

Abstract
Persistent infection with H. pylori confers an increased risk for the development of gastric cancer, however, the exact mechanism(s) whereby this bacterium causes carcinogenesis have not been completely elucidated. Recent evidence indicates that aberrant activation of the STAT3 signaling pathway may play a role in gastric carcinogenesis. Therefore, we hypothesized that H. pylori infection modulates STAT3 signalling, favouring gastric cancer development. In epithelial cells infected with H. pylori, STAT3 was activated as assessed by immunoblotting for phospho-STAT3, immunofluorescence of translocated STAT3, fluorescence recovery after photo-bleaching and luciferase activation in transfected cells. Activation was dependent on translocation, but not phosphorylation of CagA in host cells. Activation appeared to be receptor-mediated since pre-incubation of cells with the IL-6R superantagonist sant7 or inhibition of gp130 by a monoclonal antibody prevented H. pylori-mediated STAT3 activation. However, activation was not related to autocrine activation by IL-6 or IL-11. CagA+ wildtype H. pylori but not the non-carcinogenic cagA- mutant activated STAT3 in gastric epithelial cells in vivo in the gerbil model of H. pylori-mediated gastric carcinogenesis. Collectively, these results indicate that H. pylori CagA activates the STAT3 signaling pathway in vitro and in vivo, providing a potential mechanism by which chronic H. pylori infection promotes the development of gastric cancer.
Keywords: Helicobacter pylori, gastric cancer, CagA, STAT3, IL-6
Introduction
Gastric cancer is the second most common cause of cancer-related deaths worldwide, accounting for greater than one million deaths per year (1). Infection with Helicobacter pylori (H. pylori) is the major predisposing factor for the development of gastric adenocarcinomas (1, 2). Individuals infected with strains containing the cytotoxin-associated gene A (CagA) have an increased risk for developing gastric cancer in comparison to those infected with H. pylori strains lacking the CagA protein (1, 2). Upon infection, CagA is injected into host epithelial cells via a type IV secretion system (1, 3). Injected CagA localizes to the host cell membrane where it is targeted for tyrosine phosphorylation within a repeated specific 5 amino acid EPIYA motif (3). Once phosphorylated, CagA binds to a SRC Homology 2 (SH2) domain-containing protein tyrosine phosphatase SHP2, activating its phosphatase activity and forming a complex that initiates mitogenic signalling, and cell morphological changes involving rearrangement of the actin cytoskeleton (1). CagA is also able to bind, independent of its tyrosine phosphorylated EPIYA domain, to other host signalling molecules, including Grb2 and c-MET receptors (4, 5). CagA, independent of tyrosine phosphorylation, interacts with ZO-1 and junctional adhesion molecule (JAM), forming a complex that alters tight junctions (6). CagA can therefore interact with intracellular signalling pathways in both a tyrosine phosphorylation dependent and independent manner to perturb cellular homeostasis. Despite these elegant studies defining CagA-host cell interactions, the exact mechanism by which CagA promotes carcinogenesis is not known.
Current evidence indicates that Signal Transducers and Activators of Transcription 3 (STAT3) may be involved in the development of gastric cancer. STAT proteins are a family of latent transcriptional factors that exist as monomers within the cytoplasm of cells (7, 8). STAT3 can be activated by a number of different cytokines including IL-6, IL-11, ciliary neurotrophic factor, oncostatin M, and leukemia inhibitory factor, which all signal through a shared gp130 signal transducer receptor subunit. IL-6-induced STAT3 activation involves binding of cytokine molecules to a membrane bound glycoprotein IL-6 receptor (IL-6R) chain on target cells triggering hetero-dimerization with gp130, followed by activation of gp130 receptor associated JAK kinase. Latent STAT3 monomers are then recruited to the phosphorylated residues of the gp130 receptor, phosphorylated by Janus Kinases (JAKs 1,2) on single tyrosine resides (tyr705) resulting in their dimerization via reciprocal SH2 phospho-tyrosine interactions. Dimeric STAT3 then translocates to the nucleus, where it upregulates the transcription of various genes involved in growth and proliferation.
