

Abstract
Background
Fast, cost-effective and reproducible isolation of IgM from plasma is invaluable to the study of IgM and subsequent understanding of the human immune system. Additionally, vast amounts of information regarding human physiology and disease can be derived from analysis of the low abundance proteome of the plasma. In this study, methods were optimized for both the high-throughput isolation of IgM from human plasma, and the high-throughput isolation and fractionation of low abundance plasma proteins.
Materials and methods.
To optimize the chromatographic isolation of IgM from human plasma, many variables were examined including chromatography resin, mobile phases, and order of chromatographic separations. Purification of IgM was achieved most successfully through isolation of immunoglobulin from human plasma using Protein A chromatography with a specific resin followed by subsequent fractionation using QA strong anion exchange chromatography. Through these optimization experiments, an additional method was established to prepare plasma for analysis of low abundance proteins. This method involved chromatographic depletion of high-abundance plasma proteins and reduction of plasma proteome complexity through further chromatographic fractionation.
Results
Purification of IgM was achieved with high purity as confirmed by SDS-PAGE and IgM-specific immunoblot. Isolation and fractionation of low abundance protein was also performed successfully, as confirmed by SDS-PAGE and mass spectrometry analysis followed by label-free quantitative spectral analysis.
Discussion.
The level of purity of the isolated IgM allows for further IgM-specific analysis of plasma samples. The developed fractionation scheme can be used for high throughput screening of human plasma in order to identify low and high abundance proteins as potential prognostic and diagnostic disease biomarkers.
Keywords: Low abundance protein isolation, IgM, IgG, plasma proteomics
Introduction
Body fluids, especially human blood plasma and serum, are the most important sources for discovery of disease biomarkers, and are therefore the topic of intensive investigations in order to find new biomarker candidates1. There are a plethora of papers about plasma fractionation as a sample preparation step for further LC-MS/MS investigations1,2. Some protein biomarkers have been approved for regulatory agencies, and few of them are also in clinical use3. However, there have also been very critical recent statements about the application of proteomics to biomarker discovery, and even doubts if the proteomics technology has delivered sufficiently usable results, or whether a fundamental change of the concept is necessary, before clinically useful results can be accepted4,5.
Blood plasma and serum are body fluids that are most frequently used for the identification of new biomarkers1,2. These biological materials contain thousand of components in a dynamic range of concentrations up to 108 and 1012 and are very complex1,6. In human plasma, serum albumin (HAS), immunoglobulin and an additional approximately 20 proteins account for over 99% of the overall protein content. The concentrations of low abundance proteins in plasma and serum range from ng/mL down to pg/mL level1,2,6. These low abundance plasma proteins are most frequently interesting as biomarkers or biomarker candidates. Consequently, their detection and further investigation frequently demands thorough sample preparation7.
Together with electrophoretic techniques2, chromatographic and immunoaffinity chromatographic methods are used for separation of highly abundant proteins, and concentration of low abundance ones8,9. However, the need to improve separation methods still remains and the use of additional fractional methods such as combinatorial peptide libraries8, displacement chromatography9, and sample displacement chromatography10 has recently been introduced. In order to gain statistically significant results, the analyses of large number of samples are necessary. Therefore, the development of fast and reliable high-throughput methods for sample preparation, mass spectrometry and data analysis becomes one of most important challenges in future proteomic and glycomic strategy11.
In this paper, a new scheme for rapid and reliable fractionation of plasma proteins and separation of high, middle and low abundance proteins by use of a combination of chromatographic, affinity and pseudo-affinity chromatographic methods is introduced. This method can be simply adapted for use of laboratory robots and applied for high-throughput separation of a large number of samples.
Materials and methods
Plasma collection
The plasma that was used for the optimization of these techniques was cryopoor, single donor human plasma (Rhode Island Blood Center, Providence, RI, USA). Plasma samples were screened in order to exclude the presence of blood-borne viruses (hepatitis A, B and C and HIV). Cryoglobulins were removed from plasma by precipitation at 4 °C.
As a part of a larger study, plasma was also collected from patients undergoing ablation therapy for liver, renal and lung tumors (Rhode Island Hospital Department of Diagnostic Imaging, Providence, RI, USA). Blood samples from patients were collected in heparanized tubes immediately before ablation, and then subsequently after 1 hour, 4 hours, 1 week, 2 weeks, 1 month, 3 months, and 6 months depending on the patient’s ability to meet appointments. Plasma samples were screened in order to exclude the presence of blood-borne viruses (hepatitis A, B and C and HIV) and plasma was isolated from cellular components via centrifugation. This prospective study was approved by the institutional review board and was compliant with the Health Insurance Portability and Accountability Act (Rhode Island Hospital, Providence, RI, USA).
