

Summary
We investigate cytotoxicity and mechanism of action of AS703026, a novel, selective, orally bioavailable MEK1/2 inhibitor, in human multiple myeloma (MM). AS703026, more potently (9-10 fold) than AZD6244, inhibited growth and survival of MM cells and cytokine-induced osteoclast differentiation. Inhibition of proliferation induced by AS703026 was mediated by G0-G1 cell cycle arrest and was accompanied by reduction of c-maf oncogene expression. AS703026 further induced apoptosis via caspase 3 and PARP cleavage in MM cells, both in the presence or absence of bone marrow stromal cells (BMSCs). Importantly, AS703026 sensitized MM cells to a broad spectrum of conventional (dexamethasone, melphalan), novel or emerging (lenalidomide, perifosine, bortezomib, rapamycin) anti-MM therapies. Significant tumor growth reduction in AS703026- vs. vehicle-treated mice bearing H929 MM xenograft tumors correlated with downregulated pERK1/2, induced PARP cleavage, and decreased microvessels in vivo. Moreover, AS703026 (<200 nM) was cytotoxic against the majority of tumor cells tested from patients with relapsed and refractory MM (84%), regardless of mutational status of RAS and BRAF genes. Importantly, BMSC-induced viability of MM patient cells was similarly blocked within the same dose range. Our results therefore support clinical evaluation of AS703026, alone or in combination with other anti-MM agents, to improve patient outcome.
Keywords: multiple myeloma (MM), MEK1/2 inhibitor, bone marrow stromal cells (BMSCs), novel kinase inhibitor therapy
Introduction
Despite advances in the understanding of the molecular pathogenesis of multiple myeloma (MM) and promising new therapies including bortezomib, thalidomide, and lenalidomide, only 25-35% of patients respond to therapies in the relapsed and refractory settings (Richardson and Anderson 2006, Richardson, et al 2009). In addition, resistance develops in almost every patient that initially responds to these agents. Thus, there is still unmet medical need for the treatment of MM.
The mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) signal transduction pathway significantly contributes to MM cell growth and survival, as well as to angiogenesis and to the development of drug resistance within the bone marrow (BM) microenvironment (Hideshima, et al 2004, Hideshima, et al 2007, Shain, et al 2009). This major pathway is often deregulated in MM cells leading to increased proliferation and resistance to apoptosis. In parallel, the MEK/ERK signaling cascade tightly regulates cytokine and growth factor secretion within the BM milieu, which can further augment MM growth, survival, and drug resistance (Giuliani, et al 2004, Hideshima, et al 2007, Menu, et al 2004). Importantly, the key components of the Ras/Raf/MEK/ERK signaling pathway frequently mediate constitutive activation of downstream effectors in late stage MM and plasma cell leukemia (PCL) (Bezieau, et al 2002, Corradini, et al 1993, Intini, et al 2007, Liu, et al 1996, Tiedemann, et al 2008).
MEK/ERK activation in MM (9%) and PCL (31%) is due in part to the high rate of mutations of the N- and K-RAS genes (codons 12, 13 and 61), whereas the activating V600E mutation within exon 15 of the BRAF gene is relatively rare in MM and PCL (Bonello, et al 2003) despite occurrence in approximately 10-80% of melanomas and colon cancers with high constitutive MEK/ERK activity (Davies, et al 2002, Sebolt-Leopold and Herrera 2004). In these indications, the presence of the V600E BRAF mutation was suggested to predict responses to MEK inhibition (Davies, et al 2002, Friday and Adjei 2008, Pratilas and Solit 2007, Solit, et al 2006). RAS mutations, either N- or K- but not H-RAS, were found in MM patients with increasing frequency in relapsed (45-67%) versus newly diagnosed (25%) diseases, correlating with more aggressive disease features (Chng, et al 2008, Liu, et al 1996, Portier, et al 1992, Rasmussen, et al 2005). RAS mutations have been rarely detected (<7%) in pre-malignant monoclonal gammopathy of undetermined significance (MGUS) (Chng, et al 2008, Rasmussen, et al 2005), suggesting an important role of mutated RAS in malignant transformation of clonal plasma cells and MM pathogenesis. Indeed, RAS is the single most commonly mutated gene in MM and is associated with greater tumor burden and likely transforming character, especially in t(11,14) MM (Chesi, et al 2001, Chng, et al 2008). In addition, ANBL-6 MM cells containing RAS mutations exhibit increased binding to extracellular matrix protein and chemotherapeutic drug resistance via COX-2 gene upregulation (Billadeau, et al 1995, Hoang, et al 2006, Hu, et al 2003). These studies strongly support targeting MEK/ERK with a small molecule inhibitor to prevent aberrant oncogenic signaling as a novel and promising anti-MM strategy.
Our recent work demonstrated that MEK1/2 inhibition by ARRY142886/AZD6244 (Array Biopharma/AstraZeneca)(Tai, et al 2007) was directly and indirectly cytotoxic against MM cells and cytokine-induced osteoclastogenesis, respectively, suggesting potential use of MEK1/2 inhibitors in treating MM patients. In the recent solid tumor phase I/II clinical trials of AZD6244, partial responses and stable disease were seen in some patients with pancreatic cancer, non small cell lung cancer, and malignant melanoma (Adjei, et al 2008). However, the ultimate clinical benefit of AZD6244 remains to be defined. Most recently, AS703026 (N-[(2S)-2,3-dihydroxypropyl]-3-[(2-fluoro-4-iodophenyl)amino]isonicotinamide hydrochloride), a highly selective, potent, ATP non-competitive allosteric inhibitor of MEK1/2 was discovered through medicinal chemistry and cell biology efforts (Figure. 1A and (Goutopoulos, et al 2009)). AS703026 binds to MEK1/2 in an allosteric site that is distinct from, yet in close proximity to, the ATP binding site. Binding of AS703026 to this allosteric site prevents the activation of MEK1/2. AS703026 has favorable pharmacologic characteristics and completely and specifically blocks MEK1/2 activity, but does not affect activity of 217 other kinases tested. Recent studies with AS703026 in multiple solid tumor xenografts showed remarkable inhibition of both anchorage-independent growth in vitro and tumor growth in vivo (Clark, et al 2009, Machl, et al 2009), and it is currently under evaluation in Phase I clinical oncology trials in solid tumors.
Fig. 1. AS703026 inhibited growth and survival of MM cell lines as well as osteoclast formation.
