Anthuber M, et al. Injecting methylene blue into the inferior mesenteric artery assures an adequate lymph node harvest and eliminates pathologist variability in nodal staging for rectal cancer. Dis Colon Rectum 2009;52:935-41 [DOI] [PubMed] [Google Scholar]
- 29.Fan L, Levy M, Aguilar CE, et al. Lymph node retrieval from colorectal resection specimens for adenocarcinoma: is it worth the extra effort to find at least 12 nodes? Colorectal Dis 2011;13:1377-83 [DOI] [PubMed] [Google Scholar]
- 30.Shen SS, Haupt BX, Ro JY, et al. Number of lymph nodes examined and associated clinicopathologic factors in colorectal carcinoma. Arch Pathol Lab Med 2009;133:781-6 [DOI] [PubMed] [Google Scholar]
- 31.Morcos B, Baker B, Al Masri M, et al. Lymph node yield in rectal cancer surgery: effect of preoperative chemoradiotherapy. Eur J Surg Oncol 2010;36:345-9 [DOI] [PubMed] [Google Scholar]
- 32.Marks JH, Valsdottir EB, Rather AA, et al. Fewer than 12 lymph nodes can be expected in a surgical specimen after high-dose chemoradiation therapy for rectal cancer. Dis Colon Rectum 2010;53:1023-9 [DOI] [PubMed] [Google Scholar]
- 33.Lo DS, Pollett A, Siu LL, et al. Prognostic significance of mesenteric tumor nodules in patients with stage III colorectal cancer. Cancer 2008;112:50-4 [DOI] [PubMed] [Google Scholar]
- 34.Puppa G, Maisonneuve P, Sonzogni A, et al. Pathological assessment of pericolonic tumor deposits in advanced colonic carcinoma: relevance to prognosis and tumor staging. Mod Pathol 2007;20:843-55 [DOI] [PubMed] [Google Scholar]
- 35.Winawer SJ, Zauber AG, Ho MN, et al. Prevention of colorectal cancer by colonoscopic polypectomy: The National Polyp Study Workgroup. N Engl J Med 1993;329:1977-81 [DOI] [PubMed] [Google Scholar]
- 36.Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med 2012;366:687-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159-70 [DOI] [PubMed] [Google Scholar]
- 38.Shih IM, Wang TL, Traverso G, et al. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci USA 2001;98:2640-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heitman SJ, Ronksley PE, Hilsden RJ, et al. Prevalence of adenomas and colorectal cancer in average risk individuals: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2009;7:1272-8 [DOI] [PubMed] [Google Scholar]
- 40.Snover DC, Ahnen DJ, Burt RW, et al. Serrated polyps of the colon and rectum and serrated polyposis. In: WHO Classification of Tumours of the Digestive System. Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. Lyon: IARC Press, 2010:160-5. [Google Scholar]
- 41.Tsai CJ, Lu DK. Small colorectal polyps: histopathology and clinical significance. Am J Gastroenterol 1995;90:988-94 [PubMed] [Google Scholar]
- 42.Snover DC, Jass JR, Fenoglio-Preiser C, et al. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol 2005;124:380-91 [DOI] [PubMed] [Google Scholar]
- 43.Torlakovic E, Skovlund E, Snover DC, et al. Morphologic reappraisal of serrated colorectal polyps. Am J Surg Pathol 2003;27:65-81 [DOI] [PubMed] [Google Scholar]
- 44.Goldstein NS, Bhanot P, Odish E, et al. Hyperplastic-like colon polyps that preceded microsatellite-unstable adenocarcinomas. Am J Clin Pathol 2003;119:778-96 [DOI] [PubMed] [Google Scholar]
- 45.East JE, Saunders BP, Jass JR. Sporadic and syndromic hyperplastic polyps and serrated adenomas of the colon: classification, molecular genetics, natural history, and clinical management. Gastroenterol Clin North Am 2008;37:25-46 [DOI] [PubMed] [Google Scholar]
- 46.Glatz K, Pritt B, Glatz D, et al. A multinational, internet-based assessment of observer variability in the diagnosis of serrated colorectal polyps. Am J Clin Pathol 2007;127:938-45 [DOI] [PubMed] [Google Scholar]
- 47.Farris AB, Misdraji J, Srivastava A, et al. Sessile serrated adenoma: challenging discrimination from other serrated colonic polyps. Am J Surg Pathol 2008;32:30-5 [DOI] [PubMed] [Google Scholar]
- 48.Wong NA, Hunt LP, Novelli MR, et al. Observer agreement in the diagnosis of serrated polyps of the large bowel. Histopathology 2009;55:63-6 [DOI] [PubMed] [Google Scholar]
- 49.Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011;42:1-10 [DOI] [PubMed] [Google Scholar]
- 50.Vu HT, Lopez R, Bennett A, et al. Individuals with sessile serrated polyps express an aggressive colorectal phenotype. Dis Colon Rectum 2011;54:1216-23 [DOI] [PubMed] [Google Scholar]
