

Summary
Tanespimycin (17-allylamino-17-demethoxygeldanamycin, 17-AAG) disrupts heat shock protein 90 (HSP90), a key molecular chaperone for signal transduction proteins critical to myeloma growth, survival and drug resistance. In previous studies, tanespimycin monotherapy was well tolerated and active in heavily pretreated patients with relapsed/refractory multiple myeloma (MM). Preclinical data have shown antitumour synergy between tanespimycin and bortezomib, with more pronounced intracellular accumulation of ubiquitinated proteins than either drug alone, an effect attributed to the synergistic suppression of chymotryptic activity in the 20S proteasome. HSP70 induction has been observed in all Phase 1 tanespimycin studies in which it has been measured, with several separate reports of HSP70 overexpression protecting against peripheral nerve injury. In this Phase 2, open-label multicentre study, we compared 1·3 mg/m2 bortezomib + three doses of tanespimycin: 50, 175 and 340 mg/m2 in heavily pretreated patients with relapsed and refractory MM and measured HSP70 expression and proteasome activity levels in plasma of treated patients. The study was closed prematurely for resource-based reasons, precluding dose comparison. Nonetheless, antitumour activity was observed, with promising response rates and promising severity of peripheral neuropathy.
Keywords: tanespimycin, bortezomib, HSP90, HSP70, peripheral neuropathy
Multiple myeloma (MM) is the second most common blood cancer after non-Hodgkin lymphoma and recent statistics indicate both increasing incidence and earlier age of onset (Multiple Myeloma Research Foundation 2009). Annually, there are 19 920 estimated new cases of MM and 11 190 deaths in the United States (Jemal et al, 2008).
Multiple myeloma is a plasma cell malignancy characterized by extensive bone destruction, impaired haematopoiesis and dysproteinaemia leading to immune paresis and renal failure, with a key aspect of disease biology being simultaneous antiapoptotic and growth support for tumour cells from the bone marrow microenvironment (Stuhmer et al, 2008). Autologous stem cell transplantation (ASCT) and the use of novel therapies, such as bortezomib, thalidomide and lenalidomide, have improved patient outcomes, achieving event-free or progression-free survival rates from 3·5 to >10 years or remission in studies where total therapy protocols were used or in ASCT patients without chromosome 13 gene deletion (Pineda-Roman et al, 2008; Paul et al, 2009; Barlogie et al, 2010). Despite these encouraging advances, most patients relapse and become refractory to treatment (Badros et al, 2009) making the need for the continued development of new drugs and combinations paramount (Richardson et al, 2007).
The complex relationship between increased tumour cell survival in MM, cell stress response including heat, heavy metals, hypoxia and acidosis, and the subsequent overexpression of heat shock protein 90 (HSP90) are currently being explored, with cell stress response being particularly important for malignant cells stressed by chemotherapy (Takayama et al, 2003; Whitesell & Lindquist, 2005). HSP90 acts as a chaperone enabling a wide range of client proteins, including many oncoproteins (e.g. human epidermal growth factor receptor 2 [HER2], the sarcoma [SRC] family of kinase and Raf-1) (Nowakowski et al, 2006), to maintain their three-dimensional conformation (Mitsiades et al, 2007). Proper three-dimensional conformation and appropriate subcellular location of these oncoproteins promotes cell survival, proliferation and resistance to proapoptotic stimuli and is required to drive the malignant phenotype and appears to be of particular importance in MM (Zhang & Burrows, 2004).
Inhibition of HSP90 leads to degradation of client proteins by the proteasome, which results in simultaneous interruption of many signal transduction pathways that are needed for tumour survival (Burrows et al, 2004). Importantly, pharmacodynamic markers, such as the depletion of client proteins, along with the induction of the HSP70 group of chaperones, have been identified as a molecular signature for HSP90 inhibition (Maloney & Workman, 2002).
Tanespimycin (17-allylamino-17-demethoxygeldanamycin, 17-AAG), an HSP90 inhibitor, binds to the ATP-binding site of HSP90, blocking ATPase activity and resulting in degradation of HSP90 client proteins via the ubiquitin-proteasome pathway (Maloney & Workman, 2002). Tanespimycin antitumour activity and safety have been demonstrated in Phase 1 studies including patients with advanced MM (Banerji et al, 2005; Goetz et al, 2005; Grem et al, 2005; Ramanathan et al, 2007; Solit et al, 2007). HSP70 induction, a marker of HSP90 inhibition, has been observed in all Phase 1 studies in which it has been measured (Banerji et al, 2005; Goetz et al, 2005; Grem et al, 2005; Ramanathan et al, 2007; Solit et al, 2007).
In a preclinical study, administration of bortezomib, a first-in-class selective inhibitor of the 26S proteasome, induced a stress response characterized by transcription of proteasome subunits and molecular chaperones in the HSP family, including HSP90, in MM (Mitsiades et al, 2002). This study also suggested that concurrent blockade of HSP90 could enhance bortezomib-triggered apoptosis even in drug-resistant MM cells (Mitsiades et al, 2002). Subsequently, the combination of tanespimycin with bortezomib induced a more pronounced intracellular accumulation of ubiquitinated proteins than either drug alone, which can be attributed to the synergistic suppression of chymotryptic activity of the 20S proteasome (Mitsiades et al, 2006).
In a prospective, multicentre Phase 1/2 study of tanespimycin + bortezomib in patients with relapsed and refractory MM, 72 patients with a median of 5 (range, 1–15) prior regimens, including bortezomib were treated. Antitumour activity was observed with overall response rate (ORR; ≥ minimal response [MR]) in 48% of bortezomib-naive, 22% of bortezomib-pretreated and 13% of bortezomib-refractory patients (Richardson et al, 2009a). The median duration of response for all responders was 12 months (95% confidence interval [CI]: 5–19). The highest dose tested in this study, 340 mg/m2, did not demonstrate dose-limiting toxicity (DLT) and a clear dose response effect was not seen, suggesting further dose exploration was warranted. Treatment was generally well tolerated with low rates of peripheral neuropathy; in particular, no cases of grade 3 peripheral neuropathy were reported. Overall, tanespimycin + bortezomib demonstrated a strong synergistic effect on antitumour activity, with manageable toxicity and possible favourable effects on both the rate and severity of peripheral neuropathy.