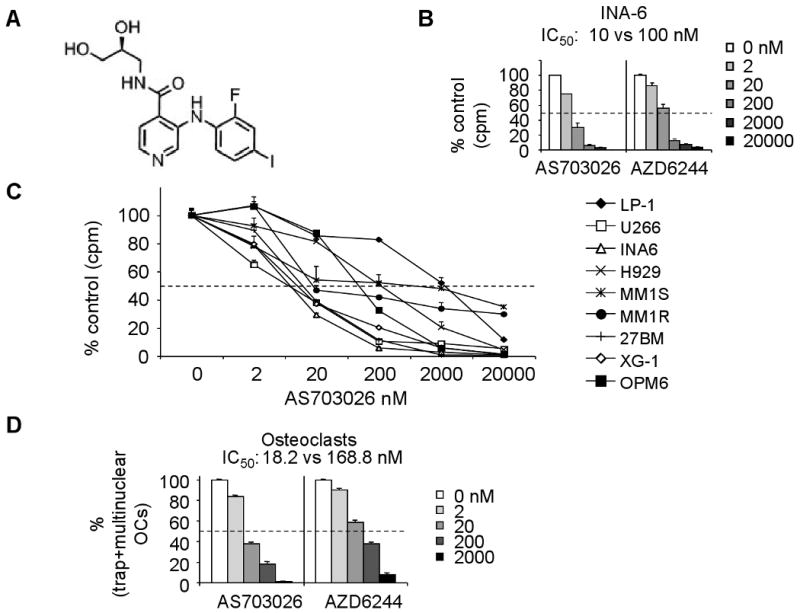
A, The chemical structure of AS703026. B, INA-6 MM cells were treated with AS703026 or AZD6244 for 2 days, followed by [3H]thymidine uptake assay. C, MM cells, sensitive or resistant to standard of care agents, were incubated with DMSO or a dose range of AS703026 (2-2000 nM) for 2 days, and subjected to DNA synthesis assay. D, PBMCs isolated from normal donors (n=3) were incubated with M-CSF and RANKL, in the presence or absence of AS703026 or AZD6244 for 14 days. The TRAP assay was performed to measure the formation of multinuclear osteoclast cells (OC).
Based on the relatively potent activity of AS703026 in various solid tumor models and the significant dependency of MM pathophysiology on the MEK/ERK signaling cascade, we investigated the cytotoxic effects of AS703026 against MM and defined its mechanisms of action in the current study.
Materials and Methods
Cell culture and bone marrow stromal cells (BMSCs)
All CD138-expressing MM cell lines were grown in RPMI1640 (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin and 100μg/ml streptomycin (Invitrogen). They were kindly provided by sources previously described (Tai, et al 2004, Tai, et al 2008, Tai, et al 2005, Tai, et al 2003), ATCC (Manassas, VA), or the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Primary CD138+ MM cells from patients were obtained after IRB-approved (Dana-Farber Cancer Institute) informed consent using positive selection with CD138 microbeads (Miltenyi Biotech, Auburn, CA). Informed consent was obtained from all patients in accordance with the Declaration of Helsinki. Residual CD138-negative bone marrow mononuclear cells (BMMCs) were cultured in RPMI 1640/10% FCS for 3 to 6 weeks to generate BM stromal cells (BMSCs), as previously described (Tai, et al 2008, Tai, et al 2009).
Reagents
The MEK1/2 inhibitor AS703026 (N-[(2S)-2,3-dihydroxypropyl]-3-[(2-fluoro-4-iodophenyl)amino]isonicotinamide hydrochloride) was provided by EMD Serono (Rockland, MA). This compound is a novel, orally bioavailable small molecule inhibitor that binds to an allosteric site in close proximity to the adenosine triphosphate binding site of MEK1/2. AS703026 was dissolved in dimethyl sulfoxide (10 mM) and stored at -20°C for in vitro study and was suspended in 0.5% carboxymethylcellulose (CMC)/0.25%Tween20 at 10 mg/ml for in vivo study. All other compounds, including AZD6244, were purchased from Selleck Chemicals Co., Ltd. (London, Canada) except dexamethasone (dex) from Sigma-Aldrich (St. Louis, MO) and rapamycin from LC Laboratories (Woburn, MA).
Cytotoxicity assays
The inhibitory effects of study compounds on MM cell growth and survival were assessed by both [3H]thymidine incorporation and by measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide (MTT; Sigma-Aldrich, St Louis, MO) dye absorbance following the manufacturer's instruction. Cells (104/well for MM cell line, in triplicates and 2-5×105/well for patient MM cells) were cultured in 96-well plates for 3 days (MM cell lines) or 5-days (patient MM cells). For the [3H]thymidine incorporation assay, cells were pulsed with 0.5 μCi (0.0185 MBq)/well [3H]thymidine (Amersham, Pittsburgh, PA) for 6 h (cell lines), harvested onto glass fiber filters, and counted in a β-scintillation counter. Due to low DNA synthesis of patient MM cells, they were pulsed with 2 μCi/well [3H]thymidine and measured during the last 36 h of culture.
Cell cycle analysis was assessed by propidium iodide (PI) staining (assessed/measured by flow cytometry), and drug-induced apoptosis was determined by annexin-V/PI staining and flow cytometric data analysis (Cytomics FC500-CXP version 3.0; Beckman Coulter, Hialeah, FL and Flowjo version 8.6.6; Tree Star, Inc., Ashland, OR).
Immunoblotting analysis
Total cell lysates were subjected to 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes, as previously reported (Tai, et al 2008, Tai, et al 2009). All antibodies (Abs) were obtained from Cell Signaling Technology (Danvers, MA).
Analyses of drug combination by the CalcuSyn program
The Calcusyn software program (Biosoft, Ferguson, MO) was used to evaluate whether treatment combinations of AS703026 with various anti-MM drugs were additive, antagonist or synergistic. This program is based upon the Chou–Talalay method to determine Combination indices (CI) (Chou 2008). CIs from three different experiments were generated for each set of combinations, in triplicate, using results obtained from each drug alone and in combination at non-constant ratios within the same experiment. CI < 1.0 indicates synergism, CI =1 suggests an additive effect, and values > 1 corresponds to potential antagonism. The Chou-Talalay plot indicates the CI on the y-axis as a function of effect level (Fa) on the X-axis.
Enzyme-linked immunosorbent assay
MM cell lines or patient MM cells were added to BMSC-coated plates (BMSCs:MM cells at 3:1), and conditioned media that were harvested from 2-day cultures were tested for VEGF and IL-6 secretion by enzyme-linked immunosorbent assay (ELISA) (R&D Systems). The minimum detectable level of both VEGF and IL-6 was 10.0 pg/mL (pM).
In vitro osteoclast culture
Peripheral blood mononuclear cells (PBMCs) were obtained from 4 normal donors after informed consent. CD14+ osteoclast precursor cells were cultured for 14 days in ISCOV/10% FCS with receptor activator of NF-κB ligand (RANKL; 50 ng/mL; Peprotech, Rocky Hill, NJ) and macrophage-colony-stimulating factor (M-CSF; 25 ng/mL; Peprotech) in the absence or presence of 0.002-2 μM of either AS703026 or AZD6244, followed by tartrate-resistant acid phosphatase (TRAP) staining according to manufacturer's protocol (Sigma). The number of TRAP+ multinucleated mature osteoclasts (>2 nuclei per cell) was measured at predetermined sites of the area of 1 × 1 mm. Five sites were measured in a well of a 96-well plate, and a mean value was calculated. Four wells were measured for each experimental condition, and these results were expressed as mean ± S.E. (standard error).
