

SUMMARY
Recent clinical and experimental evidence suggests that endoplasmic reticulum (ER) stress contributes to the life-and-death decisions of β cells during the progression of type 1 and type 2 diabetes. Although crosstalk between inflammation and ER stress has been suggested to play a significant role in β cell dysfunction and death, a key molecule connecting ER stress to inflammation has not been identified. Here we report that thioredoxin-interacting protein (TXNIP) is a critical signaling node that links ER stress and inflammation. TXNIP is induced by ER stress through the PERK and IRE1 pathways, induces IL-1β mRNA transcription, activates IL-1β production by the NLRP3 inflammasome, and mediates ER stress-mediated β cell death. Collectively, our results suggest that TXNIP is a potential therapeutic target for diabetes and ER stress-related human diseases such as Wolfram syndrome.
INTRODUCTION
A hallmark of diabetes is reduced functional β cell mass (Donath and Halban, 2004; Pipeleers et al., 2008). This can be caused by upregulation of genes inducing cell death or downregulation of genes promoting cell survival in β cells. Cell death promoting genes that are upregulated in β cells during the progression of diabetes are promising targets for the development of diabetes therapeutics.
In patients with type 1 diabetes, destruction of β cells by inflammatory cytokines including interleukin-1β (IL-1β) leads to an absolute deficiency of insulin (Atkinson et al., 2011; Bluestone et al., 2010). In contrast, type 2 diabetes is a state of relative insulin deficiency as a result of β cell dysfunction and death caused by a combination of increased circulating glucose and saturated fatty acids and development of inflammation (Butler et al., 2003; Donath and Halban, 2004). Recent evidence suggests that endoplasmic reticulum (ER) stress also plays a role in the loss of β cells during the progression of type 1 and type 2 diabetes and Wolfram syndrome, a genetic form of diabetes and neurodegeneration (Eizirik et al., 2008; Oslowski and Urano, 2011).
The crosstalk between inflammation and ER stress has been suggested to play a significant role in β cell dysfunction and death (Zhang and Kaufman, 2008). However, a key molecule that links ER stress to inflammation has not been identified. Here we report that thioredoxin-interacting protein (TXNIP), also known as thioredoxin binding protein-2 (TBP-2) and vitamin-D3 upregulated protein-1 (VDUP1) (Chen and DeLuca, 1994; Nishiyama et al., 1999), is a critical link between ER stress, inflammation and cell death.
RESULTS
TXNIP expression is increased by ER stress in β cells
It has been shown that TXNIP plays a role in β cell death in diabetes (Chen et al., 2008; Minn et al., 2005), raising the possibility that TXNIP expression is primarily expressed in β cells as compared to other cell types of the pancreas. Immunostaining of mouse pancreas sections with anti-TXNIP antibody together with anti-insulin or anti-glucagon antibody revealed that TXNIP is highly expressed in insulin-producing β cells (Figure 1A). To test whether TXNIP expression is induced by ER stress, we treated rat insulinoma cells, INS-1 832/13, with chemical ER stress inducers, thapsigargin and tunicamycin. The results illustrated in Figure 1B indicate that chemical ER stress inducers increase TXNIP mRNA expression levels. We also tested if physiological ER stress inducers in β cells could increase TXNIP expression. It has been shown that pathological β cell perturbants, such as chronic high glucose and human islet amyloid polypeptide, can cause ER stress in β cell lines and islets (Casas et al., 2007; Lipson et al., 2006). As expected, TXNIP mRNA expression was increased in INS-1 832/13 cells treated with chronic high glucose and human islet amyloid polypeptide (Figure 1C). To confirm these observations, we treated mouse and human primary islets with chemical ER stress inducers, thapsigargin and tunicamycin. As predicted, TXNIP mRNA expression was increased in both mouse and human primary islets by ER stress (Figure 1D). TXNIP protein expression levels were also increased in INS-1 832/13 cells and human primary islets treated with tunicamycin and thapsigargin (Figure 1E).
Figure 1. TXNIP is highly expressed in pancreatic beta cells and induced by ER stress.