Although preclinical and clinical studies have shown that bortezomib monotherapy can overcome resistance of MM cells to conventional or high-dose cytotoxic chemotherapy (Hideshima et al, 2001; Mitsiades et al, 2003), clinical studies have found that this treatment can induce significant peripheral neuropathy (Richardson et al, 2003). To further address this issue, additional studies have evaluated the observations made in the tanespimycin + bortezomib combination trial in which lower rates of peripheral neuropathy were observed. For example, one study showed that the inhibition of bortezomib-induced overexpression of HSP90 leads to an increase in HSP70 (Mimnaugh et al, 2004), and that selective overexpression of HSP70 leads to protection in several different models of nervous system injury (Kelly & Yenari, 2002; Yenari, 2002; Brown, 2007). In a von Frey model study of peripheral neuropathy in Sprague–Dawley rats, tanespimycin was shown to protect against bortezomib-induced peripheral neuropathy (Zhong et al, 2008), providing strong preclinical correlation for the observations from the previous Phase 1/2 study of bortezomib + tanespimycin for the treatment of patients with relapsed and refractory MM, in which no grade 3/4 peripheral neuropathy was seen (Richardson et al, 2009a).
Here, we present the results of an abbreviated Phase 2 study designed to assess safety and efficacy of three dose levels of tanespimycin in combination with bortezomib in relapsed/refractory myeloma. This study was an extension of the earlier Phase 1/2 study (Richardson et al, 2009a), but involved a different patient population and employed some differences in methodology, namely a broader tanespimycin dosage range and exclusion of bortezomib-naive patients and patients with no measurable MM. Taken together, the two studies provide a sizeable and varied patient population in which combination therapy with bortezomib and tanespimycin can be assessed. The study was closed early in its course for logistic and resource reasons unrelated to safety and efficacy.
Methods
Eligible patients were ≥18 years of age with a Karnofsky performance status of ≥70%, documented MM, and evidence of relapsed or relapsed and refractory disease after at least three prior regimens that included bortezomib and lenalidomide. Progressive disease (PD) prior to study entry was documented by the modified European Group for Bone and Marrow Transplantation (EBMT) criteria (Blade et al, 1998; Richardson et al, 2003) and confirmed by at least one repeated investigation. High-dose chemotherapy followed by bone marrow transplant (with or without maintenance therapy) counted as one line of prior therapy as long as PD by modified EBMT criteria was not observed between stem cell transplant (SCT) and initiation of maintenance therapy.
Patients were eligible who had received adequate courses of both bortezomib and lenalidomide as prior regimens and had measurable disease (defined as serum M protein >5·0 g/l or >200 mg urinary light chain excretion/24-h) assessed within 14 days prior to randomization. All adverse events (AEs) from prior therapy had to be resolved to grade ≤2, as defined by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v3.0 (http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf), with the exception of painful neuropathy and diarrhoea that were required to have resolved to grade ≤1.
Patients were excluded from the study if they were taking concurrent corticosteroids (other than oral prednisone of ≤5 mg daily or its equivalent, inhaled corticosteroids for the treatment of pulmonary disease that resulted in little or no systemic absorption, or higher doses that were tapered to this level 7 days prior to randomization); had grade 3 dyspnea or a requirement for supplemental oxygen; had known or suspected cardiac amyloidosis; had Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy and Skin Changes (POEMS) syndrome; or had plasma cell leukaemia, hepatitis, or human immunodeficiency virus infection. Patients with non-secretory or oligosecretory disease were also not eligible for the study.
Study design and treatment
This Phase 2, randomized, open-label multicentre study in patients with relapsed and refractory MM was conducted between 28 centres in the United States and compared 1·3 mg/m2 bortezomib + three different doses of tanespimycin (50, 175 and 340 mg/m2).
All patients received tanespimycin + bortezomib on days 1, 4, 8 and 11 in each 3-week cycle. Stable and responding patients were treated until PD, unacceptable toxicity, withdrawal of patient consent, or investigator judgment. Response assessments were conducted every 21 d.
Therapy consisted of bortezomib 1·3 mg/m2 administered as an intravenous (IV) rapid bolus followed by an IV infusion over 60 min of tanespimycin. Patients randomized to the 50 mg/m2 dose group who did not achieve a complete response (CR) or partial response (PR; confirmed or unconfirmed) after four cycles were allowed to cross over to the 340 mg/m2 dose group.
Objectives
The objectives of this phase 2 study were to evaluate: (i) safety and efficacy in patients with relapsed and refractory MM, and (ii) HSP70 expression and proteasome activity levels in plasma of treated patients.
Analyses
All patients were included in the safety analysis if they received at least one dose of tanespimycin and/or bortezomib. AEs were evaluated using the NCI CTCAE v3.0. Responses were assessed using the modified EBMT criteria (Blade et al, 1998; Richardson et al, 2003). Plasma was collected and lysates were analysed for HSP70 expression and 20S proteasome activity on days 1 and 11 at 0, 4, and 24 h post-dose and normalized to baseline pretreatment levels in each patient. HSP70 levels were measured by Western blot, and 20S proteasome activity was measured as a composite of chymotrypsin, trypsin and caspase activity. Due to the small number of patients enrolled in each treatment group, descriptive statistics were used to present the results.
Results
Twenty-two patients participated in the study; eight patients in the 340 mg/m2 group, eight patients in the 175 mg/m2 group and six in the 50 mg/m2 group. Two patients crossed over from the 50 mg/m2 dose group to the 340 mg/m2 dose group; one patient crossed over at cycle 5 and remained on treatment through cycle 22, then discontinued due to unacceptable toxicity, and one patient who crossed over at cycle 5 remained on treatment through cycle 19, then enrolled in the extension protocol of the current study.
Demographics and baseline characteristics are presented in Table I. The majority of patients were white (17/22, 77%) and male (14/22, 64%); the median age was 62·5 years old (range, 47–82). Cytogenetic analysis on 13 patients showed that three patients (14%) had chromosome 13 deletion, one patient each had t(11;14) or t(14;16) translocations, and one patient had TP53 mutation.
Table I.