- 51.Blackstone MO, Riddell RH, Rogers BH, et al. Dysplasia-associated lesion or mass (DALM) detected by colonoscopy in long-standing ulcerative colitis: an indication for colectomy. Gastroenterology 1981;80:366-74 [PubMed] [Google Scholar]
- 52.Butt JH, Konishi F, Morson BC, et al. Macroscopic lesions in dysplasia and carcinoma complicating ulcerative colitis. Dig Dis Sci 1983;28:18-26 [DOI] [PubMed] [Google Scholar]
- 53.Rubin PH, Friedman S, Harpaz N, et al. Colonoscopic polypectomy in chronic colitis: conservative management after endoscopic resection of dysplastic polyps. Gastroenterology 1999;117:1295-300 [DOI] [PubMed] [Google Scholar]
- 54.Engelsgjerd M, Farraye FA, Odze RD. Polypectomy may be adequate treatment for adenoma-like dysplastic lesions in chronic ulcerative colitis. Gastroenterology 1999;117:1288-94 [DOI] [PubMed] [Google Scholar]
- 55.Odze RD, Farraye FA, Hecht JL, et al. Long-term follow-up after polypectomy treatment for adenoma-like dysplastic lesions in ulcerative colitis. Clin Gastroenterol Hepatol 2004;2:534-41 [DOI] [PubMed] [Google Scholar]
- 56.Itzkowitz SH, Present DH. Consensus conference: colorectal cancer screening and surveillance in inflammatory bowel disease. Inflamm Bowel Dis 2005;11:314-21 [DOI] [PubMed] [Google Scholar]
- 57.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003;348:919-32 [DOI] [PubMed] [Google Scholar]
- 58.Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009;137:1621-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851-60 [DOI] [PubMed] [Google Scholar]
- 60.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453-6 [DOI] [PubMed] [Google Scholar]
- 61.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Björk J, Akerbrant H, Iselius L, et al. Epidemiology of familial adenomatous polyposis in Sweden: changes over time and differences in phenotype between males and females. Scand J Gastroenterol 1999;34:1230-5 [DOI] [PubMed] [Google Scholar]
- 63.Half E, Bercovich D, Rozen P.Familial adenomatous polyposis. Orphanet J Rare Dis 2009;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006;101:385-98 [DOI] [PubMed] [Google Scholar]
- 65.Bülow S, Björk J, Christensen IJ, et al. Duodenal adenomatosis in familial adenomatous polyposis. Gut 2004;53:381-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burt RW. Gastric fundic gland polyps. Gastroenterology 2003;125:1462-9 [DOI] [PubMed] [Google Scholar]
- 67.Bianchi LK, Burke CA, Bennett AE, et al. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008;6:180-5 [DOI] [PubMed] [Google Scholar]
- 68.Wu TT, Kornacki S, Rashid A, et al. Dysplasia and dysregulation of proliferation in foveolar and surface epithelia of fundic gland polyps from patients with familial adenomatous polyposis. Am J Surg Pathol 1998;22:293-8 [DOI] [PubMed] [Google Scholar]
- 69.Hassan A, Yerian LM, Kuan SF, et al. Immunohistochemical evaluation of adenomatous polyposis coli, beta-catenin, c-Myc, cyclin D1, p53, and retinoblastoma protein expression in syndromic and sporadic fundic gland polyps. Hum Pathol 2004;35:328-34 [DOI] [PubMed] [Google Scholar]
- 70.Giardiello FM, Burt RW, Jävinen HJ, et al. Familial adenomatous polyposis. In: WHO Classification of Tumours of the Digestive System. Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. Lyon: IARC Press, 2010:147-51. [Google Scholar]
- 71.Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 2004;127:444-51 [DOI] [PubMed] [Google Scholar]
- 72.Laurent-Puig P, Béroud C, Soussi T.APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 1998;26:269-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miyoshi Y, Ando H, Nagase H, et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 1992;89:4452-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci 2007;120:3327-35 [DOI] [PubMed] [Google Scholar]
- 75.Kopacova M, Tacheci I, Rejchrt S, et al. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol 2009;15:5397-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Lier MG, Wagner A, Mathus-Vliegen EM, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010;105:1258-64 [DOI] [PubMed] [Google Scholar]
- 77.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391:184-7 [DOI] [PubMed] [Google Scholar]
- 78.Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998;18:38-43 [DOI] [PubMed] [Google Scholar]
- 79.Karuman P, Gozani O, Odze RD, et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell 2001;7:1307-19 [DOI] [PubMed] [Google Scholar]