(A) Mouse pancreata were analyzed by immunohistochemistry. Merged image shows the co-localization of TXNIP and insulin (upper panel) or TXNIP and glucagon (lower panel). (B) Expression of TXNIP mRNA in INS-1 832/13 cells treated with thapsigargin (TG, 0.25 μM) or tunicamycin (TM, 5 μg/ml). (C) Expression of TXNIP mRNA in INS-1 832/13 cells treated with glucose (16.7 mM) or human islet polypeptide (hIAPP, 5μM). (D) Expression of TXNIP mRNA in mouse and human primary islets treated with thapsigargin (TG, 1 μM) or tunicamycin (TM, 5 μg/ml) for 6 h. (E) Expression of TXNIP protein in INS-1 832/13 cells treated with thapsigargin (TG, 1 μM) for 8hr or tunicamycin (TM, 5 μg/ml) for 24 h, and human primary islets treated with tunicamycin (TM, 5 μg/ml) for 24 h. n=3; values are mean ± SD
PERK and IRE1α signaling pathways required for TXNIP induction under ER stress
We next wanted to identify the components of the unfolded protein response (UPR) involved in TXNIP induction by ER stress, Inositol requiring enzyme 1 (IRE1), PKR-like ER resident kinase (PERK), and activating transcription factor 6 (ATF6).
In Ire1α knockdown INS-1 832/13 cells, TXNIP expression was modestly attenuated as compared to control cells under ER stress conditions (Figure 2A). We also observed that in Ire1α knockout mouse embryonic fibroblasts (MEFs), mRNA and protein upregulation of TXNIP were suppressed under ER stress conditions (Figure 2B). TXNIP expression under ER stress was also significantly decreased in Perk knockdown INS-1 832/13 cells and Perk knockout MEFs (Figure 2C and 2D). PERK-mediated phosphorylation of eukaryotic initiation factor 2 on Ser51 of the alpha subunit (eIF2α) is involved in the regulation of gene expression under ER stress conditions (Harding et al., 1999; Scheuner et al., 2001). To elucidate the role of eIF2α phosphorylation in TXNIP expression, we treated MEFs derived from mice homozygous for Ser51Ala mutant eIF2α (eIF2α A/A mutant) (Scheuner et al., 2001). These cells lack eIF2α phosphorylation under various stress conditions, including ER stress (Back et al., 2009; Scheuner et al., 2001). Expression levels of TXNIP mRNA and protein were significantly decreased in eIF2α A/A mutant cells under ER stress conditions (Figure 2E). Finally, we studied the possible role of ATF6α in TXNIP upregulation. However, TXNIP induction in Atf6α knockdown INS-1 832/13 cells and Atf6α knockout MEFs was comparable to wild-type control cells (Figure 2F and 2G). Collectively, these results indicate that TXNIP upregulation by ER stress is mediated by the IRE1α and especially the PERK-eIF2α arms of the UPR in β cells.
Figure 2. TXNIP expression is regulated by the IRE1 and PERK-eIF2 α pathways of the UPR.

(A) Expression of TXNIP mRNA in INS-1 832/13 cells transfected with control or IRE1α siRNA, and then treated with thapsigargin (TG, 0.5 μM) for 6 h. (B) Expression of TXNIP mRNA (left panel) and protein (right panel) in wild-type (WT) and Ire1α−/− (KO) MEFs treated with thapsigargin (0.5 μM) for 3 h or untreated. (C) Expression of TXNIP mRNA in INS-1 832/13 cells transfected with control or PERK siRNA, and then treated with thapsigargin (TG, 0.5 μM) for 6 h. (D) Expression of TXNIP mRNA (left panel) and protein (right panel) in wild-type (WT) and Perk−/− MEFs treated with thapsigargin (0.5 μM) for 3 h or untreated. (E) Expression of TXNIP mRNA and protein in wild-type eIF2αS/S or mutant eIF2αA/A MEFs treated with thapsigargin (TG, 0.5μM) for 3 h or untreated. (F) Expression of TXNIP mRNA and protein in INS-1 832/13 cells transfected with control or ATF6α siRNA, and then treated with thapsigargin (TG, 0.5 μM) for 6 h. (G) Expression of TXNIP mRNA in wild-type (WT) and Atf6α−/− MEFs treated with thapsigargin (0.5 μM) for 3 h or untreated. (H) Luciferase reporter assays in INS-1 832/13 cells transiently transfected with TXNIP promoter reporter