Summary of demographics and baseline characteristics.
| 340 mg/m2 | 175 mg/m2 | 50 mg/m2 | Total | |
|---|---|---|---|---|
| n = 8 | n = 8 | n = 6 | N = 22 | |
| Gender, n (%) | ||||
| Men | 7 (88) | 4 (50) | 3 (50) | 14 (64) |
| Age, years | ||||
| Median | 57·0 | 65·5 | 59·5 | 62·5 |
| Range | 47–82 | 57–76 | 48–74 | 47–82 |
| Race, n (%) | ||||
| White | 5 (63) | 7 (88) | 5 (83) | 17 (77) |
| Black | 3 (38) | 1 (13) | 1 (17) | 5 (23) |
| Chromosome 13 deletion, n (%) | ||||
| No | 3 (38) | 2 (25) | 2 (33) | 7 (32) |
| Yes | 0 | 3 (38) | 0 | 3 (14) |
| IGH@ translocation, n (%) | ||||
| t(4;14) | 0 | 0 | 0 | 0 |
| t(11;14) | 0 | 1 (13) | 1 (5) | |
| t(14;16) | 1 (13) | 0 | 0 | 1 (5) |
| TP53 mutation | 0 | 1 (13) | 0 | 1 (5) |
| Extramedullary disease | ||||
| No | 7 (88) | 6 (75) | 6 (100) | 19 (86) |
| Yes | 1 (13) | 2 (25) | 0 | 3 (14) |
| Prior history of PN | 7 (88) | 5 (63) | 5 (83) | 17 (77) |
PN, peripheral neuropathy.
All patients had previously received some form of therapy representative of treatment given to the general MM population in clinical practice. Overall, patients had received a median of five prior lines of therapy (range, 3–11) including thalidomide (96%), dexamethasone (96%), melphalan (77%), cyclophosphamide (73%) and doxorubicin (55%; Table II). A total of 16 patients (16/22, 73%) had had prior SCT of which two transplants were allogeneic and 16 were autologous.
Table II.
Summary of primary myeloma treatments in the as-randomized population.
| 340 mg/m2 | 175 mg/m2 | 50 mg/m2 | Total | |
|---|---|---|---|---|
| n = 8 | n = 8 | n = 6 | N = 22 | |
| Time since original diagnosis (months) | ||||
| N | 8 | 8 | 5 | 21 |
| Median | 62·5 | 95·0 | 59·0 | 81·0 |
| Range | 31–124 | 37–111 | 40–140 | 31–140 |
| Prior SCT, n (%) | ||||
| Allogeneic | 1 (13) | 0 | 1 (17) | 2 (9) |
| Autologous | 5 (63) | 6 (75) | 5 (83) | 16 (73) |
| Not considered eligible at any time since diagnosis for SCT | 2 (25) | 2 (25) | 0 | 4 (18) |
| Total number of prior myeloma regimens | ||||
| N | 8 | 8 | 6 | 22 |
| Median (range) | 4·0 (3–8) | 6·5 (4–10) | 4·5 (4–11) | 5·0 (3–11) |
| Chemotherapeutic agents, n (%) | ||||
| Bortezomib | 8 (100) | 8 (100) | 6 (100) | 22 (100) |
| Lenalidomide | 8 (100) | 8 (100) | 6 (100) | 22 (100) |
| Thalidomide | 7 (88) | 8 (100) | 6 (100) | 21 (96) |
| Dexamethasone | 7 (88) | 8 (100) | 6 (100) | 21 (96) |
| Melphalan | 6 (75) | 7 (88) | 4 (67) | 17 (77) |
| Cyclophosphamide | 6 (75) | 7 (88) | 3 (50) | 16 (73) |
| Doxorubicin | 4 (50) | 5 (63) | 3 (50) | 12 (55) |
| Best response to most recent regimen, n (%) | ||||
| Complete response | 1 (13) | 0 | 0 | 1 (5) |
| VGPR | 2 (25) | 1 (13) | 0 | 3 (14) |
| Partial response | 1 (13) | 1 (13) | 0 | 2 (9) |
| Minimal response | 1 (13) | 2 (25) | 3 (50) | 6 (27) |
| Stable disease | 1 (13) | 3 (38) | 1 (17) | 5 (23) |
| Progressive disease | 2 (25) | 1 (13) | 2 (33) | 5 (23) |
| Reason for most recent discontinuation, n (%) | ||||
| Progressive disease | 5 (63) | 5 (63) | 5 (83) | 15 (68) |
| Intolerance | 2 (25) | 0 | 0 | 2 (9) |
| Other | 1 (13) | 3 (38) | 1 (17) | 5 (23) |
SCT, stem cell transplant; VGPR, very good partial response, according to the IMWG uniform criteria (2006).
The best responses of patients to the most recent prior regimens are shown in Table II. Reasons for discontinuing the most recent prior regimen included PD in 68%, intolerance in 9% and other in 23%.
Safety
All 22 patients in the study reported one or more AEs. The most common AEs overall included: fatigue (16/22, 73%), nausea (15/22, 68%), diarrhoea (14/22, 64%), constipation (11/22, 50%) and vomiting (10/22, 45%; Table III). The most common grade 3/4 AEs reported included fatigue (32%), thrombocytopenia (27%), neutropenia (18%) and abdominal pain (9%); with all other grade 3/4 AEs reported in fewer than three patients. No severe AEs of anorexia or constipation were reported.
Table III.
Summary of AEs on study in the as-treated population*.
| AE, n | 340 mg/m2 | 175 mg/m2 | 50 mg/m2 | Total | Grade 3/4 AEs |
|---|---|---|---|---|---|
| n = 8 | n = 8 | n = 6 | N = 22 | ||
| Fatigue | 6 | 6 | 4 | 16 | 7 |
| Nausea | 4 | 7 | 4 | 15 | 0 |
| Diarrhoea | 7 | 5 | 2 | 14 | 1 |
| Vomiting | 3 | 5 | 2 | 10 | 1 |
| Constipation | 3 | 5 | 3 | 11 | 0 |
| Abdominal pain | 3 | 3 | 2 | 8 | 2 |
| Pyrexia | 3 | 3 | 1 | 7 | 0 |
| Thrombocytopenia† | 2 | 2 | 3 | 7 | 6 |
| Headache | 1 | 2 | 2 | 5 | 0 |
| Peripheral oedema | 1 | 3 | 1 | 5 | 0 |
| Peripheral neuropathy | 2 | 0 | 2 | 4 | 1 |
| Neutropenia‡ | 0 | 2 | 2 | 4 | 4 |
AEs, adverse events.
AEs are listed in decreasing frequency in all patients. AEs that occurred in ≥5 patients in the total study population are included, plus neutropenia and peripheral neuropathy.
Includes: thrombocytopenia, platelet count decreased.
Includes: neutropenia, leucopenia, neutrophil count decreased, white blood cell count decreased.
Categories of AEs that led to discontinuation from the study included gastrointestinal disturbance (18%) and musculoskeletal and connective tissue disorders (14%). Diarrhoea (9%) was the single most commonly reported related AE that led to study discontinuation.