Numerous studies indicate that STAT3 plays a key role in promoting oncogenesis. STAT3 is constitutively activated in a variety of human neoplasms (7, 8). The development of tumors in nude mice in gain-of-function studies confirms the role of STAT3 as an oncogene (9). STAT3 activation is implicated in all aspects of carcinogenesis including cell proliferation, apoptosis, angiogenesis, invasion, migration and disruption of immune surveillance (7, 8). With respect to gastric neoplasia, STAT3 is constitutively phosphorylated in various gastric cancer cell lines (10). In addition, in vivo evidence supports a role for STAT3 activation in gastric carcinogenesis. In this robust model, mice harbouring a knock-in mutation in the gp130 receptor subunit that results in constitutive upregulation of JAK-STAT3 spontaneously develop antral tumors by 4 weeks of age (11, 12). Furthermore, recent evidence indicates that a threshold level of STAT3 activation is required to promote tumor initiation in this model (13). Thus, there is an increasing body of evidence indicating that aberrant STAT3 activation is involved in the development of gastric cancer. Since the majority of gastric cancers occur as a result of H. pylori infection, we have explored the effect of H. pylori infection on STAT3 activation both in vitro and in vivo. Furthermore, the role of the cancer associated effector protein CagA and the potential molecular mechanisms involved were defined.
Materials and Methods
Cell cultures
HEp-2 cells (ATCC CCL 23) (purchased from ATCC, Rockville, MD), INT 407 and HeLa cells were cultured in Minimum Essential Medium Eagle’s with Earle’s salts and L-Glutamine (Wisent Inc, Canada) and, supplemented with 10% heat-inactivated fetal bovine serum (Gibco BRL Life Technologies, Gaithersburg, Md.) and 1% penicillin streptomycin.
H. pylori strains and infection conditions
H. pylori strain 60190 (ATCC 43526, cagA+, cagE+, vacA+) and its corresponding isogenic cagA-, cagE-, strain 251 and its TFSS mutant cagM (provided by Dr. A. Labigne, Pasteur Institute, Paris); strain G27 and the CagA EPISA mutant (provided by Dr. M. Amieva, Stanford, CA), and gerbil adapted strain 7.13 or the corresponding isogenic cagA- 7.13 mutant were employed in these studies and cultured as described previously (for strains employed see Table in supplementary material) (6, 16)._In some experiments, H. pylori was heat killed by boiling for 15 mins in sealed, sterile Eppendorf tubes. Heat killing was confirmed by lack of growth on Columbia Agar plates. HEp-2 cells or epithelial cells as noted were serum starved (24 hrs) and incubated with bacteria for up to 4 hours at multiplicities of infection (MOI) of 50:1. Where indicated, 15-30 minutes prior to infection cells were incubated with either an IL-6 receptor antagonist, sant7 (17), (50 μg/ml; provided by Dr. M. Chatterjee, Humboldt University of Berlin, Germany), human anti-gp130 receptor antibody (50-100 μg/ml; Immunonokontact, UK) or neutralizing anti-IL-6 antibody (50ng/ml, Sigma Aldrich) for 15mins prior to infection.
Cell Lysates and Western Blotting
After 4 hrs of infection, cells were washed and scraped in 200 μl of RIPA buffer containing phosphatase and protease inhibitors (all obtained from Sigma Aldrich). Cell suspensions were centrifuged and supernatants were stored at −70°C. Protein concentrations were measured using the Biorad DC protein assay. Whole cell lysates (70μg/ml) or nuclear extracts in laemelli buffer were loaded onto 10% SDS-polyacrylamide separating gels and run at 110V for 1.5hrs at RT. Proteins were then transferred onto a nitrocellulose membrane (BioTrace NT; Pall Corp., Ann Arbor, MI, USA). Following blocking, membranes were incubated overnight with either rabbit anti- phospho-STAT3 antibody (1:250; Cell Signaling, MA, USA), rabbit anti- phospho- serine 727- STAT3 antibody (1:250l; Cell Signaling, MA, USA), mouse anti-STAT3 antibody (1:500; Cell Signaling), mouse anti-phospho-tyrosine antibody (1:200; Upstate Cell Signaling Solutions, VA, USA), or mouse anti-CagA antibody (1:200; Santa Cruz Biotechnology, CA, USA) in TBST-milk._Blots were then incubated with the corresponding secondary horseradish peroxidase-conjugated antibody (Jackson Laboratories, PA) (1 h at RT). Bands were visualized by chemiluminesence using Kodak Biomax MR film.
Densitometric Analysis
Densitometric analysis was performed using FluorChem FC11 software. All blots (n values) from each independent experiment were used. Densities of pSTAT3 and STAT3 bands were measured for each treatment and expressed as a ratio (phosphorylated STAT3/STAT3). For graphic representation, the ratio of pSTAT3 to STAT3 for each treatment was expressed relative to the measured ratio for control cells, and identified as fold increase on graphs.