Human plasmacytoma xenograft model
All animal studies in the xenograft tumor model were approved by the Dana Farber Cancer Institute Animal Care and Use Committee. CB17 severe combined immunodeficiency (SCID) mice (Charles River Laboratories, Inc., Wilmington, MA) were subcutaneously inoculated with H929 (4×106) cells in 100 μL RPMI-1640 medium. Mice developed palpable tumors (∼130 mm3) approximately 3 weeks after cell injection and were randomized to receive orally twice daily either AS703026 (15 or 30 mg/kg) or control vehicle alone. Tumor size was measured every other day in 2 dimensions using calipers, and tumor volume was calculated using the following formula: V = 0.5 × a × b2, where “a” and “b” were the long and short diameter of the tumor, respectively. Animals were euthanized when their tumors reached 2 cm3 in volume, when they were moribund or showed paralysis or major compromise in their quality of life occurred. Tumor formation changes in mice treated with control vehicle vs. AS703026 were plotted using the GraphPad Prism version 4.03 for Windows (GraphPad Software, San Diego, CA) Tumors were subjected to immunoblotting and immunochemistry analyses using specific monoclonal (m)Abs, as indicated in Fig. 6. Images were examined with a Leica DM LB research microscope, captured using Leica IM50 Image Manager (Bannockburn, IL), and processed using Adobe Photoshop Software version 7.0 (Adobe, San Jose, CA).
Fig. 6. AS703026 inhibited tumor growth in a human plasmacytoma model of H929 MM cells.
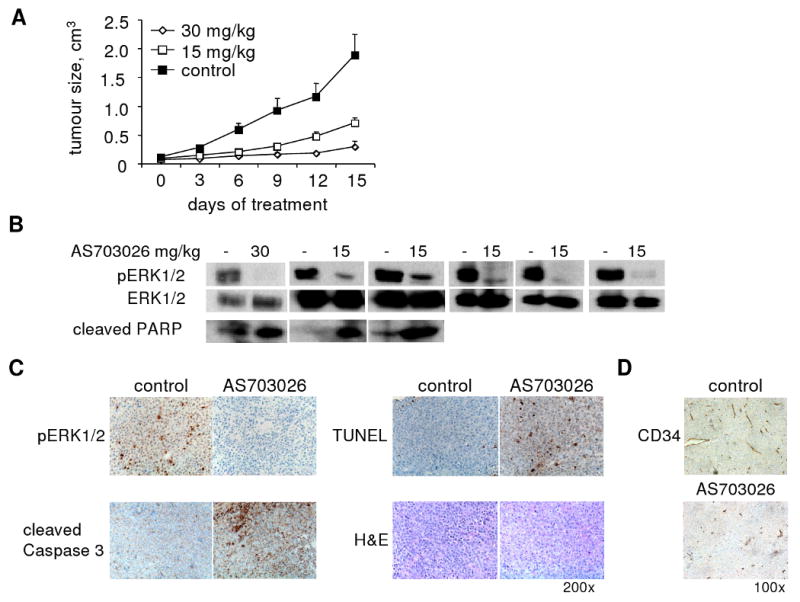
A, Mice bearing H929 MM tumors were treated with control vehicle (0.5%cmc/0.25%Tween20; n = 6) or AS703026 (n = 6 for 15 mg/kg, n = 4 for 30 mg/kg) BID orally (5 days per week for 2 weeks). Animals were euthanized when tumors reached 2 cm3 in size. Treatment with AS703026 inhibited tumor growth compared to vehicle control (RM-ANOVA & Fisher's LSD, p < 0.05) B, Four hours after the last dose, tumors were excised and subject to immunoblotting for pERK1/2 and cleaved PARP. Immunoblotting for ERK1/2 confirmed equal protein loading. C, IHC studies of tumors for pERK, cleaved caspase3, and TUNEL (200× magnification) and D, CD34 (100× magnification) Images were taken using a Leica DFC300FX with a 10/0.22 NA objective and a Leica IM50 Image Manager.
RAS and BRAF gene mutation analysis
Purified CD138+ patient MM cells were subjected to isolation of genomic DNA using the Qiagen AllPrep DNA/RNA kit (QIAGEN Inc., Valencia, CA). Genomic DNA samples were then subjected to nested PCR to amplify each exon of 3 RAS genes (N-, K-, H-) and BRAF gene (Mosaic Laboratories, Lake Forest, CA). Each nested PCR product was verified by 2% agarose gel electrophoresis to be a single band, without any visible non-specific artifacts, before subsequent denaturation and sequencing on an ABI 3100 Genetic Analyzer. DNA sequence was analyzed using ABI Sequence Analysis v3.7 and Sequencher (GeneCodes, Ann Arbor, MI).
Statistical analyses
Significant treatment effects were determined by the Student's t-test for in vitro studies. Tumor volume and body weight data from the in vivo study were analyzed by Repeated Measures ANOVA (RM-ANOVA), followed by post hoc pairwise comparisons with Fisher's Least Significant Difference (LSD) test. (All α= 0.05)
Results
Cytotoxicity of the novel MEK inhibitor AS703026 against MM cells
The effects of AS703026 on the growth of a panel of MM cell lines, either sensitive or resistant to current anti-MM therapies, were first determined by [3H]thymidine incorporation. Incubation with AS703026 for 2 days inhibited the growth of MM cell lines in a dose-dependent manner, with IC50s ranging from 0.005 to 2 μM (Figure 1B-C). Notably, AS703026, more potently than AZD6244, inhibited growth of MM cells (Fig. 1B and Supplement Fig. 1). For example, the IC50 of AS703026 and AZD6244 against INA-6 cells was 10 and 100 nM, respectively. AS703026 was also approximately 10-fold more potent than AZD6244 at inhibiting cytokine-induced osteoclast differentiation in vitro, as measured by number of TRAP-positive multinuclear cells following treatment of PBMCs with RANKL and M-CSF (Fig. 1D).
Given the differential sensitivity of the MM cell lines to AS703026, we investigated whether a correlation might exist between the potency of AS703026 on the cell lines and their respective RAS or RAF mutational status. One of the highly sensitive cell lines, U266 cells (AS703026 IC50 of 5 nM), harbors a K601N BRAF mutation but no RAS mutations (Supplement Table 1 and (Chesi, et al 2001, Portier, et al 1992)), whereas another highly sensitive cell line, INA-6 (AS703026 IC50 of 11) has a N-RAS G13D mutation. A moderately sensitive cell line, H929 (AS703026 IC50 of 200nM) harbors a N-RAS G12D mutation (Burger, et al 2001, Chesi, et al 2001). Finally, both dex-sensitive MM1S and dex-resistant MM1R cells, which are fairly resistant to AS703026 (IC50>2 μM), bear a K-RAS G12A mutation. Taken together, these data suggest that there is no discernible relationship between RAS or RAF mutational status and the sensitivity of the cell lines to AS703026.
To evaluate target modulation by AS703026 in MM cell lines, specific inhibition of pERK1/2 was measured by immunoblotting following treatment of cells with AS703026 under various culture conditions. Treatment with AS703026 significantly diminished basal levels of pERK in U266 cells in a time- and dose-dependent fashion (Fig. 2A). Interestingly, pAKT levels in these cells slightly increased over the same time period that ERK phosphorylation is inhibited (4-24 hours). Because BMSCs protect MM cells against apoptosis by activating pERK and pSTAT3 signaling pathways, we assessed the effect of AS703026 on BMSC adhesion-induced pERK and pSTAT3 in MM cells. MM1S, H929, and U266 cells were incubated with 5, 0.5, and 0.1 μM of AS703026 in the presence or absence of BMSCs overnight, followed by immunoblotting using specific Abs. AS703026 completely blocked adhesion-induced pERK but not pSTAT3, confirming its specific inhibition of ERK activation (Fig. 2B and Supplement Fig. 2). In addition, AS703026-induced PARP cleavage was seen in both MM1S and H929 cells when co-cultured with BMSCs overnight, suggesting drug-induced apoptotic signaling in MM cells even in the presence of BMSCs. These results indicate that AS703026 specifically blocks both baseline and adhesion-induced ERK1/2 phosphorylation. Furthermore, AS703026 induced apoptosis in MM cells in the presence of BMSCs.