Liver toxicity-related events were experienced by four patients (18%; three patients in the 340 mg/m2 group and one patient in the 175 mg/m2 group). One patient in the 340 mg/m2 dose had grade 4 elevated aspartate aminotransferase (AST), grade 2 elevated alanine aminotransferase (ALT), and grade 3 elevated alkaline phosphatase due to acute cholecystitis unrelated to study drug. The second patient in the 340 mg/m2 group had grade 4 hepatotoxicity, which resolved approximately 2 weeks later. Tanespimycin was held and then readministered at a lower dose of 275 mg/m2. Four days later, the patient developed grade 3 dyspnea and fatigue (considered possibly related to study drug) and was withdrawn from the study. The third patient in the 340 mg/m2 had grade 3 elevated ALT and elevated AST, both of which resolved. The patient in the 175 mg/m2 dose group had grade 2 elevated ALT that resolved 3 weeks later with no interruption or dose modification.
Thrombocytopenia was the most common haematological AE reported. Two patients (25%) in the 340 mg/m2 dose group had AEs of grade 4 thrombocytopenia, one of whom was withdrawn from the study. Thrombocytopenia as an AE was also reported for two patients (25%) in the 175 mg/m2 dose group (grade 2 and grade 3), and 1 patient (17%) in the 50 mg/m2 dose group (grade 3). All patients in the 340 mg/m2 and the 175 mg/m2 dose groups, and three patients in the 50 mg/m2 dose group (75%) had platelet count decreases to day 11 of cycle 1, with rebound to baseline by day 1 of cycle 2. This cyclical effect on platelets was observed in each subsequent cycle. The median change in platelet counts from baseline to cycle 1 d 11 was −113 × 109/l, −74 × 109/l and −74 × 109/l for the 340, 175 and 50 mg/m2 dose groups, respectively (Table IV).
Table IV.
Summary of mean change from baseline for platelets and absolute neutrophil count. 340 mg/m2
| 340 mg/m2 | 175 mg/m2 | 50 mg/m2 | Total | |||||
|---|---|---|---|---|---|---|---|---|
|
n = 8
|
n = 8
|
n = 6
|
N = 22
|
|||||
| Cycle 1 | Cycle 1 | Cycle 1 | Cycle 1 | |||||
| Day 11 | Change from BL | Day 11 | Change from BL | Day 11 | Change from BL | Day 11 | Change from BL | |
| Platelet count (×109/l) | ||||||||
| N | 5 | 5 | 7 | 7 | 4 | 4 | 16 | 16 |
| Median | 123·0 | −113·0 | 101·0 | −74·0 | 80·5 | −74·0 | 103·0 | −79·65 |
| Range | 78, 143 | −199, −51 | 68, 119 | −128, −31 | 35, 135 | −102, 11 | 35, 143 | −199, 11 |
| ANC (×109/l) | ||||||||
| N | 6 | 6 | 7 | 7 | 4 | 4 | 17 | 17 |
| Median | 2·795 | −0·025 | 2·650 | −0·020 | 1·595 | −1·110 | 2·280 | −0·150 |
| Range | 1·20, 3·92 | −2·91, 0·43 | 1·51, 4·04 | −2·09, 2·92 | 1·20, 2·40 | −2·88, 0·82 | 1·20, 4·04 | −2·91, 2·92 |
ANC, absolute neutrophil count; BL, baseline.
One patient (13%) from the 340 mg/m2 dose group had grade 2 anaemia and was withdrawn from the study. Anaemia was also reported in three patients (38%) from the 175 mg/m2 dose group (one patient grade 2; two patients grade 3). No AEs of neutropenia were reported in the 340 mg/m2 dose group; two patients (25%) had an AE of neutropenia in the 175 mg/m2 dose group (grade 3) and two patients (33%) had grade 3 AEs of neutropenia and neutrophil count decreases in the 50 mg/m2 dose group. None of these patients experiencing neutropenia had dose reductions, delays, or discontinuations. In cycle 1, two patients in the 340 mg/m2 dose group (33%), two patients in the 175 mg/m2 dose group (29%), and three patients in the 50 mg/m2 dose group (75%) had decreases in absolute neutrophil count (ANC) on day 11. In cycle 2, two patients in the 175 mg/m2 group (50%) and four patients in the 50 mg/m2 group (100%) had decreases in ANC on day 11. Only two of the 22 patients in the study (one each in the 175 and 50 mg/m2 groups) received concomitant administration of granulocyte growth factors. The median change in ANC from baseline to cycle 1 day 11 was not significant with observed decreases of −0·025 × 109/l, −0·02 × 109/l and −1·11 × 109/l in the 340, 175 and 50 mg/m2 dose groups, respectively (Table IV). Three patients (one in the 340 mg/m2 dose group and two in the 150 mg/m2 dose group) with ANC laboratory values through cycle 3 d 11 did not have decreases in ANC on day 11 during the first two cycles. However, all 3 patients did have decreases in ANC on cycle 3 day 11 relative to cycle 3 day 1.
Treatment-emergent peripheral neuropathy was reported by six patients (28%), all of whom had a history of prior peripheral neuropathy (Table V). Five of the six patients had grade ≤2 peripheral neuropathy and only one patient had grade 3 peripheral neuropathy. The latter patient had a history of significant peripheral neuropathy and had stopped tanespimycin on day 4 due to elevated liver function tests (LFTs), but continued to receive bortezomib. Grade 3 peripheral neuropathy occurred on day 13 and the patient was subsequently withdrawn from the study.
Table V.
Patients with peripheral neuropathy on treatment.
| Dose group | Neuropathy history | Last dose of tanespimycin | Last dose of bortezomib | Onset date | Worst grade on treatment |
|---|---|---|---|---|---|
| 340 mg/m2 | Mild PN in the past 12 months/no painful PN | Day 4, stopped due to elevated LFTs but continued bortezomib on days 8 and 11 | Day 11 | Day 13 | Grade 3 PN |
| 340 mg/m2 | Mild PN in the past 12 months/no painful PN | Day 379, administered weekly due to patient decision | Day 379, administered weekly due to patient decision | Day 92 | Grade 1 PN |
| 340 mg/m2 | Mild painful PN in the past 12 months | Day 8, discontinued due to DIC | Day 8, discontinued due to DIC | Day 17 | Grade 2 worsening sensory PN |
| 50 mg/m2 | Moderate PN in the past 12 months/no painful PN | Crossed over to high dose on day 85 (cycle 5), continued up to day 403 (cycle 19) | Dose reduced to 1·0 mg/m2 bortezomib on day 4 due to grade 3 hypotension and continued up to day 403 (cycle 19) | Day 64 | Grade 1 PN |
| 50 mg/m2 | Mild PN in the past 12 months/no painful PN | Crossed over to high dose on day 92, continued receiving up to day 466 (cycle 22) | Continued receiving 1·3 mg/m2 bortezomib up to day 466 (cycle 22) | Day 372 | Grade 1 neuralgia, painful PN |
| 50 mg/m2 | Mild painful PN in the past 12 months | Continued up to day 53 | Continued up to day 53 | Day 42 | Grade 2 PN |
DIC, disseminated intravascular coagulation; LFT, liver function test; PN, peripheral neuropathy.