constructs. Cells were untreated (UT) or treated with thapsigargin (TG, 0.5 μM) for 6 h. (I) Luciferase reporter assays in INS-1 832/13 cells transfected with TXNIP promoter reporter construct carrying 1.5 Kb of TXNIP promoter together with control, PERK expression, or GADD34 expression vector. Cells were untreated (UT) or treated with thapsigargin (TG, 0.5 μM) for 6 h. (J) Luciferase reporter assay in INS-1 832/13 cells transfected with a TXNIP promoter reporter construct carrying 1.5 Kb of TXNIP promoter. Cells were untreated (UT) or treated with Salubrinal (25 μM) for 24 h. (K) Luciferase reporter assays in INS-1 832/13 cells transfected with a TXNIP promoter reporter construct carrying 1.5 Kb of TXNIP promoter together with control or ChREBP siRNA. Cells were untreated (UT) or treated with thapsigargin (TG, 0.5 μM) for 6 h. (L) ChIP assays monitoring binding of ChREBP and Mlx to the TXNIP promoter in INS-1 832/13 cells treated with or without thapsigargin (TG, 0.5 μM) for 6 h. NA, Non-specific IgG Antibody. (M) Expression of ChREBP mRNA in INS-1 cells transfected with control or PERK siRNA (left panel), and wild-type and Perk−/− MEFs (right panel) treated with or without thapsigargin (TG, 0.5 μM) for 6 h. (N) Luciferase reporter assays in INS-1 832/13 cells transfected with a TXNIP promoter reporter construct carrying 1.5 Kb of TXNIP promoter together with control or ATF5 siRNA. Cells were untreated (UT) or treated with thapsigargin (TG, 0.5 μM) for 6 h. (O) ChIP assay monitoring binding of ATF5 to the TXNIP promoter in INS-1 832/13 cells treated with or without thapsigargin (TG, 0.5 μM) for 6 h. NA, Non-specific IgG Antibody. (P) Expression of ATF5 mRNA in INS-1 832/13 cells transfected with control or PERK siRNA (left panel), and wild-type and Perk−/− mouse embryonic fibroblasts (right panel) treated with or without thapsigargin (TG, 0.5 μM) for 6 h. n=3; values are mean ± SD. * p<0.05, ** p<0.01, *** p<0.001. n.s., not significant
As described above, PERK signaling is critical for TXNIP expression under ER stress conditions, which prompted us to identify the downstream signaling molecules of PERK that regulate TXNIP expression. The UPR regulates gene expression at the transcriptional and post-transcriptional levels (Walter and Ron, 2011). Luciferase reporter constructs containing 1 kb and 1.5 kb of the TXNIP promoter, but not a shorter version containing 0.5 kb of the TXNIP promoter, were activated by an ER stress inducer thapsigargin in INS-1 832/13 cells, suggesting that ER stress-mediated TXNIP upregulation is controlled at the transcription level through a critical site within 1.5 kb of the TXNIP promoter (Figure 2H). To confirm that PERK signaling is involved in the transcriptional regulation of TXNIP, the luciferase reporter construct containing 1.5 kb of the TXNIP promoter was co-transfected with PERK or GADD34, a negative regulator of PERK signaling, in INS-1 832/13 cells (Novoa et al., 2001). As expected, PERK stimulated luciferase production, whereas GADD34 substantially suppressed luciferase production, especially under ER stress conditions (Figure 2I). In addition, ectopic expression of PERK stimulated luciferase production in 293T cells as well (data not shown). Consistent with these findings, salubrinal, a chemical inhibitor for eIF2α dephosphorylation, also activated the luciferase reporter construct containing 1.5 kb of the TXNIP promoter (Figure 2J). To identify transcription factors that regulate TXNIP transcription, we screened transcription factors known to be regulated by PERK as well as those that are involved in TXNIP transcription. Based on this screening, we concluded that ATF4, CHOP, XBP-1, Mondo A, and Foxo1 are not significant regulators of TXNIP upregulation under ER stress conditions (data not shown) and elected to focus on ChREBP and ATF5 (Cha-Molstad et al., 2009; Zhou et al., 2008).