Fig. 2. AS703026 specifically blocked ERK1/2 activation in MM cells, cultured alone or with BMSCs.
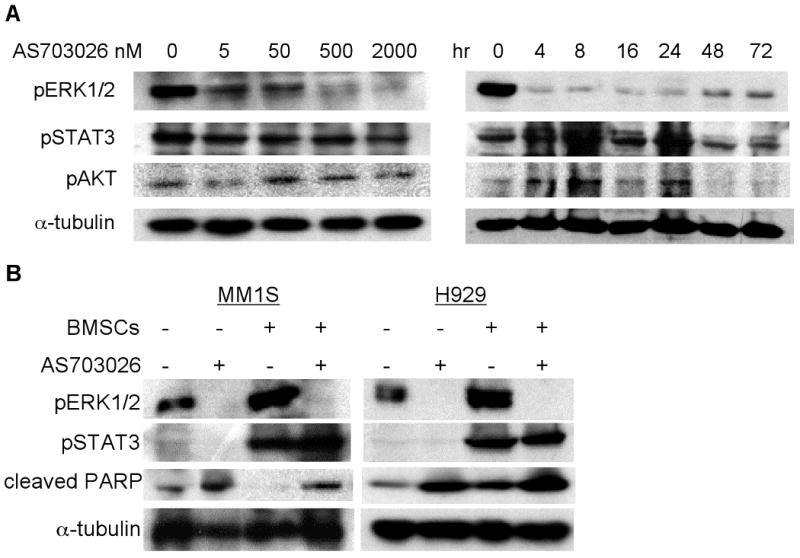
A, U266 cells were incubated with DMSO or AS703026 (5-2000 nM) for 1h (left) or 200 nM for 0-72h (right) and then subjected to immunoblotting. B, MM cell lines cultured alone or with BMSCs were treated with (+) or without (-) AS703026 (5 μM for MM1S; 0.5 μM for H929) overnight, followed by immunoblotting of cell lysates with indicated Abs. Anti-α-tubulin was used as a loading control.
Mechanisms of action of AS703026 against MM cells and BM microenvironment
We next analyzed molecular cascades following treatment of H929 MM cells with AS703026 to determine the compound's mechanism of cytotoxicity. As shown in Figure 3A, AS703026 (0.5 μM) triggered time-dependent apoptosis, as measured by both PARP and caspase 3 cleavage. Marked induction of PARP and caspase 3 cleavages was observed as early as 4 hours following treatment with AS703026, whereas inhibition of pERK was sustained up to 72 hours post-treatment. MM cells overexpress the c-maf oncogene, resulting in increased cell proliferation and survival via upregulation of cell cycle and adhesion to BMSCs (Hurt, et al 2004). We therefore examined the effects of AS703026 on c-maf expression in H929 cells. AS703026 reduced c-maf expression in a time-dependent fashion (Fig. 3A), suggesting MEK/ERK-dependent regulation of c-maf regulates MM cell growth and further supports the therapeutic targeting of this pathway. Interestingly, phosphorylation of AKT was upregulated in AS703026-treated H929 MM cells during the same time period that ERK phosphorylation was inhibited, similar to U266 cells. Quantitatively, the ratio between pAKT vs. α-tubulin was increased by 3-4 fold following treatment with AS703026 for 4 hours. In contrast to complete blockade of pERK, pAKT remained upregulated 72 hours after drug treatment. As expected, AS703026 modulated the cell cycle in MM cells, increasing subG0 and decreasing S phase cell population in both H929 and U266 cells in a time-dependent manner (Fig. 3B & Supplement Fig. 3). Therefore, AS703026 blocks cell proliferation via induction of G0-G1 cell cycle arrest in these MM cell lines.
Fig. 3. AS703026 induced apoptosis and modulated the cell cycle profile.
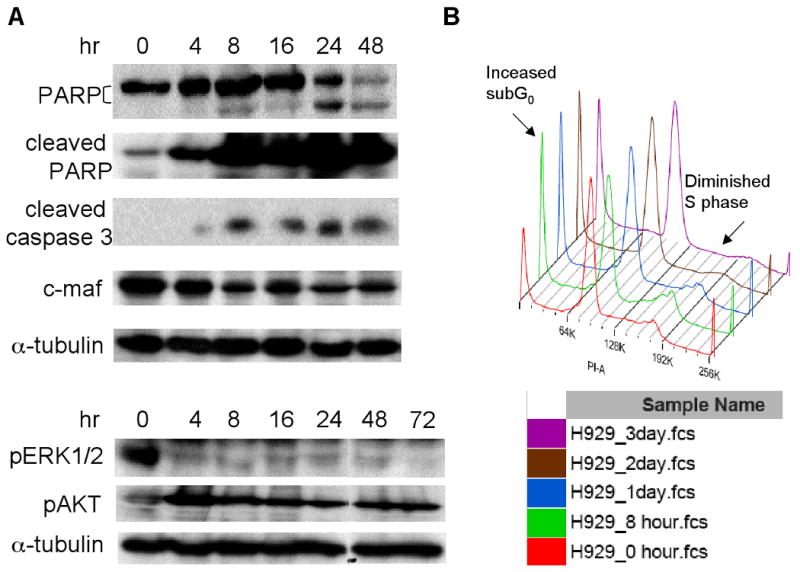
A, H929 MM cells were treated with AS703026 (0.5 μM) for 0-72h; cell lysates were then subjected to immunoblotting using specific Abs. B, H929 MM cells were treated with AS703026 (0.1 μM) and stained with PI; cell cycle profiles were evaluated using flow cytometry.
Mechanistic effects of AS703026 in the BM microenvironment were assessed by measuring the secretion of MM growth and survival factors IL-6 and VEGF from BMSCs, as well as osteoclast formation. Conditioned media were harvested 1 day following treatment of U266 and H929 MM cells, alone or in coculture with BMSCs. IL-6 and VEGF were measured by ELISA. AS703026 significantly decreased secretion of both cytokines induced by adhesion of MM cells to BMSCs in a dose-dependent manner (Fig. 4A-B, p<0.05 for AS703026 at >20 nM vs control). Although AS703026 (2, 20 nM) inhibited MM cell adhesion-induced IL-6 from BMSCs (p<0.02), same concentrations of AS703026 did not significantly alter constitutive IL-6 secretion from BMSCs derived from 3 normal donors (Fig. 4C). Because osteoclasts in the BM microenvironment promote progression of MM and osteolytic bone destruction, we also measured the effects of AS703026 on cytokine-induced osteoclast differentiation in vitro. AS703026 blocked osteoclastogenesis in a dose-dependent manner, as measured by number of TRAP-positive multinuclear cells following 14 days of treatment of CD14+monocytes from 3 normal donors with RANKL and M-CSF (p< 0.02, Fig. 1D). These results demonstrate effects of AS703026 on the BM microenvironment that may further block MM growth and survival.