Efficacy
In the present study, the best responses observed with tanespimycin + bortezomib were one MR in the 340 mg/m2 dose group and two PR in the 175 mg/m2 dose group for an ORR of 14%, and an additional 10 patients across all three dose groups with a best response of stable disease (45%; Table VI).
Table VI.
Antitumour response to study treatment.
| Dose group | Best response | PFS (months) | Response to prior bortezomib/no. of priors | Disposition |
|---|---|---|---|---|
| 340 mg/m2 | Minimal response | 12·7+ | Responder/3–4 | Moved to rollover study |
| 175 mg/m2 | Partial response | 2·9+ | Nonresponder/3–4 | Terminated on day 95 due to unacceptable toxicity (prolonged grade 2 diarrhoea) |
| 175 mg/m2 | Unconfirmed partial response* | 0·9+ | Responder/>4 | Terminated due to unacceptable toxicity (grade 2 abdominal pain) |
| 50 mg/m2 | No change but long PFS after crossover; bone marrow from 60% plasma cell at baseline to 5% plasma cell at end of study | 2·9+, censored prior to crossover | Responder/3–4 | Crossed over to high dose on day 85 (cycle 5) and continued up to day 403 (cycle 19) |
| 50 mg/m2 | No change but long PFS after crossover† | 3·1+, censored prior to crossover | Responder/>4 | Crossed over to high dose on day 92, continued receiving up to day 466 (cycle 22) |
PFS, progression-free survival.
With a > 90% reduction in paraprotein observed, consistent with VGPR, by uniform criteria (2006).
Based on M protein alone.
Two patients with stable disease in the 50 mg/m2 dose group crossed over to the 340 mg/m2 group per protocol. One patient remained on study up to cycle 22; the other stayed on study up to cycle 19 and had a decrease in the percentage of plasma cells in the bone marrow from 60% at baseline to 5% by the end of the study. The patient in the 175 mg/m2 dose group with a PR had received 10 prior regimens, including three bortezomib-containing regimens and was refractory to the last regimen of bortezomib + vorinostat, with PD observed after three cycles of treatment.
Pharmacodynamic results
From day 1 h 0 to day 11 h 0, HSP70 levels increased 3·1-, 4·6- and 26·5-fold in the 50 (n = 3), 175 (n = 3), and 340 mg/m2 (n = 4) dose groups, respectively. Plasma chymotrypsin activity per μg proteasome at day 1 h 4 was 106%, 72% and 69% of baseline (day 1 h 0) for the 50, 175 and 340 mg/m2 dose groups respectively; at day 11 h 4, chymotrypsin activity was 96%, 33% and 40% of baseline for the three doses, respectively.
A similar trend was observed at day 1 h 4 for caspase activity per μg proteasome: 103%, 90% and 91% relative to baseline for the 50, 175 and 340 mg/m2 dose groups, respectively. At day 11 h 4, caspase activity was 49%, 36% and 43% of baseline for the three doses.
Trypsin-like activity per μg proteasome showed a different pattern with 126%, 89% and 112% of baseline for the 50, 175 and 340 mg/m2 groups at day 1 h 4, increasing to 241%, 147% and 160% of baseline respectively on day 11 h 4.
Discussion
This study enrolled 22 heavily pretreated patients with relapsed and refractory MM who had received a median of five prior lines of therapy. All patients had been exposed to prior bortezomib and lenalidomide, with 18% and 27% refractory to these prior agents, respectively. Responses (MR or better) were observed in 14% of patients and 50% of patients achieved stable disease, suggesting that the tanespimycin and bortezomib combination had antitumour activity in this patient population, consistent with prior studies.
While all enrolled patients had prior MM therapy that included bortezomib and lenalidomide, five participants (23%) did not receive melphalan, a long-accepted first-line treatment typically given in combination with prednisone. Reasons for this departure from standard practice are speculative, but may have been due to preference for SCT as initial therapy or based on recent studies supporting bortezomib and lenalidomide as primary therapy for patients with relapsed or refractory MM, the type of patient population enrolled in our study (Fonseca & Rajkumar, 2008; Richardson et al, 2009b).
The AE profile for tanespimycin + bortezomib was also consistent with that observed previously (Richardson et al, 2009a), with fatigue, nausea and diarrhoea as the most commonly reported treatment-emergent AEs. Overall, tanespimycin + bortezomib was generally well tolerated with manageable toxicity and the majority of AEs were mild to moderate.
The liver toxicity observed with tanespimycin treatment is noteworthy, but appears to be manageable and reversible. Liver toxicity resolved within 1–2 weeks and some patients were then successfully rechallenged with tanespimycin. Results of this study were similar to two earlier Phase 1 studies of tanespimycin in which severe LFT abnormalities were rare (Modi et al, 2007; Richardson et al, 2009a).
Thrombocytopenia was observed at all dose levels, with platelet decreases on day 11 of each cycle, consistent with the pattern observed with bortezomib monotherapy (Lonial et al, 2008). Characterization of bortezomib-associated thrombocytopenia in previous trials, specifically the Study of Uncontrolled Multiple Myeloma managed with proteasome Inhibition Therapy (SUMMIT) (Richardson et al, 2003) and the Clinical Response and Efficacy Study of Bortezomib in the Treatment of Relapsing Multiple Myeloma (CREST) (Jagannath et al, 2004), revealed the toxicity to be transient, cyclical, and predictable with platelet counts decreasing during treatment with bortezomib and then rapidly recovering during the 10-d rest period at the end of each cycle (Richardson et al, 2003). This pattern was attributed to a reversible effect on megakaryocytes, possibly due to transient inhibition of platelet budding (Lonial et al, 2005).