IgM isolation technique
Protein A chromatographic separation
For immunoglobulin separation from plasma, glass columns with an inner diameter of 6.5 mm packed with 1.0 mL of Toyopearl AF-rProtein A-650F support were used (Tosoh Bioscience, Stuttgart, Germany). All chromatographic separations in this study were loaded and eluted manually using All Plastic Luer-Slip Syringes (National Scientific Company, Rockwood, TN, USA) at room temperature with a flow rate of roughly 2 mL/min.
Before loading sample, columns were washed with 2 column volumes (CV) of 0.2 M citric acid (Aqua Solutions, Deer Park, TX, USA), regenerated with 2 CV of 0.2 M tris HCl pH 7.4 (Fischer Biotech, Fair Lawn, NJ, USA) and washed with 5 CV of 10 mM tris HCl pH 7.4. Before separation on Protein A columns, 200 μl of plasma was diluted five-fold in 10 mM tris pH 7.4 and centrifuged at 14,000 rpm for 10 minutes (Centrifuge 5417 R, Eppendorf, Hamburg, Germany). The resulting supernatant was then applied to the column and unbound proteins were collected. After sample application, the column was washed with 4 CV of 10 mM tris HCl pH 7.4. Elution was performed with 0.2 M citric acid, during which 1.4 mL of eluate was collected and immediately neutralised to a pH of approximately 7.4 with 0.6 mL of 1 M tris base, pH 10.9.
Columns were then regenerated with 2 CV of 0.2 M tris HCl pH 7.4, and washed with 5 CV of 10 mM tris HCl, pH 7.4. To increase the amount of IgM that was isolated from the plasma, the unbound protein fractions were then reapplied to the column. Again, unbound proteins were collected and the column was washed and eluted again as described above. Before storage, columns were again regenerated with 0.2 M tris HCl pH 7.4 and stored in 20% ethanol (Acros Organics, Geel, Belgium).
Concentration and buffer exchange
Protein A elutions were concentrated to approximately 100 μL using Amicon Ultra-0.5 Ultracel-10 Membrane 10 kDa cutoff centrifugal filter units (Millipore, Billerica, MA, USA) according to the manufacturer’s procedure. A buffer exchange was then performed by diluting the concentrated sample 5X in 0.05 M NaCl, 20 mM tris pH 7.2 buffer and concentrating the samples again to approximately 100 μL. This buffer exchange procedure was repeated twice more before the final resulting 100 μL sample was transferred out of the centrifugal filter units and diluted to 500 μL with 0.05 M NaCl, 20 mM tris pH 7.2, resulting in a final buffer dilution of 624:1.
QA strong anion exchange
CIM QA Disk Monolithic Columns, 0.34 mL, and corresponding housing (BIASeparations, Klagenfurt, Austria) were used to further fractionate the immunoglobulin-rich Protein A elution. The disks were first cleaned with 6 CV of 0.2 M NaOH, neutralised with 6 CV of 0.5 M tris HCl pH 7.2, then washed with 6 CV of 1 M NaCl 20 mM tris HCl pH 7.2, and finally equilibrated with 10 CV of 0.05 M NaCl 20 mM tris HCl pH 7.2. Concentrated and desalted Protein A elutions were then applied to the column and the unbound proteins were caught. The monolith was then washed with 10 CV of 0.05 M NaCl 20 mM tris HCl pH 7.2. Two elutions were performed with 10 CV of 0.225 M NaCl and 0.50 M NaCl in 20 mM tris HCl pH 7.2, and protein peaks of each were caught and analysed. Disks were stored in 20% ethanol.
Low abundance protein isolation/fractionation technique
In isolation of low-abundance plasma proteins, plasma was first separated using the Protein A affinity chromatography procedure described above. Low abundance protein isolation was performed using the separation’s resulting unbound proteins (IgM isolation was most successfully performed using the eluate of this separation).
AF-Blue chromatographic separation
For albumin depletion, glass columns with an inner diameter of 6.5 mm packed with 1.0 mL of Toyopearl AF-Blue HC-650M support were used (Tosoh Bioscience). Before loading sample, columns were washed with two CV of 2.0 M NaCl (Fischer Scientific, Fair Lawn, NJ), followed by 10 CV of 10 mM tris HCl pH 7.4. Samples of human plasma that had been depleted of IgG via Protein A separation as described above were applied to the column, and manually recirculated through the column three times before unbound proteins were analysed. After sample application, the column was washed with 5 CV of 10 mM tris HCl pH 7.4. Elution was performed with 2.0 M NaCl, and 2.0 ml of eluate was collected. Columns were further washed with 5 CV of 2 M guanidine in 10 mM tris. Columns were stored in 20% ethanol.