Immunofluorescence and Fluorescence Microscopy
HEp-2 cells were seeded on glass coverslips and serum starved for 24hrs. Cells were then incubated with either IL-6 (100ng/ml) for 30mins or H. pylori for 4hrs. Following fixation, cells were permeabilized, blocked with 10% normal goat serum and STAT3 detected using a mouse anti-STAT3 antibody (1:100; Cell Signaling, MA, USA) and Cy3 donkey anti-mouse conjugated antibody (1:1000; Jackson Laboratories, PA). Images were captured using an inverted Carl Zeiss META Laser Scanning Confocal Microscope. Analysis of relative fluorescence intensity was performed on cells using Image J software1.
DNA Transfections
Transient transfection of GFP, STAT3-GFP plasmids (a gift from Dr. K.Watanabe, Hokkaido University, Japan) and wildtype CagA-GFP constructs were performed in HEp-2 cells using the AMAXA Nucleofector System according to the procedure outlined (AMAXA Biosystems, MD, USA). Twenty hrs post transfection, whole cell lysate extracts were collected.
ELISA
HEp-2 cells were grown to confluency in six well plates and infected with H. pylori wt strain 60190 for 4 hrs. Supernatants were collected and assayed for IL-6, IL-11 or human LIF using specific ELISAs as per the manufacturers instructions (R&D Systems, MN, USA).
Fluorescence Recovery after Photo-bleaching (FRAP)
FRAP was performed using an inverted confocal LSM 510 (Carl Zeiss, Jena, Germany) microscope equipped with an argon Laser. Coverslips with cells expressing STAT3- GFP constructs were mounted on the stage of the confocal microscope. After acquiring 2 baseline fluorescence measurements, the nucleus was bleached and the recovery in fluorescence of both areas measured over time. The fractional fluorescence recovery was expressed relative to the initial fluorescence of the nucleus, and the changes in fluorescence that occurred in unbleached cytoplasm post bleaching. Mathematical treatment and modeling was performed using Graph Pad Prism (version 3.02).
Luciferase Assays
The pSTAT3-luciferase reporter, which carries two tandem repeats of the acute phase response element of the alpha-2-macroglobulin and the p950M4/luciferase reporter, characterized by four copies of the STAT3-binding DNA element from the human angiotensinogen promoter (5′-CGTTTCTGGGAACCT-3′) cloned into pBL2Luc (provided by Dr. Sehgal, New York Medical College, New York) were transfected into HEp-2 cells using the Amaxa system (18). Transfected cells were incubated with IL-6 or infected with H. pylori, cell lysates collected and luciferase activity measured using a Promega Dual Luciferase Kit. Assays were carried out at least in triplicate, and luciferase activity expressed in arbitrary units after normalization against renilla luciferase activity
In vivo infection of Mongolian gerbils
Male Mongolian gerbils (Harlan Labs, Indianapolis) 4-8 wks of age were orogastrically challenged with either sterile Brucella broth, H. pylori strain 7.13 or an isogenic 7.13 cagA mutant as described before (16). After 6 and 12 wks, animals were euthanized and the stomachs collected and homogenized in RIPA buffer containing phosphatase and protease inhibitors. Gastric tissue lysates were probed via immunoblotting for changes in STAT3 activation using a rabbit anti- phospho-STAT3 antibody (1: 250; Cell Signaling, Massachusetts, USA). All procedures were carried out with the approval of the Institutional Animal Care Committee of Vanderbilt University.
Immunohistochemistry
Four micrometer thick sections were cut from paraffin embedded sections from control or H. pylori infected gerbil stomachs. Paraffin embedded tissue was rehydrated and antigen unmasking performed by boiling slides in 10mM Tris (pH 10.0). Sections were fixed with 4% PFA and blocked in 5% donkey serum in 0.1% triton X. Overnight incubations in rabbit anti- phospho-STAT3 antibody (1:50; Cell Signaling, MA, USA), followed by 1hr secondary Cy3 A donkey anti- rabbit conjugated antibody incubations (1:500; Jackson Laboratories, PA) were performed. DAPI (1:1000) staining was done to identify nuclear compartments. Slides were mounted using DAKO. Consecutive tissue sections were stained with haemotoxylin and eosin and graded for chronic antral inflammation and colonization density as described by Franco et al., 2008 (16).