Bortezomib-induced, cyclical neutropenia with a decrease on day 11 has also been reported (Lonial et al, 2008). In the APEX (Assessment of Proteasome Inhibition for Extending Remissions) trial, ≥ grade 3 neutropenia was more common with bortezomib (14%) than with dexamethasone (1%) (Lonial et al, 2008). However, in the present study of tanespimycin + bortezomib, only a few patients had decreases in ANC on day 11 during the first two cycles. This is consistent with the low incidence of grade ≥3 neutropenia AEs in this study (3%) and with similar observations from an earlier study (Richardson et al, 2009a). The numbers of patients are nonetheless small and this potential protective effect needs further clinical evaluation in large trials.
In the 12 months preceding study entry, most patients (17/22%) had a history of peripheral neuropathy suggesting that peripheral neuropathy is common in heavily pretreated patients with MM. Peripheral neuropathy is the most common DLT reported with bortezomib treatment. In fact, when bortezomib was used as part of a combination approach first-line treatment in MM, drug administration was discontinued because of peripheral neurotoxicity in 4% of the patients, with an overall incidence of peripheral neuropathy of 30% including 6% grade 3 in one study (Harousseau et al, 2006), while 31% of the patients had grade 2 or higher (16% grade 3) peripheral neuropathy reported in another study (Jagannath et al, 2005). The incidence, severity and clinical features of bortezomib-induced peripheral neuropathy were confirmed in other studies of patients with haematological malignancies, as well as MM, but it appears especially important in MM (Orlowski et al, 2005; Fisher et al, 2006).
In the present study, two patients with a history of painful peripheral neuropathy developed grade 2 peripheral neuropathy while on study. One patient randomized to the 340 mg/m2 dose group developed grade 3 peripheral neuropathy; however, this patient had discontinued tanespimycin and, interestingly, was on bortezomib alone during the 7 days prior to the onset of significant peripheral neuropathy. Despite the high incidence of peripheral neuropathy prior to study entry, no other patients discontinued due to peripheral neuropathy. These observations are consistent with the lack of grade ≥ 3 peripheral neuropathy observed in a previous study of tanespimycin + bortezomib in 72 heavily pretreated patients, which was then corroborated in a preclinical rodent model of peripheral neuropathy demonstrating that tanespimycin was neuroprotective against bortezomib-induced peripheral neuropathy (Zhong et al, 2008; Richardson et al, 2009a).
Although not well understood, the mechanisms underlying this neuroprotective response appear to be multifactorial, encompassing a range of cellular chaperone functions from the prevention of protein aggregation to interfering with various cell death cascades (Yenari, 2002). In the nervous system, HSP70 overexpression in cultured hippocampal and peripheral neurons and glia protects against insults such as heat shock and metabolic stress (Yenari, 2002). HSP70 overexpression in various non-neuronal cell lines also protects against oxidative injury, apoptotic stimuli and ischaemia, as well as neurodegenerative disease, epilepsy and trauma (Yenari, 2002).
The induction of HSP70 in peripheral blood mononuclear cells (PBMCs) has been found to represent a classic stress response to heat (Banerji et al, 2005; Goetz et al, 2005; Grem et al, 2005; Modi et al, 2007; Ramanathan et al, 2007; Solit et al, 2007). In one study, HSP70 induction occurred 6 h after a dose of ≤80 mg/m2 of tanespimycin per week (Banerji et al, 2005). At the highest dose levels, 320 and 450 mg/m2 per week, HSP70 induction in PBMCs persisted for 48 and 96 h, respectively. In two earlier tanespimycin studies in patients with relapsed and refractory MM, PBMCs were obtained on day 1 and day 11 in cycle 1 before infusion and 4 h after infusion of tanespimycin. Induction of HSP70 was observed in lysates from PBMCs following a single dose of tanespimycin on day 1 as well as before the fourth dose on day 11, suggesting that the heat stress response is sustained over a 3- to 4-d dosing interval (Richardson et al, 2005). In the present study, plasma HSP70 levels increased on day 11 at the 340 mg/m2 dose (Fig 1) suggesting that biologically active plasma concentrations of tanespimycin were achieved and tanespimycin effectively inhibited its target, HSP90.
Fig 1.
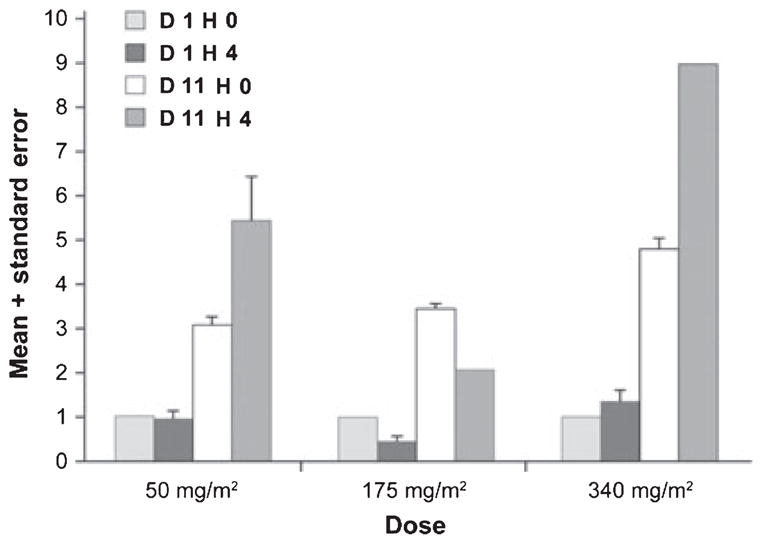
HSP70 Relative to Cycle 1 Day 1 Hour 0 mean + standard error. HSP70 expression, a measure of HSP90 inhibition, was assessed on days 1 and 11 at h 0 and h 4. From day 1 h 0 to day 11 h 0, HSP70 levels increased 3·1-, 4·6- and 26·5-fold in the 50, 175 and 340 mg/m2 dose groups, respectively.
Proteasome inhibition was also observed in this study indicating that tanespimycin does not interfere with the effects of bortezomib on its target. Evidence of antimyeloma activity of tanespimycin + bortezomib in this study included observed responses of an MR, PR, and very good PR (VGPR) in the 340 and 175 mg/m2 dose groups with no responses seen in the 50 mg/m2 group. Most notably, the patient with a PR in the 340 mg/m2 dose group had progressed on the last prior regimen of bortezomib + vorinostat.