Tryptophan chromatographic separation
For tryptophan separation, glass columns with an inner diameter of 6.5 mm packed with 1.0 mL of Toyopearl Tryptophan Immobilized Resin were used (Tosoh Bioscience). Before loading sample, columns were washed with 3 CV of 0.5 M NaOH (Fischer Scientific), followed by 5 CV of 0.3 M NaCl in 0.1 M tris HCl pH 8.5 and 10 CV of 0.05 M sodium acetate pH 4.7 (Fischer Scientific). Samples of human plasma that had been depleted of both IgG and albumin via Protein A and AF-Blue separation as described above were diluted in a 1:1 ratio with 0.05 M sodium acetate pH 4.7, and adjusted to pH 4.7 by addition of 1 M HCl (Fischer), measured with colorpHast pH 0–6 Test Strips (EMD, Gibbstown, NJ, USA). Samples were applied to the column, and unbound proteins were collected. After sample application, the column was washed with 5 column volumes of 0.05 M sodium acetate pH 4.7. A first elution was performed with 5 column volumes of 0.1 M NaCl pH 4.7 in 0.05 M sodium acetate, and a second elution was performed with 5 column volumes of 0.3 M NaCl in 0.1 M tris HCl pH 8.5. Protein peaks from each elution were collected and further analysed. Columns were stored in 20% ethanol.
SDS-PAGE
Protein concentrations in collected fractions and starting material were determined with the Bicinchoninic Acid Protein Assay kit (Pierce, Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s procedure. For SDS-PAGE, samples were solubilised in NuPAGE LDS sample buffer, reduced with NuPAGE Sample Reducing Agent and loaded on NuPAGE Novex 4–12% tris-glycine pre-cast gels (Life Technologies, Carlsbad, CA, USA). The electrophoresis was performed using XCELL Sure Lock Mini-Cell in MES SDS Running Buffer with NuPAGE Antioxidant at 180 V for 50 min (Life Technologies). Particularly dilute samples were concentrated briefly via evaporation at 100 °C prior to loading. Gels were stained with GelCodeBlue dye (Pierce) and visualised by a VersaDoc Imaging System (BioRad, Hercules, CA).
IgM specific Immunoblot
Proteins were separated via SDS PAGE as described above, then instead of staining, transferred electrophoretically to an Invitrogen Nitrocellulose Membrane, 0.2 μm pore size using the XCell II Blot Module according to the manufacturer’s procedure (Life Technologies). Membranes were then washed and blotted using the Immuno-Blot Assay Kit with Goat Anti-Rabbit IgG (H+L) secondary antibody (BioRad) according to the manufacturer’s procedure. The primary antibody used was Anti-Human IgM (μ-chain specific), produced in rabbit (Sigma Aldrich, St. Louis MO, USA). Membranes were the developed for approximately 2 minutes using the AP Conjugate Substrate Kit (BioRad) according to the manufacturer’s procedure.
As a control, IgM from bovine serum (Sigma Aldrich) was used.
Sample preparation for MS analysis
In-Solution Digestions
For “in-solution” digestions, 50 μg of protein was cleaned up using the BioRad 2D Clean-Up Kit according to the manufacturer’s procedure. The resulting pellet was solubilised in 10 μL of 4 M urea, 0.05 M NH4HCO3 (Fischer). The solubilised proteins were reduced in 20 mM dithiothreitol (Thermo Scientific, Rockford, IL, USA) for 45 min at 37 °C and then alkylated in 50 mM iodoacetamide (Sigma Aldrich) at room temperature for 30 min in the dark, by addition of 1 μL of 200 mM DTT and 0.5 M IAA, respectively. Before tryptic digestion, 36 μL of 100 mM NH4HCO3 buffer was added to reduce the concentration of urea to approximately 0.8 M. Proteomics Grade Trypsin from porcine pancreas (Sigma Aldrich) was added to the protein mixture at an enzyme to substrate ratio of 1:50 (w/w). Half of the total trypsin solution was added to the protein mixture, and after 3 hours of incubation at 37 °C, the second half was added. After incubating at 37 °C overnight, the tryptic peptides were dried in a vacuum centrifuge (Vacufuge, Eppendorf).
In-Gel Digestions
For “in-gel” digestions12, specific bands were excised from gels using a OneTouch Plus Spotpicker, 1.5 mm (The Gel Company, San Francisco, CA, USA) and were consecutively washed in roughly twice their volume of 1:1 0.1 M NH4HCO3:Acetonitrile (Sigma Aldrich), ddH2O, 1:1 0.1 M NH4HCO3:Acetonitrile, and ddH2O. Each wash was performed with agitation for 15 minutes, and after each wash, wash solution was removed via pipette. Gel pieces were then washed with a volume of acetonitrile approximately equal to their volume, and after 5 minutes an equal volume of 0.1 M NH4HCO3 was added to each sample. Gel pieces were agitated for 15 more minutes, followed by removal of wash solutions via pipette, and samples were dried down using a vacuum centrifuge (Vacufuge, Eppendorf). Samples were then swelled in 10 mM dithiothreitol, 0.1 M NH4HCO3 and incubated at 56 °C for 45 minutes. Tubes were chilled back to room temperature, and the reducing solution was removed via pipette and quickly replaced with an equal volume of 55 mM iodoacetamide. Samples were then incubated at room temperature in the dark for 30 minutes. All liquid was removed and gel pieces were again washed using ddH2O, acetonitrile, and 0.1 M NH4HCO3 following the same steps as the wash performed prior to reduction and alkylation. Gel pieces were dried again, and were subjected to trypsin digestion using Sigma Proteomics Grade Trypsin from porcine pancreas according to the manufacturer’s in-gel digestion procedure.