Fig. 4. AS703026 targeted MM cells in the BM microenvironment.

MM cells, cultured alone or with BMSCs, were treated with DMSO or a dose range of AS703026 (0.002-20 μM) and conditioned media were collected for measurement of VEGF (A) and IL-6 (B) concentrations by ELISA. p<0.05 for AS703026 >20 nM vs control. C, Conditioned media from MM patient cells cultured with or without BMSCs were similarly examined for IL-6. p<0.02 (left panel). IL-6 from BMSCs alone was reduced when higher concentrations (>2000 nM) of AS703026 were used (right panel).
Combination of AS703026 with conventional and novel therapies further augmented cytotoxicity against MM cells
Due to the differential sensitivities of the MM cell lines to AS703026, and because MEK/ERK can cross-talk with other important signaling cascades, we next studied whether the combination of AS703026 with chemo- or targeted agents would show additive or synergistic anti-proliferative activities in MM cell lines and patient-derived MM cells. Using a DNA synthesis assay following treatment of the cells with either the agents alone or in combination at non-constant ratios of individual drugs, we determined the combination indices (CI) and confirmed that AS703026 has apparent synergistic activities against MM cells when combined with dex and perifosine (Supplement Fig. 4A-B). Synergy (as determined by the CI value) was also seen when AS703026 was combined with rapamycin in MM1S cells that were relatively insensitive to AS703026 (Fig. 5A, CI<1). Importantly, AS703026 markedly enhanced the cytotoxicity of dex, bortezomib, lenalidomide, and rapamycin against patient plasma cell leukemia, as evidenced by either [3H]thymidine uptake or MTT assays (Supplement 5 Fig 5 & 6) and subsequent CI determination (CI of these combinations was ≤1, indicating synergistic/additive effects).
Fig. 5. AS703026 combined with anti-MM drugs augmented MM cell death.
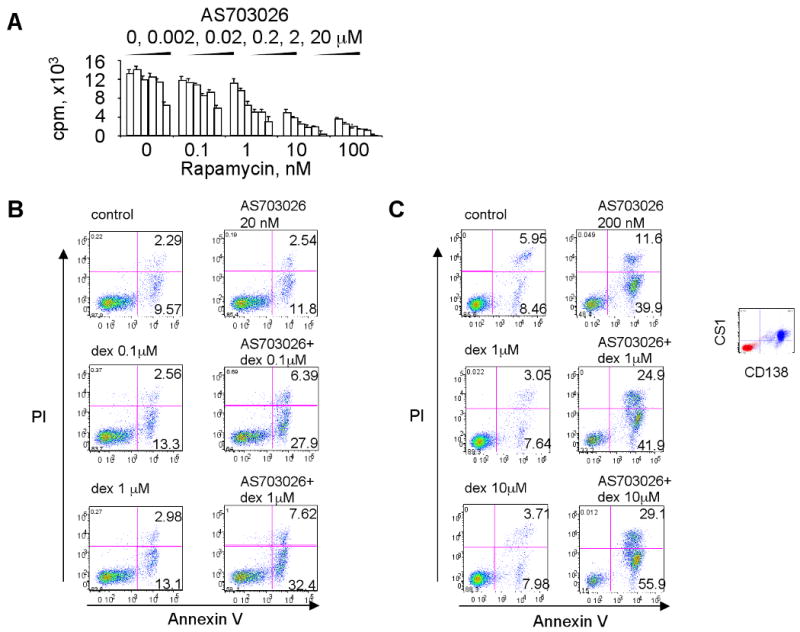

A, MM1S cells, which are relatively insensitive to AS703026, were treated with DMSO or a dose range of AS703026 (0.02-20 μM) and rapamycin (0.1-100 nM) for 2 days, followed by DNA synthesis assay. B, U266 cells were treated with AS703026 (20 nM) or dexamethasone (dex; 0.1, 1 μM) alone or in combination for 3 days, followed by annexin V/PI staining and flow cytometric analysis. C, Dex-resistant CD138+ patient MM cells were cocultured with BMSCs for 5 days with AS703026 (200 nM), dex (1, 10 μM), or the combination, and then subjected to apoptosis assay. Dex- and AS703026-resistant MM1R cells were treated with AS703026 (200 nM) with or without melphalan (mel 0.2, 2 μM; D) or lenalidomide (len; E), alone or together for 5 days. Control medium, thin line; treatment, bold line
To investigate the mechanism of synergistic cytotoxicity of AS703026 in combination with dex, dex-resistant U266 cells were incubated with AS703026 (20 nM) and dex (0.1, 1 μM), alone or together, for 2 days and apoptosis was quantified. Significant increases in combined annexin-V+ and annexin-V+/PI+ cells were observed in cultures of U266 cells treated with both drugs vs. each alone for 2 days; 34.8±4.5% vs. 11.9±3.4% (Fig. 5B). Importantly, an increase in apoptosis triggered by combined AS703026 and dex treatment was also measured in dex-resistant CD138-purified patient MM cells cocultured with BMSCs (Fig. 5C). Following 5-days of treatment, combined annexin V+ and annexin V+/PI+ patient MM cells were 14.4%, 10.7%, 11,7%, 51.5%, 66.8%, and 85% in control, dex (1 μM), dex (10 μM), AS703026 (200 nM), AS703026+dex (1 μM), and AS703026+dex (10 μM)-treated cells, respectively. Dose-dependent increases in MM1S and MM1R cell death were also observed when AS703026 was combined with lenalidomide or melphalan (Fig. 5D-E, Supplement Fig 7). These data suggest that the combination of these agents with AS703026 augmented apoptosis and cell death in MM cells compared to that induced by each agent alone, confirming enhanced cytotoxicity against MM cells when in combination.
AS703026 inhibited human MM cell growth in vivo
The antitumor activity of AS703026 was next investigated in vivo in the human H929 MM xenograft model in CB17 SCID mice. AS703026 was administrated twice daily orally when H929 tumors became palpable (∼ 130 mm3). Significant (P < 0.05) time-dependent tumor growth inhibition was seen in AS703026-treated (n=4 at 30 mg/kg, n=6 at 15 mg/kg) vs. control mice (n=6)) (Fig. 6A). No significant body weight loss was observed in AS703026-treated vs. control mice (P>0.05; data not shown). Importantly, specific target inhibition was seen in tumors harvested from AS703026-treated mice, as evidenced by significant downregulation of pERK measured by immunoblotting and immunohistochemistry (IHC) (Fig. 6B-C). Immunoblotting studies further showed PARP cleavage induced by AS703026 in H929 tumors from treated mice, indicating that AS703026-induced apoptosis in vivo (Fig. 6B). In accordance with these results, AS703026-treated H929 tumors from mice exhibited increased cleaved caspase 3 and TUNEL in IHC analyses (Fig. 6C). In addition, AS703026 treatment significantly reduced the percentage of CD34+ cells and microvascular density in H929 tumors (Fig. 6D), suggesting a possible inhibitory effect on angiogenesis in vivo.