ChREBP is a transcription factor that binds to the carbohydrate response element of the LPK gene (Yamashita et al., 2001) and has been shown to be involved in TXNIP upregulation by chronic hyperglycemia in INS-1 cells (Minn et al., 2005). To investigate the role of ChREBP in TXNIP transcription under ER stress conditions, we transfected INS-1 832/13 cells with the luciferase reporter construct containing 1.5 kb of the TXNIP promoter and siRNA directed against ChREBP and treated the cells with thapsigargin. RNAi-mediated knockdown of ChREBP suppressed luciferase production under ER stress condition (Figure 2K). The efficiency of ChREBP knockdown was confirmed by real-time PCR (Figure S1). We next investigated whether ChREBP directly regulates TXNIP expression. Chromatin immunoprecipitation (ChIP) analysis demonstrated that ChREBP binding to the TXNIP promoter was significantly enhanced by thapsigargin treatment (Figure 2L and Figure S2). Mlx has been shown to be a functional heteromeric partner of ChREBP in regulating the expression of ChREBP target genes (Stoeckman et al., 2004), raising the possibility that Mlx is also involved in TXNIP expression. As we expected, Mlx binding to the TXNIP promoter was enhanced by thapsigargin treatment (Figure 2L). We further confirmed the relationship between PERK signaling and ChREBP expression. We found that ChREBP expression was induced by ER stress and was significantly decreased in Perk knockdown INS-1 832/13 cells and Perk knockout MEFs (Figure 2M). The efficiency of Perk knockdown was confirmed by real-time PCR (Figure S1). It has been shown that stimulus-coupling nuclear translocation of ChREBP contributes to expression of its target genes, raising the possibility that ChREBP nuclear translocation might be increased by ER stress. Accordingly, we treated INS-1 832/13 cells with thapsigargin and measured ChREBP levels in cytoplasmic and nuclear fractions by immunoblot. As we surmised, nuclear ChREBP levels were slightly increased by thapsigargin treatment (Figure S3).
Next we investigated the possible role of ATF5 in ER stress-mediated TXNIP expression. ATF5 is a member of the ATF/cAMP response-element binding protein (CREB) family of transcription factors and makes life-or-death decisions in cells (Persengiev and Green, 2003). It has been shown that PERK-mediated eIF2α phosphorylation directs ATF5 protein translation as well as ATF5 mRNA transcription, and ATF5 is integral to the eIF2α kinase response (Zhou et al., 2008), raising the possibility that ATF5 plays a role in TXNIP upregulation under ER stress conditions. To test this possibility, we transfected INS-1 832/13 cells with the luciferase reporter construct containing 1.5 kb of the TXNIP promoter and siRNA directed against ATF5 and treated the cells with thapsigargin. RNAi-mediated knockdown of ATF5 suppressed luciferase production under ER stress conditions (Figure 2N). The efficiency of ATF5 knockdown was confirmed by real-time PCR (Figure S1). We further investigated whether ATF5 is recruited to the TXNIP promoter. ChIP analysis demonstrated that ATF5 binding to the TXNIP promoter was enhanced by thapsigargin treatment (Figure 2O and Figure S2). In addition, ATF5 expression under ER stress was decreased in Perk knockdown INS-1 832/13 cells and Perk knockout MEFs (Figure 2P). The efficiency of Perk knockdown was confirmed by real-time PCR (Figure S1). Taken together, our results demonstrate that ChREBP and ATF5 are major regulators of TXNIP expression under ER stress conditions.
TXNIP plays a role in ER stress-mediated inflammasome activation, IL-1β production and apoptosis
It has previously been shown that TXNIP is essential for activation of the NLRP3 inflammasome and IL-1β production under oxidative stress (Zhou et al., 2010), raising the possibility that TXNIP is involved in IL-1β production through NLRP3 under ER stress conditions. To test this idea, we first analyzed expression levels of IL-1β and its major downstream target, IL-6, in ER stressed β cells. We treated INS-1 832/13 cells with thapsigargin or tunicamycin and measured IL-1β and IL-6 expression levels by real-time PCR. Figure 3A illustrates that IL-1β and IL-6 expression was increased by thapsigargin and tunicamycin. Notably, ectopic expression of GADD34, a negative regulator for PERK signaling under ER stress, strongly suppressed thapsigargin-mediated IL-1β and IL-6 induction (Figure 3B). Consistent with these findings was our observation that IL-1β and IL-6 mRNA expression levels were increased by tunicamycin treatment in human primary islets (Figure 3C). To test that IL-1β produced from islets by ER stress was functional, we treated human primary islets with an IL-1 receptor antagonist together with tunicamycin and then measured IL-1β and IL-6 expression levels. IL-1 receptor antagonist has been shown to suppress IL-1β signaling and decrease IL-1β and IL-6 mRNA expression levels in β cells (Ehses et al., 2009). We found that IL-1β and IL-6 upregulation was attenuated in human primary islets treated with IL-1 receptor antagonist (Figure 3C). These results indicate that ER stress induces secretion of functional IL-1β in β cells. To further investigate whether ER stress-mediated IL-1β production was dependent on TXNIP, we suppressed TXNIP expression using shRNA directed against TXNIP in INS-1 832/13 cells, treated these cells with thapsigargin, and then measured expression levels of IL-1β and IL-6. As we expected, IL-1β and IL-6 upregulation was attenuated in TXNIP knockdown cells as compared to control cells (Figure 3D). To further confirm this observation, we measured IL-β and IL-6 expression in primary islets from TXNIP knockout and control mice treated with thapsigargin. As expected, IL-1β and IL-6 upregulation by ER stress was attenuated in islets from TXNIP knockout mice as compared to islets of control mice (Figure 3E). In support of this conclusion, RNAi-mediated suppression of NLRP3, a major component of inflammasome involved in TXNIP-mediated IL-1β activation (Zhou et al., 2010), blocked IL-1β and IL-6 upregulation under ER stress conditions (Figure 3F).