Identification of proteins with LC-ESI-MS/MS
Following in-gel and in-solution enzymatic digestion of proteins, their tryptic peptides were analysed by mass spectrometry. Tryptic digests were separated with a reversed-phase (RP) column (C-18 PepMap 100, LC Packings/Dionex, Sunnyvale, CA, USA) as previously described7,12. The separation gradient applied was starting with 5% (v/v) acetonitrile (ACN) in 0.1% (v/v) formic acid (FA) (Solvent A) to 95% ACN in 0.1% FA (Solvent B) for 75 min. The column eluate was introduced directly onto a QSTAR XL mass spectrometer (Sciex and Applied Biosystems, Concord, Ontario, Canada) via ESI. Candidate ion selection, fragmentation and data collection were performed as previously described13. Protein identifications were performed with ProteinPilot software (Sciex and Applied Biosystems), using a mouse “RefSeq” database from NCBI (http://www.ncbi.nim.nih.gov/RefSeq/) as previously described13. In parallel experiments, tryptic peptides were separated on a 12 cm x 75 μm I.D. C18 RP column (Column Engineering, Ontario, CA, USA) and eluted using a linear gradient starting with 100% solvent A (0.1 M acetic acid in water) to 70% solvent B (ACN) over 30 min (Agilent Technologies, Paolo Alto, CA, USA). Eluted peptides were introduced onto a LTQ linear ion trap mass spectrometer (Thermo Electron Corporation, San Jose, CA, USA) and analysed as previously described7. Peptide spectrum matching was performed against a IPI human database using the peak lists in the SEQUEST program as described14 and protein identifications were derived from the peptide matches using in-house software.
Results
In an effort to purify IgM, several parameters, including chromatography resin, mobile phases, and order of separations performed were varied and results were analysed. Successful purification of IgM was ultimately achieved by sequential separation of plasma using Protein A chromatography followed by QA strong anion exchange chromatography. Our quest for IgM isolation also yielded the optimization of a method for the separation of plasma into fractions depleted of high abundance proteins and decreased sufficiently in proteome complexity to allow for successful analysis of the plasma’s low abundance proteome with mass spectrometry.
Tryptophan binding ability
Immobilised tryptophan chromatography was first explored as a method of IgM isolation. The first parameter investigated was the binding buffer that would be ideal for IgM binding to this resin. Tryptophan columns were first tested for ability to bind proteins from human plasma that was diluted 5x in a binding buffer of A.) 0.1 M NaCl in 0.05 M sodium acetate pH 4.7, and B.) 0.15 M NaCl in 0.05 M sodium acetate pH 4.7. An elution was performed using 0.1 M tris pH 8.5. While much more protein bound to the column when plasma was applied in the first binding buffer, in both cases almost all of the plasma proteins did not bind to the tryptophan column (Figure 1).
Figure 1.

Optimisation of NaCl concentration of buffers for pseudo-affinity chromatography with immobilised tryptophan. SDS-PAGE of loaded and eluted plasma proteins.
CP: calibration proteins; lanes 1 and 2 (run A): plasma diluted with 0.1 M NaCl; 1: unbound proteins; 2: eluted proteins; lanes 3 and 4 (run B): plasma diluted with 0.15 M NaCl; 1: unbound proteins; 2: eluted proteins. For chromatographic conditions see Material and methods.
Next, to identify the column’s ability to bind IgM, purified bovine IgM rather than human plasma was separated on a tryptophan column. While it was noted that human and bovine IgM would not act identically during chromatography, it was assumed that bovine IgM would be a good preliminary indicator of the separation expected with human IgM. Using 0.1 M NaCl in 0.05 M sodium acetate pH 4.7 as binding buffer, pure bovine IgM samples were separated using a tryptophan column. The majority of the IgM did not bind to the column using this binding buffer (not shown here). The experiment was repeated, this time using a binding buffer of 0.05 M sodium acetate pH 4.7 (no NaCl), and diluted IgM was pH adjusted to below 4.7 with HCl before loading. In this experiment, approximately 70% of bovine IgM bound to the column.