Cytotoxicity of AS703026 against tumor cells from patients with relapsed and refractory MM
We next evaluated cytotoxicity of AS703026 against CD138+ tumor cells from patients with relapsed and refractory MM (n=18), including cells from one patient with plasma cell leukemia, alone or in coculture with BMSCs. AS703026, at concentrations below 200 nM, induced dose-dependent cytotoxicity against patient MM cells from 15 of 18 patients (83%) as assessed by [3H]thymidine update and MTT assays (Fig 7A-B). A greater sensitivity to AS703026 was measured by DNA synthesis compared to MTT, as patient MM cells did not proliferate in cell culture. For example, greater sensitivity to AS703026, as measured by DNA synthesis (IC50 of 6 nM) vs. MTT (IC50 of 52 nM), was observed for MM8 cells. Furthermore, AS703026 (IC50s ranging from 20-200 nM) blocked the increased viability of patient MM cells that was due to coculture with BMSCs, suggesting that MEK inhibition by AS703026 can overcome the protective effect of BMSCs (Fig. 7C). Because MEK1/2 inhibitors have shown activity in solid tumor models harboring mutations in BRAF or RAS, we further analyzed mutations in every exon of the K-, N-, and H-RAS and BRAF genes in genomic DNA from this cohort of MM patient samples (n=18). Approximately 34% of cells from patients with relapsed and/or refractory MM (6/18) harbored either N- or K-, but not, H-RAS mutations (Table 1). In addition, BRAF mutations were found in 2 of 18 samples, one with the D594G mutation and the other with the activating V600E mutation that is frequently reported in solid tumors. Interestingly, the mutational status of the RAS and BRAF genes did not associate with sensitivity to MEK inhibition by AS703026 in cells from these patients with relapsed and refractory MM, and therefore correlated nicely with the sensitivity data from the MM cell lines presented earlier.
Fig. 7. AS703026 is cytotoxic against CD138-purified MM cells from patients with relapsed and refractory MM.

CD138-purified patient tumor cells were treated with AS703026 for 5 days and then subjected to MTT (A) or [3H]thymidine uptake (B) assays. Data in A and B were derived from the same patient cells (MM1-8). Two patient MM cells (MM11 & MM14) were also cocultured with BMSCs in the presence or absence of drug for 5 days (C).
Table 1. Effects of AS703026 on CD138-purified tumor cells from patients with relapsed/refractory MM.
| Patient | RAS mutation | BRAF mutation | % Inhibition at 200 nM | IC50 (nM) |
|---|---|---|---|---|
| MM1 | N- (G12T) | - | 57% | 200 |
| MM2 | - | - | 75% | 2 |
| MM3 | - | - | 60% | < 200 |
| MM4 | - | - | 79%* | < 20 |
| MM5 | - | D594G | 76%* | < 20 |
| MM6 | - | - | 61%* | < 20 |
| MM7 | K- (G12A) | - | 51% | 20 |
| MM8a | N- (G13D) | - | 95% | 20 |
| MM9 | - | - | 98% | 2-20 |
| MM10 | - | - | 98% | 2-20 |
| MM11 | N- (R97G) | V600E | 99% | < 20 |
| MM12 | - | - | 64%* | < 20 |
| MM13 | - | - | 73% | 20-200 |
| MM14 | N- (Q61K) | - | 30%* | >200 |
| MM15 | - | - | 8%* | >200 |
| MM16 | K- (G12A, A61G) | - | 21% | >2000 |
| MM17 | - | - | 23% | >2000 |
| MM18 | - | - | 74% | 20-200 |
MM patient cells were purified by positive selection with CD138 microbeads, treated in vitro with AS703026 for 5 days and then analyzed by cytotoxicity assays using [3H] update (MM1-18) and MTT assays (MM1-8).
-, wild type of the gene;
% Inhibition at 20 nM drug;
MM8a: a plasma cell leukemia
Discussion
Some patients treated with small molecule inhibitors of MEK have achieved stable disease in recent solid tumor phase I and II clinical trials (Friday and Adjei 2008). Although the ultimate clinical proof-of-concept remains to be demonstrated, the central role of MEK signaling in cancer physiology, including MM, suggests potential efficacy of selective inhibitors. We here showed that AS703026 was more potent than AZD6244 at inhibiting MM growth and inducing cell death. Importantly, AS703026 was potently cytotoxic against tumor cells from the majority of patients with relapsed and refractory MM, regardless of prior treatments or mutational status of BRAF and RAS genes. AS703026 also blocked cytokine-induced osteoclast formation more potently than AZD6244 and overcame BMSC-induced MM growth, survival, and drug-resistance by down-regulating the MM growth and survival factors IL-6 and VEGF. Importantly, AS703026 blocked tumor growth and induced apoptosis in vivo, as evidenced by increased caspase 3 and PARP cleavage and TUNEL staining in H929 tumors from AS703026-treated xeno-engrafted mice. In addition, microvessel density in H929 tumors from mice treated with AS703026 was reduced, possibly due to anti-angiogenic action of the compound. Finally, AS703026 potently induced cell death in patient MM cells, alone or in coculture with BMSCs, and synergistically enhanced apoptosis in combination with a broad spectrum of conventional, recently approved, or emerging anti-MM therapies.
MM patients require multiple lines of therapy with combination regimens. The novel MEK inhibitor AS703026 holds great promise for MM treatment because the MEK/ERK signaling cascade is both central to, and cross-talks with, related growth and survival pathways, including the PI3K-AKT-mTOR and the JAK/STAT pathways. Here we confirmed enhanced cytotoxicity by AS703026 when combined with dexamethasone, lenalidomide, perifosine, bortezomib, melphalan, or rapamycin. For example, AS703026 synergistically induced cytotoxicity with rapamycin in AS703026-insensitive MM1S cells. Mechanistically, the enhanced combination effect of rapamycin (or perifosine that inhibits phosphorylation of AKT(Hideshima, et al 2006)) and AS703026 was supported by the measured increase of AKT phosphorylation following MEK inhibition alone in U266 and H929 cells. Upregulated pAKT was also seen in MM1S cells following the MEK inhibitor U0126, whereas pERK was completely blocked (Hideshima, et al 2006). Moreover, synergistic/additive cytoxicity and induction of apoptosis of AS703026 with anti-MM drugs was observed in both dex-resistant and dex-sensitive MM cell lines and patient MM cells, even in coculture with BMSCs. These results strongly provide the framework for clinical evaluation of AS703026-based combination trials in MM.