In an earlier phase 1/2 study of tanespimycin + bortezomib, the ORR was 64% in bortezomib-naive patients who had ≤3 prior regimens. The median duration of response was 12 months (n = 18), including three bortezomib-refractory patients each with durable PRs through months 12, 22 and 27, respectively. Of these three bortezomib-refractory patients with long-term responses, two patients had progressed while being treated with prior bortezomib (bortezomib/dexamethasone and bortezomib/thalidomide/dexamethasone). Median progression-free survival for bortezomib-naive, pretreated and refractory patients was 7·2, 3·7 and 1·6 months respectively (Richardson et al, 2009a). Patients received a tanespimycin dosage of 100–340 mg/m2.
In our study, tanespimycin was given at a broader dosage range (50–340 mg/m2), with the 50 mg/m2 dose administered to more than a quarter (n = 6) of the patients. The 50% total response rate to bortezomib-tanespimycin was an encouraging result in patients that had relapsed or were refractory after at least three prior bortezomib and lenalidomide treatment regimens. The relatively small patient population was a limitation of the study. However, our study was perhaps a more rigorous evaluation of the therapy in that all patients had measurable MM as determined by M protein values, all were relapsed and refractory cases involving at least three prior treatment regimens, and 27% (6/22) received a low (50 mg/m2) tanespimycin dose that detracted from overall response rates. The favourable outcomes of this study in relapsed and refractory patients together with results of the earlier, larger Phase 1/2 study supports continued evaluation of patients with MM given bortezomib at 1·3 mg/m2 in combination with tanespimycin at 340 mg/m2.
Conclusions
This study, evaluating the combination of tanespimycin + bortezomib in heavily pretreated patients with MM, showed encouraging antitumour activity and a low frequency of peripheral neuropathy. The combination of tanespimycin and bortezomib not only may provide improved antitumour efficacy, but may also limit potential side effects including peripheral neuropathy. Additional studies are needed to further elucidate the possible connection between HSP70 and reduced peripheral neuropathy in patients treated with tanespimycin.
Acknowledgments
The authors wish to thank all of the patients and their families, and the research nurses, physicians, coordinators, and other staff at the study sites. We would also like to thank Katie Redman for administrative assistance; and Anne Lambert for editorial assistance. This study was sponsored by Bristol-Myers Squibb (and previously Kosan Biosciences).
Footnotes
Conflicts of Interest
Dr. Richardson serves on advisory boards for Millennium Pharmaceuticals and Celgene Corporation and receives research funding from Millennium Pharmaceuticals. Drs. Badros, Jagannath, Tarantolo, Wolf and Albitar have no other relevant conflicts of interest to disclose. Dr. Berman and Ms. Messina are employees of Bristol-Myers Squibb and have no other relevant conflicts of interest to disclose. Dr. Anderson is a consultant and has received honoraria and research funding from Celgene Corporation, Novartis, Millennium Pharmaceuticals, and Bristol-Myers Squibb.
References
- Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zwiebel J, Wright JJ, Espinoza-Delgado I, Baer MR, Holleran JL, Egorin MJ, Grant S. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clinical Cancer Research. 2009;15:5250–5257. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, Walton M, Lakhani S, Kaye S, Workman P, Judson I. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. Journal of Clinical Oncology. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Attal M, Crowley J, van Rhee F, Szymonifka J, Moreau P, Durie BG, Harousseau JL. Long-term follow-up of autotransplantation trials for multiple myeloma: update of protocols conducted by the Intergroupe Francophone du Myelome, Southwest Oncology Group, and University of Arkansas for Medical Sciences. Journal of Clinical Oncology. 2010;28:1209–1214. doi: 10.1200/JCO.2009.25.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blade J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S, Vesole D. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. British Journal of Haematology. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- Brown IR. Heat shock proteins and protection of the nervous system. Annals of the New York Acadademy of Sciences. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- Burrows F, Zhang H, Kamal A. Hsp90 activation and cell cycle regulation. Cell Cycle. 2004;3:1530–1536. doi: 10.4161/cc.3.12.1277. [DOI] [PubMed] [Google Scholar]
- Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Journal of Clinical Oncology. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Rajkumar SV. Consolidation therapy with bortezomib/lenalidomide/dexamethasone versus bortezomib/dexamethasone after a dexamethasone-based induction regimen in patients with multiple myeloma: a randomized phase III trial. Clinical Lymphoma and Myeloma. 2008;8:315–317. doi: 10.3816/CLM.2008.n.046. [DOI] [PubMed] [Google Scholar]
- Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, Adjei AA, Sloan J, Atherton P, Vasile V, Salazaar S, Adjei A, Croghan G, Erlichman C. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Journal of Clinical Oncology. 2005;23:1078–1087. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- Grem JL, Morrison G, Guo XD, Agnew E, Takimoto CH, Thomas R, Szabo E, Grochow L, Grollman F, Hamilton JM, Neckers L, Wilson RH. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. Journal of Clinical Oncology. 2005;23:1885–1893. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- Harousseau JL, Attal M, Leleu X, Troncy J, Pegourie B, Stoppa AM, Hulin C, Benboubker L, Fuzibet JG, Renaud M, Moreau P, Avet-Loiseau H. Bortezomib plus dexamethasone as induction treatment prior to autologous stem cell transplantation in patients with newly diagnosed multiple myeloma: results of an IFM phase II study. Haematologica. 2006;91:1498–1505. [PubMed] [Google Scholar]
- Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Research. 2001;61:3071–3076. [PubMed] [Google Scholar]
- Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. British Journal of Haematology. 2004;127:165–172. doi: 10.1111/j.1365-2141.2004.05188.x. [DOI] [PubMed] [Google Scholar]
- Jagannath S, Durie BG, Wolf J, Camacho E, Irwin D, Lutzky J, McKinley M, Gabayan E, Mazumder A, Schenkein D, Crowley J. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. British Journal of Haematology. 2005;129:776–783. doi: 10.1111/j.1365-2141.2005.05540.x. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer Statistics, 2008. CA Cancer Journal for Clinicians. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Kelly S, Yenari MA. Neuroprotection: heat shock proteins. Current Medical Research and Opinion. 2002;18(Suppl 2):s55–s60. [PubMed] [Google Scholar]
- Lonial S, Waller EK, Richardson PG, Jagannath S, Orlowski RZ, Giver CR, Jaye DL, Francis D, Giusti S, Torre C, Barlogie B, Berenson JR, Singhal S, Schenkein DP, Esseltine DL, Anderson J, Xiao H, Heffner LT, Anderson KC. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood. 2005;106:3777–3784. doi: 10.1182/blood-2005-03-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonial S, Richardson PG, San Miguel J, Sonneveld P, Schuster MW, Blade J, Cavenagh J, Rajkumar SV, Jakubowiak AJ, Esseltine DL, Anderson KC, Harousseau JL. Characterisation of haematological profiles and low risk of thromboembolic events with bortezomib in patients with relapsed multiple myeloma. British Journal of Haematology. 2008;143:222–229. doi: 10.1111/j.1365-2141.2008.07321.x. [DOI] [PubMed] [Google Scholar]
- Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opinion on Biological Therapy. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, Gius D, Neckers L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances anti-tumor activity. Molecular Cancer Therapeutics. 2004;3:551–566. [PubMed] [Google Scholar]
- Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, Munshi NC, Richardson PG, Hideshima T, Anderson KC. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14374–14379. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Richardson PG, Treon SP, Anderson KC. Novel biologically based therapies for Waldenstrom’s macroglobulinemia. Seminars in Oncology. 2003;30:309–312. doi: 10.1053/sonc.2003.50065. [DOI] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R, Munshi NC, Richardson PG, Hideshima T, Chauhan D, Gu X, Bailey C, Joseph M, Libermann TA, Rosen NS, Anderson KC. Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006;107:1092–1100. doi: 10.1182/blood-2005-03-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades C, Richardson PG, Munshi NC, Anderson KC. Inhibition of heat shock proteins: therapeutic perspectives. Haematologica. 2007;92(Suppl 2) Abstract S4.5. [Google Scholar]
- Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. Journal of Clinical Oncology. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- Multiple Myeloma Research Foundation. Newly Diagnosed Patients: what is multiple myeloma. [Accessed August 28 2009];Causes and incidence. 2009 WWW document. URL. http://www.themmrf.org/living-with-multiplemyeloma/newly-diagnosed-patients/what-is-multiple-myeloma/causes-and-incidence.html.