Tryptophan fractionation of DEAE fractionated plasma
After establishing the ability of the tryptophan column to bind IgM, focus was then shifted to isolation of IgM from low-abundance protein enriched human plasma using this tryptophan separation technique. Samples of plasma diluted 5x in 10 mM tris pH 7.4 were first depleted of albumin via AF-Blue affinity chromatography, and then applied to a diethylaminoethyl weak anion exchange column (Toyopearl DEAE-650S, Tosoh Bioscience), and eluted with PBS. The PBS elution of this separation was then diluted 5x in 0.05 M sodium acetate pH 4.7, adjusted to a pH of 4.7 with HCl, and applied to the tryptophan column. Elution from the tryptophan column was performed with 0.1 M tris pH 8.5. Originally, fractionated PBS elutions were largely too dilute to analyse, but these fractions were concentrated down via centrifugal filtration, and the entirety of the concentrated samples were loaded on an SDS PAGE gel. Both the DEAE elution and the unbound proteins from the tryptophan separation appeared to have a large band that could potentially be IgM, but both contained many other large bands of other proteins as well. It was also noted in this experiment that very few proteins bound to the tryptophan column following DEAE separation (not shown here).
Tryptophan fractionation of low-abundance protein enriched plasma
Four unique samples of normal plasma were then separated sequentially by Protein A (IgG depletion), AF-Blue (albumin depletion), and tryptophan chromatography. The amount of difference between the binding of bovine IgM to the tryptophan column in a binding buffer of 0.05 M sodium acetate pH 4.7 compared to a binding buffer of 0.1 M NaCl in 0.05 M sodium acetate pH 4.7 in our previous experiment was noted, and this information was utilised for IgM isolation. Tryptophan separations were performed using a two step elution system: samples were loading using 0.05 M sodium acetate pH 4.7 and eluted in the first step with 0.1 M NaCl in 0.05 M sodium acetate pH 4.7 and in the second with 0.3 M NaCl in 0.1 M tris pH 8.5.
Samples from ablation patients were then separated using the methods described above. IgM specific immunoblots were performed on the elutions from these tryptophan column separations to search for changes in IgM concentration over the course of patient enrollment. After development, IgM bands were imaged using QuantityOne software, and volumetric analysis was utilised to compare the IgM bands of each sample. Samples were analysed by both the thickness of their IgM band and the volume of their IgM band (not shown here). It was also evident from these immunoblots that IgM was present, at least in part, in both elutions after pseudo-affinity separation with immobilised tryptophan (Figure 2).
Figure 2.
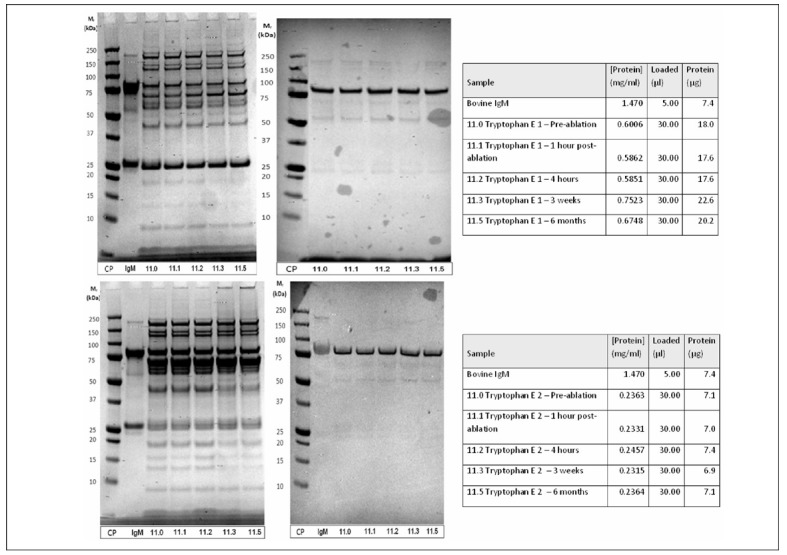
Optimisation of pseudo-affinity chromatography with immobilised tryptophan for plasma protein fractionation and IgM concentration. Samples of patient 11 before and at several time points after tumor ablation were separated on tryptophan pseudo-affinity column.
Upper part: elution 1 (with 0.1 M NaCl in 0.05 M sodium acetate buffer, pH 4.7) SDS-PAGE (left) and immunoblot with anti-IgM polyclonal antibodies (right).
Lower part: elution 2 (with 0.3 M NaCl in 0.1 M tris buffer pH 8.5).
SDS-PAGE (left) and immunoblot with anti-IgM polyclonal antibodies (right); lane 1: calibration proteins; lane 2: Bovine IgM standard; lanes 3–7: patient samples at several time points before and after tumor ablation.
For more details see Material and methods.
Sample separation for mass spectrometry preparation
It was noted that this separation scheme was a very good way to fractionate plasma to reduce the complexity of each sample such that they could be analysed for changes in the low abundance proteome via mass spectrometry. SDS PAGE analysis of normal whole plasma compared to fractionated plasma showed that the amount of proteins visible, and thus likely to be detected with mass spectrometry after in-solution digestion, was greatly increased by the reduction of IgG and albumin and further fractionation through tryptophan chromatography (Figure 3). Patient plasma samples fractionated with this scheme and tryptically digested in-solution were able to yield very extensive mass spectrometry data. Of the patient plasma samples prepared using these methods, an average of 361 proteins were able to be confirmed with at least 90% certainty using ProteoIQ (NuSep, Bogart, GA, USA) and an Orbitrap mass spectrometer (Thermo Scientific). Some identified proteins are listed in Table I. Label-free quantification was used to look for proteins of changing concentrations over the course of the patient enrollment in this study. Additionally, proteins noted in SDS PAGE gels that appeared to be changing over the course of a patient trial were able to be and digested in-gel to yield proteomic identification (examples indicated in Figure 4 and Table II).