Phosphorylated ERK (pERK) suppression alone was not sufficient to predict the cytotoxic effects of AS703026, as this compound effectively blocked pERK in all MM lines that showed differential sensitivities to the drug. We therefore examined RAS and BRAF mutations in MM cell lines and patient MM cells to see whether mutational status correlated with responsiveness to MEK inhibition by AS703026. A cohort of 18 patients with MM, which had relapsed and was refractory to several prior therapies, was studied. In accordance with previous publications, we found that BRAF mutations within exon 15 of the gene were either undetectable or occurred at a low frequency in MM and PCL, regardless of the presence of mutations in N- or K-RAS genes. We found that only the U266 MM cell line had a mutation in BRAF (K601N) and only 2 of 18 (10%) of patients with relapsed MM harbored BRAF mutations: one in exon 15 and the other in exon 11. Although the V600E mutation is the most prevalent activating BRAF mutation seen in solid tumors, only one patient (MM11) with relapsed refractory MM had mutations in both N-RAS (R97G) and BRAF (V600E) genes. Importantly, AS703026 was potently cytotoxic (IC50 ∼2-20 nM) against this patient's MM that had progressed following combination therapies of Revlimid (lenalidomide) with Velcade and then with dexamethasone. The tumor from another patient (MM5) that had relapsed after Revlimid with bortezomib therapy harbored the D594G mutation of BRAF and was sensitive to AS703026 cytotoxicity (IC50 <20 nM).
To our knowledge, this is the first report in MM of a BRAF mutation other than the V600E variant. A recent report demonstrated attenuated sensitivity to the MEK inhibitor U0126 in one melanoma cell line with D594G BRAF mutation (Smalley, et al 2009). However, growth and survival of CD138+ cells from patient MM5 with the D594G BRAF mutation was significantly inhibited by treatment with AS703026. Importantly, the detection of two different BRAF mutations in 18 of our patient samples suggested that these mutations may be relatively rare in MM, but more studies are needed to determine whether BRAF mutations confer anti-tumor sensitivity to MEK inhibition in MM.
RAS mutations that were identified in these 18 relapsed MM patients (> 33%) were in K- and N-RAS but not H-RAS, which is in agreement with all published studies in MM to date (Bezieau, et al 2002, Chng, et al 2008, Kalakonda, et al 2001, Liu, et al 1996). RAS mutations frequently occur in codons 12, 13, and 61 of the gene, which are previously identified hot spots. We also identified the R97G N-RAS mutation in one sample. Additional studies with larger sample sizes will help to support or refute K- or N-RAS mutational status or MEK/ERK activity as reliable biomarkers for anti-MEK1/2 therapy in MM. Nevertheless, these results strongly support targeting MEK inhibition by AS703026 in relapsed and refractory MM regardless of mutational status.
In conclusion, AS703026 not only directly targeted MM cell growth and survival, but also acted in the BM milieu to block cytokine secretion and osteoclastogenesis. There was no significant association of RAS and BRAF mutations with sensitivity of MM to AS703026. Most importantly, AS703026 was synergistic/additive when administered in combination with multiple conventional, novel, and emerging anti-MM therapies, providing the framework for clinical studies of this agent, alone and in combination, to improve patient outcome in MM.
Supplementary Material
Acknowledgments
We thank Dr. William Dalton at Lee Moffitt Cancer Center, Tampa, FL; Dr. Steven Rosen at Northwestern University, Chicago, IL; Dr. Takemi Otsuki at Kawasaki Medical School, Okayama, Japan; Dr. Steven Rosen at Northwestern University, Chicago, IL; Dr. Renate Burger at University of Erlangen-Nuernberg, Erlangen, Germany; Dr. Megumu Ogawa at Osaka University, Osaka, Japan for MM lines used in the study. The authors thank Dr Ender Soydan, Giovanni Tonon, and Sophia Adamia for help and suggestions on animal work, RAS gene mutation, and microphotography. We thank the nursing staff and clinical research coordinators of the LeBow Institute for Myeloma Therapeutics and Jerome Lipper Multiple Myeloma Center of Dana-Farber Cancer Institute for their constant help in providing primary tumor specimens for this study.
Grant support: Multiple Myeloma Research Consortium (Y.-T.Tai); National Institutes of Health Grants RO-1 50947, PO1-78378 and SPORE P50CA100707; and the Lebow Fund to Cure Myeloma (K.C.A.).
Footnotes
Contributions: Y.-T.T., A.C., A.G., K.K., J.O. designed and analyzed data; Y.-T.T. wrote the manuscript; M.F., X.-F.L., P.B., K.K. performed animal studies; X.-F. L., P.B., S.N., M.J.R., W.S. performed research and analyzed data; K.P., T.H., D.C. provided reagents; A.C., X.-F. L., S.-Y.K. performed statistical analysis; N.C.M., P.R. provided patient BM and blood samples; A.C., A.G., L.R. designed in vivo studies and provided AS703026; J.O. performed and analyzed mutational data; and A.C., L.R., J.O., A.G. and K.C.A. critically evaluated and edited the manuscript.
Conflict-of-interest disclosure: A.C., J.O., A.G., L.R. are employees of EMD Serono Research Institute and provided the MEK inhibitor AS703026 for these studies. All other authors declare no competing financial interests.
References
- Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S, Poch G, Kane K, Haney J, Eckhardt SG. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezieau S, Avet-Loiseau H, Moisan JP, Bataille R. Activating Ras mutations in patients with plasma-cell disorders: a reappraisal. Blood. 2002;100:1101–1102. doi: 10.1182/blood-2002-03-0795. author reply 1103. [DOI] [PubMed] [Google Scholar]
- Billadeau D, Jelinek DF, Shah N, LeBien TW, Van Ness B. Introduction of an activated N-ras oncogene alters the growth characteristics of the interleukin 6-dependent myeloma cell line ANBL6. Cancer Res. 1995;55:3640–3646. [PubMed] [Google Scholar]
- Bonello L, Voena C, Ladetto M, Boccadoro M, Palestro G, Inghirami G, Chiarle R. BRAF gene is not mutated in plasma cell leukemia and multiple myeloma. Leukemia. 2003;17:2238–2240. doi: 10.1038/sj.leu.2403116. [DOI] [PubMed] [Google Scholar]
- Burger R, Guenther A, Bakker F, Schmalzing M, Bernand S, Baum W, Duerr B, Hocke GM, Steininger H, Gebhart E, Gramatzki M. Gp130 and ras mediated signaling in human plasma cell line INA-6: a cytokine-regulated tumor model for plasmacytoma. Hematol J. 2001;2:42–53. doi: 10.1038/sj.thj.6200075. [DOI] [PubMed] [Google Scholar]
- Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, Kuehl WM, Bergsagel PL. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- Chng WJ, Gonzalez-Paz N, Price-Troska T, Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S, Greipp P, Van Ness B, Fonseca R. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22:2280–2284. doi: 10.1038/leu.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49:2059–2080. doi: 10.1080/10428190802353591. [DOI] [PubMed] [Google Scholar]