Figure 3. IL1-β is induced by ER stress and regulated by TXNIP.
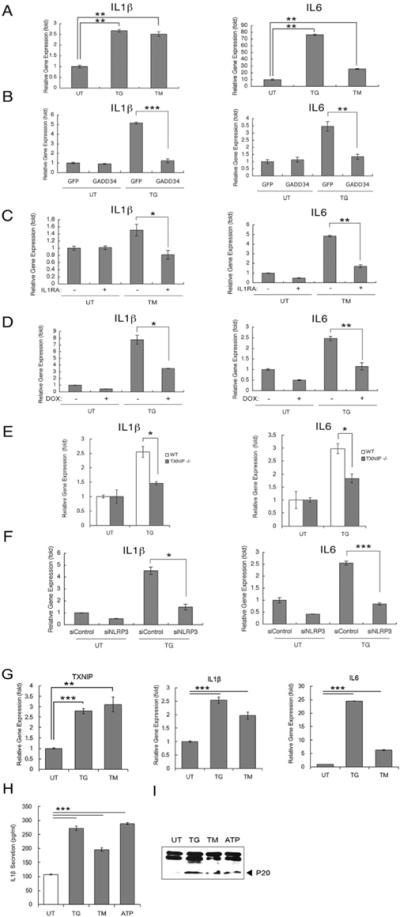
(A) IL-1βb and IL-6 mRNA expression in human islets treated with thapsigargin (TG, 1 μM) for 6h, tunicamycin (TM, 5 μg/ml) for 6 h or untreated. (B) IL-1β and IL-6 mRNA expression in INS-1 832/13 cells transfected with control GFP or GADD34 expression plasmid. After 36 h, cells were untreated (UT) or treated with thapsigargin (TG, 0.5 μM) for 6 h. (C) IL-1b and IL-6 mRNA expression in human islets pretreated with interleukin-1 receptor antagonist (IL1RA, 1 ug/ml) for 24 h, and then treated with tunicamycin (TM, 5 ug/ml) for 8 h. (D) IL-1β and IL-6 mRNA expression in INS-1 832/13 cells stably transduced with pLenti-TO/shTXNIP, inducible lentivirus expressing shTXNIP. Cells were cultured with doxycycline (2 μg/ml) to induce shTXNIP or without doxycycline for 48 h, then challenged with thapsigargin (TG, 0.5 μM) for 6 h. (E) IL-1β and IL-6 mRNA expression in primary islets from Txnip−/− and control (WT) mice treated with thapsigargin (TG, 0.5 μM) for 6 h. (F) IL-1β and IL-6 mRNA expression in INS-1 832/13 cells transfected with control or NLRP3 siRNA, and then treated with thapsigargin (TG, 0.5 μM) for 6 h or untreated. (G) TXNIP, IL-1β and IL-6 mRNA expression in THP-1 cells transformed with phorbol 12-myristate 13-acetate (0.5 μM), and then treated with thapsigargin (TG, 1 μM), tunicamycin (TM, 20 μg/ml) for 6 h or untreated. (H)(I) THP-1 cells transformed with phorbol 12-myristate 13-acetate (0.5 μM) were treated with thapsigargin (TG, 1 μM), tunicamycin (TM, 20 μg/ml), ATP (5 mM) for 6 h or untreated. Secreted IL-1β was measured by ELISA (H) and caspase-1 cleavage was measured by immunoblot. P20, cleaved caspase-1 (I). n=3; values are mean ± SD. * p<0.05, ** p<0.01, *** p<0.001. n.s., not significant.