Figure 3.

Optimisation of plasma fractionation. SDS-PAGE of samples before and after chromatographic separation.
Upper part: whole patient plasma at different time points before and after tumor ablation.
Lower part: SDS PAGE analysis of samples separated by affinity chromatography after removal of immunoglobulins (protein A affinity chromatography) and HSA (affi-blue pseudo-affinity chromatography). Patient samples at different time points before and after tumor ablation.
Elution 1 (with 0.1 M NaCl in 0.05 M sodium acetate buffer, pH 4.7); elution 2 (with 0.3 M NaCl in 0.1 M tris buffer pH 8.5).
Table I.
Proteins identified in samples after fractionation and “in-gel digestion” (see also Figures 3 and 4).
| Apolipoprotein B-100 |
| Complement C3 |
| Alpha-2-macroglobulin |
| VDAC1 32 kDa protein, mitochondrial |
| AHNAK Neuroblast differentiation associated protein |
| ATP5A1 ATP synthase subunit alpha, mitochondrial |
| Prohibitin |
| ATP1A1 Sodium/potassium transporting ATPase alpha-1-chain |
| SLC25A5 ADP/ATP translocase 2 |
| VDAC2 36 kDa protein |
| Actg 1 33 kDa protein |
| DYNC1H1 Cytoplasmic dynein 1 heavy chain 1 |
| Inter-alpha inhibitor heavy chain H2 |
| EEF1A1 Elongation factor 1-alpha 1 |
| HRNR Hornerin |
| ANXA1 Annexin A1 |
| ANXA2 Annexin A2 |
| Inter-alpha inhibitor heavy chain H1 |
| RAB10RAB10, member RAS oncogene family |
| HTRA1 serine protease HTRA1 |
| HSPA8 71 kDa proteinGADPH Glyceraldehyde-3-phosphate dehydrogenase |
| NNT NAD(P) transhydrogenase, mitochondrial |
Figure 4.

SDS-PAGE of different patient samples after preparation (see Figure 3). Bands of interest that show different density were excised and digested by trypsin (“in solution digestion”). Proteins were identified by LC-MS/MS (see Materials and methods). These proteins were listed in Table I.
Table II.
Some proteins identified in samples after fractionation and “in-solution digestion” (see also Figures 3 and 4).
| APOA1 Apolipoprotein A-I |
| APOA4 Apolipoprotein A4 |
| Serrotransferrin |
| C4b Complement component 4B |
| Prothrombin |
| Inter-alpha inhibitor heavy chain H1 |
| Inter-alpha inhibitor heavy chain H2 |
| APOC2 Apolipoprotein C2 |
| Zgc:86725 42 kDa protein |
| CP ceruloplasmin |
| Transthyretin |
| Coagulation factor XIII |
| Lpha-2-macroglobulin |
| Clusterin isoform 3 |
| Vitronectin |
| Myosin 6 |
| ATP2A2 Sarcoplasmatic/endoplasmatic reticulum ATPase 2 |
| Vitamin K-dependent protein C |
| Inter-alpha inhibitor heavy chain H4 |
| HADH Isoform 2 of Hydroxyacyl-CoA dehydrogenase, mitochondrial |
| Vitamin K-dependent protein S |
| Vinculin isoform 2 |
| Creatine kinase S-type, mitochondrial |
| ATP5B ATP synthase, subunit beta, mitochondrial |
| Histidine-rich glycoprotein |
| HSPD1 60 kDa heat-shock protein, mitochondrial |
| NDUFS1 NADH-ubiquinone oxidoreductase 75 kDa subunit |
| Cops2 33 kDa protein |
| Znf142 Zinc finger protein 142 |
QA strong anion exchange separation of IgG depleted plasma
Next, QA strong anion exchange chromatography was explored as a method of IgM isolation. An elution system of 0.1 M NaCl, 0.25 M NaCl and 0.5 M NaCl each in a binding buffer of 20 mM tris pH 7.2 was originally adopted. Both whole plasma and plasma depleted of IgG via Protein A separation were then tested via separation on the QA disk, and sequentially eluted with the three buffers mentioned above. Resulting samples were analysed with SDS PAGE, IgM immunoblot and IgA immunoblot. IgM specific immunoblot confirmed that all of the IgM ended up in the final 2 elutions of the separation; however, the IgM in these fractions was overshadowed by additional proteins (not shown here).