- Clark AM, Ma J, Qiu D, Lin J, Syed S, Romanelli A, Spooner E, Shaw J, Rocha C, Tian H, A G. Pharmacokinetics, efficacy and target pathway inhibition in vivo of AS703026, a small molecule inhibitor of MEK. AACR Annual meeting. 2009 Abstract #3694. [Google Scholar]
- Corradini P, Ladetto M, Voena C, Palumbo A, Inghirami G, Knowles DM, Boccadoro M, Pileri A. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias. Blood. 1993;81:2708–2713. [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–346. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- Giuliani N, Lunghi P, Morandi F, Colla S, Bonomini S, Hojden M, Rizzoli V, Bonati A. Downmodulation of ERK protein kinase activity inhibits VEGF secretion by human myeloma cells and myeloma-induced angiogenesis. Leukemia. 2004;18:628–635. doi: 10.1038/sj.leu.2403269. [DOI] [PubMed] [Google Scholar]
- Goutopoulos A, Askew BC, Bankston D, Clark A, Dhanabal M, Dong R, Fischer D, Healey B, Jiang X, Josephson K, Lin J, Ma J, Noonan T, Qiu D, Rocha C, Romanelli A, Shutes A, Spooner E, Tian H, H Y. AS703026: a novel allosteric MEK inhibitor. AACR Annual meeting. 2009 Abstract#4776. [Google Scholar]
- Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004;104:607–618. doi: 10.1182/blood-2004-01-0037. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, Mitsiades C, Podar K, Munshi NC, Chauhan D, Richardson PG, Anderson KC. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107:4053–4062. doi: 10.1182/blood-2005-08-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- Hoang B, Zhu L, Shi Y, Frost P, Yan H, Sharma S, Goodglick L, Dubinett S, Lichtenstein A. Oncogenic RAS mutations in myeloma cells selectively induce cox-2 expression, which participates in enhanced adhesion to fibronectin and chemoresistance. Blood. 2006;107:4484–4490. doi: 10.1182/blood-2005-09-3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Shi Y, Hsu JH, Gera J, Van Ness B, Lichtenstein A. Downstream effectors of oncogenic ras in multiple myeloma cells. Blood. 2003;101:3126–3135. doi: 10.1182/blood-2002-08-2640. [DOI] [PubMed] [Google Scholar]
- Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, Bergsagel PL, Kuehl WM, Staudt LM. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5:191–199. doi: 10.1016/s1535-6108(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Intini D, Agnelli L, Ciceri G, Ronchetti D, Fabris S, Nobili L, Lambertenghi-Deliliers G, Lombardi L, Neri A. Relevance of Ras gene mutations in the context of the molecular heterogeneity of multiple myeloma. Hematol Oncol. 2007;25:6–10. doi: 10.1002/hon.801. [DOI] [PubMed] [Google Scholar]
- Kalakonda N, Rothwell DG, Scarffe JH, Norton JD. Detection of N-Ras codon 61 mutations in subpopulations of tumor cells in multiple myeloma at presentation. Blood. 2001;98:1555–1560. doi: 10.1182/blood.v98.5.1555. [DOI] [PubMed] [Google Scholar]
- Liu P, Leong T, Quam L, Billadeau D, Kay NE, Greipp P, Kyle RA, Oken MM, Van Ness B. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: analysis of the Eastern Cooperative Oncology Group Phase III Trial. Blood. 1996;88:2699–2706. [PubMed] [Google Scholar]
- Machl AW, Ogden JA, Romanelli A. Efficacy of MEK inhibitor AS703026 in various primary tumor explants. AACR Annual meeting. 2009 abstract#3698. [Google Scholar]
- Menu E, Kooijman R, Van Valckenborgh E, Asosingh K, Bakkus M, Van Camp B, Vanderkerken K. Specific roles for the PI3K and the MEK-ERK pathway in IGF-1-stimulated chemotaxis, VEGF secretion and proliferation of multiple myeloma cells: study in the 5T33MM model. Br J Cancer. 2004;90:1076–1083. doi: 10.1038/sj.bjc.6601613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier M, Moles JP, Mazars GR, Jeanteur P, Bataille R, Klein B, Theillet C. p53 and RAS gene mutations in multiple myeloma. Oncogene. 1992;7:2539–2543. [PubMed] [Google Scholar]
- Pratilas CA, Solit DB. Therapeutic strategies for targeting BRAF in human cancer. Rev Recent Clin Trials. 2007;2:121–134. doi: 10.2174/157488707780599393. [DOI] [PubMed] [Google Scholar]
- Rasmussen T, Kuehl M, Lodahl M, Johnsen HE, Dahl IM. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood. 2005;105:317–323. doi: 10.1182/blood-2004-03-0833. [DOI] [PubMed] [Google Scholar]
- Richardson P, Anderson K. Thalidomide and dexamethasone: a new standard of care for initial therapy in multiple myeloma. J Clin Oncol. 2006;24:334–336. doi: 10.1200/JCO.2005.03.8851. [DOI] [PubMed] [Google Scholar]
- Richardson P, Jagannath S, Hussein M, Berenson J, Singhal S, Irwin D, Williams SF, Bensinger W, Badros AZ, Vescio R, Kenvin L, Yu Z, Olesnyckyj M, Zeldis J, Knight R, Anderson KC. Safety and efficacy of single-agent lenalidomide in patients with relapsed and refractory multiple myeloma. Blood. 2009;114:772–778. doi: 10.1182/blood-2008-12-196238. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Shain KH, Yarde DN, Meads MB, Huang M, Jove R, Hazlehurst LA, Dalton WS. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–1015. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28:85–94. doi: 10.1038/onc.2008.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YT, Catley LP, Mitsiades CS, Burger R, Podar K, Shringpaure R, Hideshima T, Chauhan D, Hamasaki M, Ishitsuka K, Richardson P, Treon SP, Munshi NC, Anderson KC. Mechanisms by which SGN-40, a humanized anti-CD40 antibody, induces cytotoxicity in human multiple myeloma cells: clinical implications. Cancer Res. 2004;64:2846–2852. doi: 10.1158/0008-5472.can-03-3630. [DOI] [PubMed] [Google Scholar]
- Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, Lee AI, Podar K, Hideshima T, Rice AG, van Abbema A, Jesaitis L, Caras I, Law D, Weller E, Xie W, Richardson P, Munshi NC, Mathiot C, Avet-Loiseau H, Afar DE, Anderson KC. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112:1329–1337. doi: 10.1182/blood-2007-08-107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF, Rumizen M, Burger P, Morrison A, Podar K, Chauhan D, Tassone P, Richardson P, Munshi NC, Ghobrial IM, Anderson KC. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood. 2007;110:1656–1663. doi: 10.1182/blood-2007-03-081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YT, Li X, Tong X, Santos D, Otsuki T, Catley L, Tournilhac O, Podar K, Hideshima T, Schlossman R, Richardson P, Munshi NC, Luqman M, Anderson KC. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res. 2005;65:5898–5906. doi: 10.1158/0008-5472.CAN-04-4125. [DOI] [PubMed] [Google Scholar]
- Tai YT, Podar K, Catley L, Tseng YH, Akiyama M, Shringarpure R, Burger R, Hideshima T, Chauhan D, Mitsiades N, Richardson P, Munshi NC, Kahn CR, Mitsiades C, Anderson KC. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3′-kinase/AKT signaling. Cancer Res. 2003;63:5850–5858. [PubMed] [Google Scholar]
- Tai YT, Soydan E, Song W, Fulciniti M, Kim K, Hong F, Li XF, Burger P, Rumizen MJ, Nahar S, Podar K, Hideshima T, Munshi NC, Tonon G, Carrasco RD, Afar DE, Anderson KC. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood. 2009;113:4309–4318. doi: 10.1182/blood-2008-10-183772. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, Chng WJ, Ketterling RP, Gertz MA, Henderson K, Greipp PR, Dispenzieri A, Lacy MQ, Rajkumar SV, Bergsagel PL, Stewart AK, Fonseca R. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.