Three experiments verified that ER stress and TXNIP are involved in inflammasome activation. For this purpose, we elected to use THP-1 cells in which activation levels of inflammasome can be quantified by multiple methods (Zhou et al., 2010). First, TXNIP, IL-1β, and IL-6 mRNA expression levels were increased by both thapsigargin and tunicamycin treatments in THP-1 cells (Figure 3G). Second, IL-1β secretion was increased by thapsigargin and tunicamycin treatments (Figure 3H). Third, caspase-1 cleavage, a major downstream event of inflammasome activation (Kuida et al., 1995), was also increased by thapsigargin and tunicamycin treatment (Figure 3I). Collectively, these results indicate that ER stress is involved in inflammasome activation and TXNIP plays a crucial role in ER stress-mediated upregulation and maturation of IL-1β.
TXNIP and IL-1β signaling has a role in β cell death in type 1 and type 2 diabetes, raising the possibility that TXNIP-mediated IL-1β induction plays a role in ER stress mediated cell death (Dinarello et al., 2010; Larsen et al., 2007). Therefore, we next examined the role of TXNIP in β cell death under ER stress conditions. For this purpose, we derived INS-1 832/13 cells in which we could induce shRNA-mediated suppression of TXNIP using doxycycline (Figure 4A, left panel). Figure 4A (right panel) demonstrates that suppression of TXNIP protected INS-1 832/13 cells from ER stress-mediated cell death. We also measured the amount of PI positive cells (i.e., dead cells) when we treated TXNIP knockdown cells with thapsigargin and tunicamycin. As expected, suppression of TXNIP decreased the amount of PI positive cells under ER stress conditions (Figure 4B). Consistent with this finding was our observation that shRNA-mediated knockdown of TXNIP increased the viability of INS-1 832/13 cells under ER stress conditions (Figure 4C). To test the involvement of IL-1β signaling in ER stress-induced cell death, we treated human islets with an IL-1 receptor antagonist together with or without thapsigargin and then monitored cell death. As shown in Figure 4D, caspase-3 activity induced by thapsigargin was modestly reduced by the treatment with an IL-1 receptor antagonist. Taken together, these results indicate that TXNIP-mediated IL-1β production plays a role in ER stress-mediated β cell death and suppression of TXNIP signaling confers protection against ER stress.
Figure 4. TXNIP is an apoptotic component of the unfolded protein response.

(A) TXNIP, caspase-3, and actin protein expression in INS-1 832/13 cells stably transduced with pLenti-TO/shTXNIP, inducible lentivirus expressing shTXNIP. Cells were cultured with doxycycline (Dox, 2 μg/ml) to induce shTXNIP or without doxycycline for 48 h, then challenged with thapsigargin (TG, 0.05 μM) or tunicamycin (TM, 5 μg/ml) for 24 h. Single and double asterisks indicate uncleaved and cleaved caspase-3, respectively. (B) INS-1 832/13-TetR-shTXNIP cells were incubated with doxycycline (Dox, 2 μg/ml) for 48 h, then treated with thapsigargin (TG, 10 nM), tunicamycin (TM, 5 μg/ml) for 24 h or untreated. Cells were stained with propidium iodide solution (PI) followed by flow cytometry analysis. (TG, n=6 and TM, n=9; values are mean ± SD). (C) Viability of INS-1 832/13-TetR-shTXNIP cells incubated with doxycycline (Dox, 2 μg/ml) for 48 h, and then treated with thapsigargin (TG, 50 nM) for 24 h or untreated (n=3; values are mean ± SD). (D) Human primary islets (28 year old male, BMI 21, HbA1C 5.4) were treated with thapsigargin (TG, 1 μM) in the presence of BSA(−, 1 μg/ml) or interleukin-1 receptor antagonist (+, 1 μg/ml) for 24 h. Caspase 3/7 activity was measured by Caspase-Glo 3/7 assay (Triplicated, values are mean ± SD). * p<0.05, ** p<0.01.
DISCUSSION
The data here demonstrate that TXNIP is a critical signaling node that links ER stress and IL-1β production. TXNIP expression is induced by ER stress under the IRE1α and PERK-eIF2α pathways of the UPR. Transcriptionally TXNIP expression is regulated by ChREBP and ATF5. An accompanying manuscript from Drs. Feroz Papa and Scott Oakes revealed that IRE1α regulates TXNIP expression at the post-transcriptional level, which may be complementary to PERK's transcriptional control. As a result TXNIP induces IL-1β production through the activation of NLRP3 inflammasome and IL-1β mRNA transcription, leading to β cell death.