Tryptophan column separation of plasma was revisited by being coupled with QA separation. Our previous immunoblots of tryptophan separations had indicated that IgM ended up in both tryptophan elutions. In this experiment, our separation was reverted to a single elution system, using only 0.3 M NaCl in 0.1 M tris pH 8.5 as an elution buffer. IgG/albumin depleted plasma was subjected to this tryptophan separation, and elutions were concentrated, subjected to buffer exchange via centrifugal filtration, and were separated on a QA anion exchange monolithic disk using the previously described method eluting with 0.1, 0.25 and 0.5 M NaCl. While these results did not produce a fraction that had IgM in a high degree of purity, it was noted in these results that much of the IgM was still present in the Protein A elution fraction, which had not been previously suspected (not shown here).
QA strong anion exchange separation of Protein A elutions
Accordingly, the elution resulting from Protein A separation of normal plasma was subjected to buffer exchange via centrifugal filtration into 20 mM tris pH 7.2 (binding buffer of QA separation) and applied to a QA monolith. Again, elutions were performed with 0.1, 0.25 and 0.5 M NaCl. The resulting third elution appeared to produce very pure IgM, but a large amount of IgM remained in the second elution (not shown here).
It was presumed that we would be able to obtain more IgM in the third elution by lowering the molarity of the second elution buffer. The QA separation procedure was repeated, varying the buffer used for the second elution from 0.25 M NaCl to 0.175 M NaCl, 0.200 M NaCl, and 0.225 M NaCl in three parallel experiments. It was decided that a second elution of 0.225 M NaCl produced optimal results that would result in a large amount of IgM that would not be overshadowed by impurities (not shown here).
It was also noted that the majority of IgG ended up in the unbound proteins fraction after QA separation of Protein A elution proteins, but a considerable amount was still eluted in the first elution of 0.1 M NaCl (not shown here). In our experiments, we were most interested in IgG and IgM, so in order to increase the amount of IgG in the unbound protein fraction and to cut down on unnecessary steps, the binding buffer was switched to 0.05 M NaCl in 20 mM tris pH 7.2, and the first elution of 0.1 M NaCl in 20 mM tris pH 7.2 was eliminated. We were therefore left with three fractions: the unbound proteins fraction which contained mainly IgG, the first elution fraction which contained proteins that we were not interested in, and a second elution containing mainly IgM.
Maximising IgM in Protein A elution
With confirmation that the Tosoh Protein A columns that we had been using did indeed bind IgM, we investigated a different type of Protein A column in hopes that it could potentially bind IgG without binding IgM, allowing for an easy purification of IgM from the Tosoh Protein A eluate. Normal plasma was sequentially separated using Tosoh Protein A and GE Hitrap Protein A columns to look for differential binding of IgM. Normal plasma was separated on a GE Hitrap Protein A column. The elution from this separation was desalted via buffer exchange in centrifugal filtration units and applied to a Tosoh Protein A column. It was evident from these results that IgM bound to the GE Hitrap Protein A column in a similar fashion to the Tosoh Protein A columns, and thus IgM purification using this method would not be possible (not shown here).
Continuing on with Tosoh Protein A columns, recirculation of flow through was tested to see if more IgM would be retained on columns during subsequent recirculation of plasma through the columns. In two parallel experiments, recirculation was performed 3x without elution, as well as 3 times with elution between each reapplication. From these results it was decided that recirculating the plasma through the column twice, with elution after each application, would achieve the highest amount of IgM while introducing a minimal amount of impurities into the final IgM fraction.
IgM isolation of patient plasma
Using the procedures that were optimised in these experiments, IgM was then separated from patient plasma. IgM isolations showed a high degree of purity and reproducibility (see Figures 5 and 6).
Figure 5.

Optimised fractionation scheme for fast-throughput isolation of IgM and enrichment of low-abundance proteins.
Figure 6.

Isolation of IgM from patient plasma before and after tumor ablation. For the isolation scheme - see Figure 5.
Discussion
Low abundance protein isolation/fractionation
The low abundance protein fractionation method that we have optimised in these experiments has allowed us to successfully perform an in-depth label free quantitative analysis of the low abundance plasma proteome via mass spectrometry. The developed fractionation scheme is shown in Figure 5. In order to successfully prepare samples for such an analysis, first the low abundance proteome must not be hidden by high abundance proteins; this is accomplished by IgG and albumin reduction. Second, proteins must be fractionated enough to decrease the total complexity of each fraction while remaining concentrated enough to be detected by mass spectrometry. This is provided by tryptophan fractionation of low abundance protein enriched samples into three relatively equally complex, but different fractions (see Figures 3 and 4 and Tables I and II).
The benefits of being able to successfully perform label free quantitation of the low abundance proteome of human plasma are wide ranging and can be used in many experiments. We will utilise these methods to continue analysis of ablation patient plasma in search of new potential prognostic and diagnostic tumor biomarkers, inflammatory mediators and immunological stimuli.