- 80.Brosens LA, Langeveld D, van Hattem WA, et al. Juvenile polyposis syndrome. World J Gastroenterol 2011;17:4839-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007;56:965-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Hattem WA, Brosens LA, de Leng WW, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut 2008;57:623-7 [DOI] [PubMed] [Google Scholar]
- 83.Nugent KP, Talbot IC, Hodgson SV, et al. Solitary juvenile polyps: not a marker for subsequent malignancy. Gastroenterology 1993;105:698-700 [DOI] [PubMed] [Google Scholar]
- 84.Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet 2002;30:227-32 [DOI] [PubMed] [Google Scholar]
- 85.Farrington SM, Tenesa A, Barnetson R, et al. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet 2005;77:112-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lipton L, Tomlinson I.The multiple colorectal adenoma phenotype and MYH: a base excision repair gene. Clin Gastroenterol Hepatol 2004;2:633-8 [DOI] [PubMed] [Google Scholar]
- 87.Cheadle JP, Sampson JR. MUTYH-associated polyposis: from defect in base excision repair to clinical genetic testing. DNA Repair (Amst) 2007;6:274-9 [DOI] [PubMed] [Google Scholar]
- 88.Sampson JR, Jones N. MUTYH-associated polyposis. Best Pract Res Clin Gastroenterol 2009;23:209-18 [DOI] [PubMed] [Google Scholar]
- 89.Boparai KS, Dekker E, Van Eeden S, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 2008;135:2014-8 [DOI] [PubMed] [Google Scholar]
- 90.Lewin MR, Fenton H, Burkart AL, et al. Poorly differentiated colorectal carcinoma with invasion restricted to lamina propria (intramucosal carcinoma): a follow-up study of 15 cases. Am J Surg Pathol 2007;31:1882-6 [DOI] [PubMed] [Google Scholar]
- 91.Ramirez M, Schierling S, Papaconstantinou HT, et al. Management of the malignant polyp. Clin Colon Rectal Surg 2008;21:286-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cooper HS. Pathologic issues in the treatment of endoscopically removed malignant colorectal polyps. J Natl Compr Canc Netw 2007;5:991-6 [DOI] [PubMed] [Google Scholar]
- 93.Butte JM, Tang P, Gonen M, et al. Rate of residual disease after complete endoscopic resection of malignant colonic polyp. Dis Colon Rectum 2012;55:122-7 [DOI] [PubMed] [Google Scholar]
- 94.Ogino S, Goel A.Molecular classification and correlates in colorectal cancer. J Mol Diagn 2008;10:13-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bufill JA. Colorectal cancer: evidence for distinct genetic categories based on proximal or distal tumor location. Ann Intern Med 1990;113:779-88 [DOI] [PubMed] [Google Scholar]
- 96.Beart RW, Melton LJ, III, Maruta M, et al. Trends in right and left-sided colon cancer. Dis Colon Rectum 1983;26:393-8 [DOI] [PubMed] [Google Scholar]
- 97.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759-67 [DOI] [PubMed] [Google Scholar]
- 98.Portela A, Esteller M.Epigenetic modifications and human disease. Nat Biotechnol 2010;28:1057-68 [DOI] [PubMed] [Google Scholar]
- 99.Yamashita K, Dai T, Dai Y, et al. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003;4:121-31 [DOI] [PubMed] [Google Scholar]
- 100.Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell 2002;1:299-305 [DOI] [PubMed] [Google Scholar]
- 101.Norrie MW, Hawkins NJ, Todd AV, et al. The role of hMLH1 methylation in the development of synchronous sporadic colorectal carcinomas. Dis Colon Rectum 2002;45:674-80 [DOI] [PubMed] [Google Scholar]
- 102.Anacleto C, Leopoldino AM, Rossi B, et al. Colorectal cancer “methylator phenotype”: fact or artifact? Neoplasia 2005;7:331-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ahuja N, Issa JP. Aging, methylation and cancer. Histol Histopathol 2000;15:835-42 [DOI] [PubMed] [Google Scholar]
- 104.Xiong Z, Wu AH, Bender CM, et al. Mismatch repair deficiency and CpG island hypermethylation in sporadic colon adenocarcinomas. Cancer Epidemiol Biomarkers Prev 2001;10:799-803 [PubMed] [Google Scholar]
- 105.Chen WY, Zeng X, Carter MG, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet 2003;33:197-202 [DOI] [PubMed] [Google Scholar]
- 106.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997;57:808-11 [PubMed] [Google Scholar]
- 107.Ma AH, Xia L, Littman SJ, et al. Somatic mutation of hPMS2 as a possible cause of sporadic human colon cancer with microsatellite instability. Oncogene 2000;19:2249-56 [DOI] [PubMed] [Google Scholar]