Recent experimental, clinical, and genetic evidence suggests that ER stress-mediated β cell dysfunction and death contribute to the pathogenesis of type 1 and type 2 diabetes as well as genetic forms of diabetes such as Wolfram syndrome and permanent neonatal diabetes (Eizirik et al., 2008; Kaufman, 2011; Oslowski and Urano, 2011; Tersey et al., 2012). However, the mechanisms involved and pathological contributions remain elusive. Here we propose that during diabetes, TXNIP expression is induced through the UPR and leads to β cell inflammation and apoptosis.
Inflammation is a critical component leading to β cell dysfunction and death in type and type 2 diabetes. (Atkinson et al., 2011; Bluestone et al., 2010; Corbett et al., 1993; Eizirik et al., 2009; Larsen et al., 2007). There has been substantial interest in identifying pathways initiating inflammation. A variety of inflammatory pathways have been associated with IL-1β production. However, essential factors initiating inflammation by IL-1β production in β cells have been elusive. Our findings unexpectedly revealed that the UPR regulates IL-1β production through TXNIP. ER stress-mediated upregulation of TXNIP leads to IL-1β secretion through inflammasome activation. Secreted IL-1β is capable of binding to IL-1 receptor on β cell surface, which may lead to induction of its own expression. It is also possible that TXNIP upregulation by ER stress may be involved in the transcriptional activation of IL-1β (Koenen et al., 2011). Our findings suggest that environmental and genetic factors that can elicit ER stress are promising candidates for crucial factors initiating inflammation in β cells.
It has been shown that TXNIP expression is increased by high glucose in human islets and plays an important role in glucose toxicity (Hui et al., 2008; Oka et al., 2009; Shalev et al., 2002; Yoshihara et al., 2010). We have previously shown that chronic high glucose leads to unresolvable ER stress in β cells, leading to β cell dysfunction and death (Lipson et al., 2006; Lipson et al., 2008). Thus, TXNIP upregulation by high glucose could be mediated by the UPR. TXNIP has been also shown to be a critical signaling molecule linking oxidative stress to inflammasome activation (Zhou et al., 2010). These findings, combined with our data, indicate that a variety of stress signaling pathways converge at TXNIP, leading to inflammasome activation and IL-1β production.
ER stress and inflammation are critical pathogenic components of β cell dysfunction and death in diabetes. Our data combined with recent findings indicate that there exists a tight link between ER stress, oxidative stress, glucose toxicity, and inflammation, suggesting that a therapeutic strategy that aims to target the common molecular processes that are altered in stressed β cells might be effective. TXNIP provides such a target.
EXPERIMENTAL PROCEDURES
Cell culture
INS-1 832/13 cells were a gift from Dr. Christopher Newgard (Duke University Medical Center). THP-1 cells were obtained from ATCC and cultured in RPMI 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, and 0.05 mM 2-mercaptoethanol. These cells were transformed with phorbol 12-myristate 13-acetate (0.5 μM for 3 h) to measure IL-1β secretion and caspase-1 cleavage. Mouse islets were cultured in RPMI 1640 supplemented with 5% FBS and 5 mM glucose. Human islets were cultured in CMRL media supplemented with 5 mM glucose and grown on laminin V coated plates. Mouse embryonic fibroblasts were maintained in DMEM with 10% fetal bovine serum. Ireα−/− and Perk−/− fibroblasts were a gift from Dr. David Ron (University of Cambridge). Atf6α−/−, eIF2αS/S and eI2F2αA/A fibroblasts were gifts from Dr. Randal Kaufman (Sanford-Burnham Medical Research Institute)
Lentivirus system
For generation of cells stably suppressing TXNIP, INS-1 832/13 cells were transduced with a lentivirus expressing shRNA against rat TXNIP.
Antibodies used for immunoblots
Blots were probed with the following antibodies: anti-TXNIP (MBL, Nagoya, Japan), anti-CHOP (Pierce Biotechnology, Rockford, IL), anti-eIF2α and anti-ChREBP (Santa Cruz Biotechnology, Santa Cruz, CA), anti-caspase-1, anti-CREB, anti-eIF2α-P, and anti-GAPDH (Cell Signaling, Danvers, MA), and anti-actin (Sigma-Aldrich, St. Louis, MO).
Luciferase Assay
For reporter assays, INS1 cells were cotransfected with a TXNIP promoter-luciferase construct and various constructs as indicated. The culture medium was changed to fresh medium with 2.5mM glucose after 6 hours. Prior to lysis at 24 hours after transfection, cells were treated with or without 0.5uM of thapsigargin for 6h. Luciferase activity was measured with a Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and transfections were normalized with the pRL-TK vector (Promega) as an internal control.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the Chromatin IP Kit (Cell Signaling, Beverly, MA) according to the manufacturer's instructions. The cartoon of promoter region used for ChiP was shown in Figure S2.