IgM isolation
Contrary to our original expectations, when plasma is applied to a Protein A column, a large amount of IgM binds to the column and is able to be collected via elution. However, it is also true that a significant, reproducible amount appears in the separation’s unbound protein fraction (even after recirculation). In our experiments, purification of IgM from the Protein A elution fraction was able to be done more efficiently than purification of IgM from the Protein A unbound proteins fraction. This is most likely because the elution of the Protein A separation has a much more simple protein profile than that of the unbound protein fraction. In short, very few different proteins bind to the Protein A column, and it is easier to purify IgM out from a relatively small group of proteins than a larger group.
Isolation of IgM from plasma is crucial in studying the body’s immune response to different stimuli. With IgM successfully purified, we will be able to move on to other aspects of our research of the body’s immunological responses to tumor treatment with ablation. Having purified IgM allows us to study changes in IgM glycosylation over time, as well as look at the changes of IgM immunospecificity.
The presented method for isolation of high (IgG), medium (IgM) and low abundance proteins, as potential prognostic and diagnostic candidates for patients after non-invasive tumor removal (such as ablation15,16) is highly reproducible, inexpensive, simple and also able to be adapted for high-throughput sample preparation by use of laboratory robots10,11. We have demonstrated an ability to isolate very low abundance proteins and subsequently identify them by LC-MS/MS. We have also demonstrated that it is possible to isolate glycosylated proteins IgG and IgM in amounts sufficient enough to allow for further high-throughput analyses in a large amount of patient samples11.
Footnotes
Conflicts of interest disclosure
Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number P20GM103421. The previous segment of this project was supported by the National Center for Research Resources (NCRR) under P20 RR 017695. Martina Srajer Gajdosik was supported by Fullbright scholarship.
References
- 1.Surinova S, Schiess R, Hüttelhain R, et al. On the development of plasma protein biomarkers. J Proteome Res. 2011;10:5–16. doi: 10.1021/pr1008515. [DOI] [PubMed] [Google Scholar]
- 2.Bandow E. Comparison of protein enrichment strategies for proteome analysis of plasma. Proteomics. 2010;10:1416–25. doi: 10.1002/pmic.200900431. [DOI] [PubMed] [Google Scholar]
- 3.Addona TA, Shi H, Keshishian H, et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat Biotechnol. 2011;29:639–41. doi: 10.1038/nbt.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitaeker JR, Lin C, Kennedy J, et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29:625–34. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell P. Proteomics retrenches. Nat. Biotechnol. 2011;28:665–70. doi: 10.1038/nbt0710-665. [DOI] [PubMed] [Google Scholar]
- 6.Anderson NL, Anderson ND. The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol Cell Proteomics. 2002;1:845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 7.Clifton J, Huang F, Rucevic M, et al. Protease inhibitors as possible pitfalls in proteomic analyses of complex biological samples. J Proteomics. 2011;74:935–41. doi: 10.1016/j.jprot.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Righetti PG, Fasoli E, Boschetti E. Combinatorial peptide ligand libraries: The conquest of the ‘hidden proteome’ advances and great strides. Electrophoresis. 2011;32:960–6. doi: 10.1002/elps.201000589. [DOI] [PubMed] [Google Scholar]
- 9.Evans ST, Holstein M, Cramer SM. Detection of trace proteins in multicomponent mixtures using displacement chromatography. Anal Chem. 2011;83:4184–92. doi: 10.1021/ac200486e. [DOI] [PubMed] [Google Scholar]
- 10.Srajer Gajdosik M, Clifton J, Josic Dj. Sample displacement chromatography as a method for purification of proteins and peptides from complex mixtures. J Chromatogr. 2012 doi: 10.1016/j.chroma.2012.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pucic M, Knezevic A, Vidic J, et al. High throughput isolation and glycosylation analysis of IgG-variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics. 2012. mcp. M111.010090. [DOI] [PMC free article] [PubMed]
- 12.Josic Dj, Brown MK, Huang F, et al. Use of selective extraction and fast chromatographic separation combined with electrophoreticmethods formapping of membrane proteins. Electrophoresis. 2005;26:2809–922. doi: 10.1002/elps.200500060. [DOI] [PubMed] [Google Scholar]
- 13.Huang F, Clifton J, Yang X, et al. SELDI-TOF as a method for biomarker discovery in the urine of aristolochic-acidtreated mice. Electrophoresis. 2009;30:1168–74. doi: 10.1002/elps.200800548. [DOI] [PubMed] [Google Scholar]
- 14.Yates JR, 3rd, Eng JK, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 15.Dupuy DE, Zagoria RJ, Akerley W, et al. Percutaneous radiofrequency ablation of malignancies in the lung. AJR Am J Roentgenol. 2000;174:57–9. doi: 10.2214/ajr.174.1.1740057. [DOI] [PubMed] [Google Scholar]
- 16.Dupuy DE. Image-guided thermal ablation of lung malignancies. Radiology. 2011;260:633–55. doi: 10.1148/radiol.11091126. [DOI] [PubMed] [Google Scholar]