- Nowakowski GS, McCollum AK, Ames MM, Mandrekar SJ, Reid JM, Adjei AA, Toft DO, Safgren SL, Erlichman C. A phase I trial of twice-weekly 17-allylamino-demethoxygeldanamycin in patients with advanced cancer. Clinical Cancer Research. 2006;12:6087–6093. doi: 10.1158/1078-0432.CCR-06-1015. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105:3058–3065. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- Paul E, Sutlu T, Deneberg S, Alici E, Björkstrand B, Jansson M, Lerner R, Wallblom A, Gahrton G, Nahi H. Impact of chromosome 13 deletion and plasma cell load on long-term survival of patients with multiple myeloma undergoing autologous transplantation. Oncology Reports. 2009;22:137–142. doi: 10.3892/or_00000416. [DOI] [PubMed] [Google Scholar]
- Pineda-Roman M, Barlogie B, Anaissie E, Zangari M, Bolejack V, van Rhee F, Tricot G, Crowley J. High-dose melphalanbased autotransplants for multiple myeloma: the Arkansas experience since 1989 in 3077 patients. Cancer. 2008;112:1754–1764. doi: 10.1002/cncr.23327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan RK, Egorin MJ, Eiseman JL, Ramalingam S, Friedland D, Agarwala SS, Ivy SP, Potter DM, Chatta G, Zuhowski EG, Stoller RG, Naret C, Guo J, Belani CP. Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with refractory advanced cancers. Clinical Cancer Research. 2007;13:1769–1774. doi: 10.1158/1078-0432.CCR-06-2233. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. New England Journal of Medicine. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Chanan-Khan AA, Alsina M, Doss D, Landrigan B, Kettner D, Albitar M, Mitsiades C, Cropp GF, Johnson RG, Hannah AL, Anderson KC. Safety and activity of KOS-953 in patients with relapsed refractory multiple myeloma (MM): interim results of a phase 1 trial. Blood (ASH Annual Meeting Abstracts) 2005;106:361. [Google Scholar]
- Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K. New drugs for myeloma. Oncologist. 2007;12:664–689. doi: 10.1634/theoncologist.12-6-664. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Chanan-Khan A, Lonial S, Krishnan A, Carroll M, Alsina M, Albitar M, Berman D, Kaplita S, Anderson KC. Tanespimycin + bortezomib in patients with relapsed and refractory multiple myeloma: final results of a phase 1/2 study. Journal of Clinical Oncology, 2009 ASCO Annual Meeting Proceedings. 2009a;27(15S):8503. [Google Scholar]
- Richardson PG, Weller E, Jagannath S, Avigan DE, Alsina M, Schlossman RL, Mazumder A, Munshi NC, Ghobrial IM, Doss D, Warren DL, Lunde LE, McKenney M, Delaney C, Mitsiades CS, Hideshima T, Dalton W, Knight R, Esseltin DL, Anderson KC. Multicenter, phase I, dose-escalation trial of lenalidomide plus bortezomib for relapsed and relapsed/refractory multiple myeloma. Journal of Clinical Oncology. 2009b;27:5713–5719. doi: 10.1200/JCO.2009.22.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Ivy SP, Kopil C, Sikorski R, Morris MJ, Slovin SF, Kelly WK, DeLaCruz A, Curley T, Heller G, Larson S, Schwartz L, Egorin MJ, Rosen N, Scher HI. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clinical Cancer Research. 2007;13:1775–1782. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer T, Zollinger A, Siegmund D, Chatterjee M, Grella E, Knop S, Kortum M, Unzicker C, Jensen MR, Quadt C, Chene P, Schoepfer J, Garcia-Echeverria C, Einsele H, Wajant H, Bargou RC. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia. 2008;22:1604–1612. doi: 10.1038/leu.2008.111. [DOI] [PubMed] [Google Scholar]
- Takayama S, Reed JC, Homma S. Heat-shock proteins as regulators of apoptosis. Oncogene. 2003;22:9041–9047. doi: 10.1038/sj.onc.1207114. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature Reviews Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Yenari MA. Heat Shock Proteins and Neuroprotection. In: Alzheimer C, editor. Molecular and Cellular Biology of Neuroprotection in the CNS. Vol. 513. Landes Bioscience, Kluwer Academic/Plenum Publishers; New York: 2002. pp. 281–300. [Google Scholar]
- Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. Journal of Molecular Medicine. 2004;82:488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Simmons J, Timmermans P. Prevention and treatment of bortezomib-induced peripheral neuropathy by the Hsp90 inhibitor tanespimycin (KOS-953) in the rat. European Journal of Cancer Supplements. 2008;6 Abstract 152. [Google Scholar]