Animal Experiments
Txnip knockout mice were generated as previously described (Hui et al., 2008). All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School.
Statistical analysis
Welch's t-test on log transformed data was used for determining the significance between two treatments. Data are presented as the mean ± SD.
Supplementary Material
RESEARCH HIGHLIGHTS
TXNIP is a critical signaling node that links ER stress and inflammation.
TXNIP is induced by ER stress through the PERK and IRE1 pathways.
TXNIP mediates ER stress-mediated β cell death.
Acknowledgments
We thank Karen Sargent, Mai Kanekura, Linda Leehy, and Elaine Norowski for technical assistance. This work was supported by grants from NIH (DK067493, DK016746, P60 DK020579, RR024992, and UL1 TR000448) and JDRF (11-2011-40) to F. Urano, NIH (HL057346, HL052172, DK042394, DK088227, and DK93074) to R. Kaufman and NIH (DK080339) for S. Hui.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, and Rhodes CJ. How does type 1 diabetes develop?: the notion of homicide or beta-cell suicide revisited. Diabetes. 2011;60:1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, Novials A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes. 2007;56:2284–2294. doi: 10.2337/db07-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Saxena G, Chen J, Shalev A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. The Journal of biological chemistry. 2009;284:16898–16905. doi: 10.1074/jbc.M109.010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. 2008;57:938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, DeLuca HF. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochimica et biophysica acta. 1994;1219:26–32. doi: 10.1016/0167-4781(94)90242-9. [DOI] [PubMed] [Google Scholar]
- Corbett JA, Sweetland MA, Wang JL, Lancaster JR, Jr., McDaniel ML. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1731–1735. doi: 10.1073/pnas.90.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:314–321. doi: 10.1097/MED.0b013e32833bf6dc. [DOI] [PubMed] [Google Scholar]
- Donath MY, Halban PA. Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia. 2004;47:581–589. doi: 10.1007/s00125-004-1336-4. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Lacraz G, Giroix MH, Schmidlin F, Coulaud J, Kassis N, Irminger JC, Kergoat M, Portha B, Homo-Delarche F, Donath MY. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13998–14003. doi: 10.1073/pnas.0810087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, Chen AZ, Sachithanantham S, Jung DY, Kim JK, Davis RA. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3921–3926. doi: 10.1073/pnas.0800293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Beta-cell failure, stress, and type 2 diabetes. N Engl J Med. 2011;365:1931–1933. doi: 10.1056/NEJMcibr1109442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, Tack CJ, Netea MG. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes. 2011;60:517–524. doi: 10.2337/db10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lipson KL, Ghosh R, Urano F. The Role of IRE1alpha in the Degradation of Insulin mRNA in Pancreatic beta-Cells. PLoS ONE. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. The Journal of biological chemistry. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. The Journal of cell biology. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Yoshihara E, Bizen-Abe A, Liu W, Watanabe M, Yodoi J, Masutani H. Thioredoxin binding protein-2/thioredoxin-interacting protein is a critical regulator of insulin secretion and peroxisome proliferator-activated receptor function. Endocrinology. 2009;150:1225–1234. doi: 10.1210/en.2008-0646. [DOI] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. The binary switch that controls the life and death decisions of ER stressed beta cells. Curr Opin Cell Biol. 2011;23:207–215. doi: 10.1016/j.ceb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persengiev SP, Green MR. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis. 2003;8:225–228. doi: 10.1023/a:1023633704132. [DOI] [PubMed] [Google Scholar]
- Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional beta-cell mass in diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):54–62. doi: 10.1111/j.1463-1326.2008.00941.x. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Shalev A, Pise-Masison CA, Radonovich M, Hoffmann SC, Hirshberg B, Brady JN, Harlan DM. Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology. 2002;143:3695–3698. doi: 10.1210/en.2002-220564. [DOI] [PubMed] [Google Scholar]
- Stoeckman AK, Ma L, Towle HC. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. The Journal of biological chemistry. 2004;279:15662–15669. doi: 10.1074/jbc.M311301200. [DOI] [PubMed] [Google Scholar]
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D, Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, Masutani H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nature communications. 2010;1:127. doi: 10.1038/ncomms1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. The Journal of biological chemistry. 2008;283:7064–7073. doi: 10.1074/jbc.M708530200. [DOI] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.