- 108.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA 1998;95:8698-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shen L, Kondo Y, Rosner GL, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005;97:1330-8 [DOI] [PubMed] [Google Scholar]
- 110.Bevilacqua RA, Simpson AJ. Methylation of the hMLH1 promoter but no hMLH1 mutations in sporadic gastric carcinomas with high-level microsatellite instability. Int J Cancer 2000;87:200-3 [DOI] [PubMed] [Google Scholar]
- 111.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998;95:6870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008;135:1079-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007;50:113-30 [DOI] [PubMed] [Google Scholar]
- 114.Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res 2012;18:1506-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248-57 [PubMed] [Google Scholar]
- 116.Schneider R, Schneider C, Kloor M, et al. Lynch syndrome: clinical, pathological, and genetic insights. Langenbecks Arch Surg 2012;397:513-25 [DOI] [PubMed] [Google Scholar]
- 117.Weissman SM, Burt R, Church J, et al. Identification of individuals at risk for Lynch Syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer Joint Practice Guideline. J Genet Couns 2012,21:484-93 [DOI] [PubMed] [Google Scholar]
- 118.Brink M, de Goeij AF, Weijenberg MP, et al. K-ras oncogene mutations in sporadic colorectal cancer in the Netherlands Cohort Study. Carcinogenesis 2003;24:703-10 [DOI] [PubMed] [Google Scholar]
- 119.Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992-5 [DOI] [PubMed] [Google Scholar]
- 120.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230-7 [DOI] [PubMed] [Google Scholar]
- 121.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626-34 [DOI] [PubMed] [Google Scholar]
- 122.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091-6 [DOI] [PubMed] [Google Scholar]
- 123.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Colon Cancer v.2.2009. Fort Washington, PA: National Comprehensive Cancer Network. 2009. [DOI] [PubMed] [Google Scholar]
- 124.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989;49:4682-9 [PubMed] [Google Scholar]
- 125.Pritchard CC, Akagi L, Reddy PL, et al. COLD-PCR enhanced melting curve analysis improves diagnostic accuracy for KRAS mutations in colorectal carcinoma. BMC Clin Pathol 2010;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 2009;101:715-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Clayton SJ, Scott FM, Walker J, et al. K-ras point mutation detection in lung cancer: comparison of two approaches to somatic mutation detection using ARMS allele-specific amplification. Clin Chem 2000;46:1929-38 [PubMed] [Google Scholar]
- 128.Elenitoba-Johnson KS, Bohling SD, Wittwer CT, et al. Multiplex PCR by multicolor fluorimetry and fluorescence melting curve analysis. Nat Med 2001;7:249-53 [DOI] [PubMed] [Google Scholar]
- 129.Monzon FA, Ogino S, Hammond ME, et al. The role of KRAS mutation testing in the management of patients with metastatic colorectal cancer. Arch Pathol Lab Med 2009;133:1600-6 [DOI] [PubMed] [Google Scholar]
- 130.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J Mol Diagn 2005;7:413-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dufort S, Richard MJ, de Fraipont F. Pyrosequencing method to detect KRAS mutation in formalin-fixed and paraffin-embedded tumor tissues. Anal Biochem 2009;391:166-8 [DOI] [PubMed] [Google Scholar]
- 132.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063-9 [DOI] [PubMed] [Google Scholar]
- 133.Ogino S, Shima K, Meyerhardt JA, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res 2012;18:890-900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54 [DOI] [PubMed] [Google Scholar]
- 135.Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002;418:934. [DOI] [PubMed] [Google Scholar]
- 136.Koinuma K, Shitoh K, Miyakura Y, et al. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer 2004;108:237-42 [DOI] [PubMed] [Google Scholar]
- 137.Lea A, Allingham-Hawkins D, Levine S.BRAF p.Val600Glu (V600E) Testing for assessment of treatment options in metastatic colorectal cancer. Version 2. PLoS Curr 2010;2:RRN1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006;38:787-93 [DOI] [PubMed] [Google Scholar]
- 139.Cunningham JM, Christensen ER, Tester DJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 1998;58:3455-60 [PubMed] [Google Scholar]
- 140.Lagerstedt Robinson K, Liu T, Vandrovcova J, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst 2007;99:291-9 [DOI] [PubMed] [Google Scholar]