Results
H. pylori triggers tyrosine phosphorylation of STAT3
To determine if H. pylori induces tyrosine phosphorylation of STAT3 in epithelial cells, immunoblotting of cell lysates using a phosphoSTAT3 antibody was performed. As described previously, AGS gastric epithelial cells display constitutively active STAT3 (10). Therefore HEp-2 cells, which have been employed previously as a model epithelium of signal transduction responses during H. pylori infection, which do not constitutively express activated STAT3 were employed for the remainder of the studies. HEp-2 cells treated with IL-6 (positive control) displayed increased phospho-STAT3 in comparison with uninfected cells (Fig. 1). Similarly, HEp-2 cells infected with wildtype H. pylori strains 60190 (Fig. 1) and 7.13 (suppl Fig. 1) for 4 hrs showed increased STAT3 phosphorylation in comparison with uninfected cells. In addition, the degree of phosphorylation of STAT3 increased with higher multiplicities of infection with strain 60190 (suppl. Fig. 2). H. pylori also induced STAT3 phosphorylation in INT 407 cells (suppl Fig. 3) and HeLa cells (data not shown) indicating that this finding was not restricted to HEp-2 cells.
Figure 1. H. pylori promotes phosphorylation of STAT3 pathway.
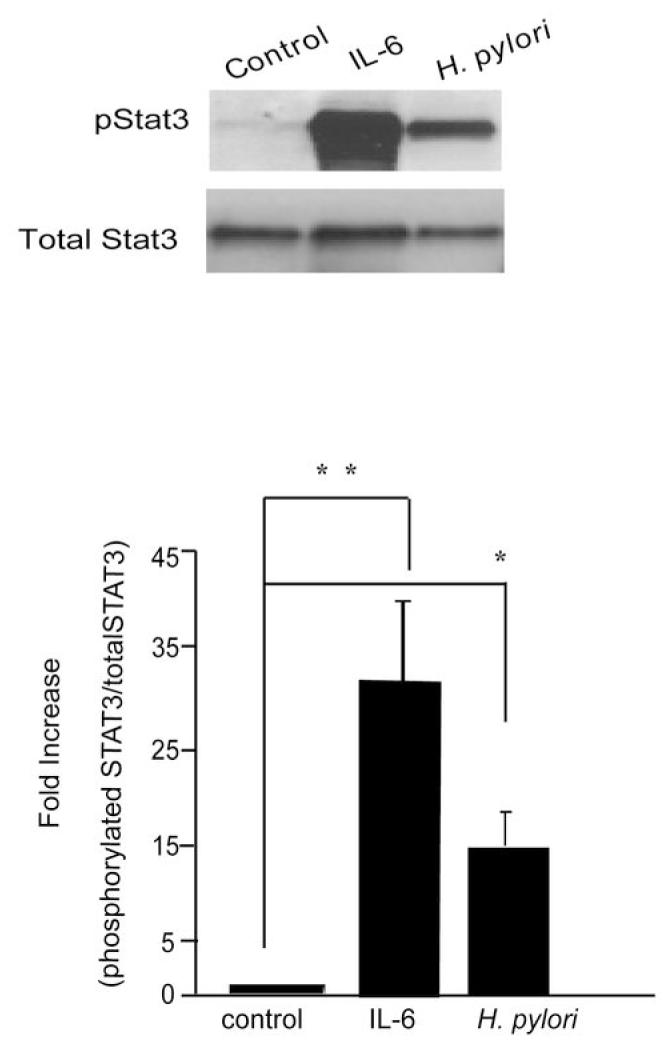
Lysates obtained from untreated HEp-2 cells, cells incubated with IL-6 (100ng/ml, 30mins), or cells infected with H. pylori 60190 (wt) (MOI of 100:1) for 4hrs were used for immunoblotting to detect changes in phosphorylated and native STAT3. The graph represents densitometric analysis of the bands obtained for each protein signal normalized to native STAT3. Fold increase given as a ratio of control pSTAT3/STAT3. Results expressed as mean +/- S.E. (** p < 0.01 and * p < 0.05 using the Student’s t test, n=8)
H. pylori infection induces nuclear translocation of STAT3
Next, nuclear translocation of STAT3 was determined by STAT3 immunolabelling. Control cells displayed a greater distribution of STAT3 in the cytoplasm, with ghost-like nuclei (Fig. 2A). In contrast, IL-6 treated cells displayed highly fluorescent nuclei (Fig. 2B). Similarly, nuclear translocation of STAT3 was observed in cells infected with H. pylori strain 7.13 (Fig. 2C). To assess the dynamics of the process, fluorescence recovery after photobleaching was employed. Cells transfected with GFP-tagged STAT3 proteins were targeted with high intensity laser light, which bleaches GFP, but does not affect the function of endogenous STAT3 protein. The movement of non-bleached molecules into the bleached region was then monitored over time to measure the percentage recovery post-bleaching. Following bacterial infection, the nuclei of STAT3-GFP transfected cells were bleached using 100% laser photon output at 488nm, and the recovery kinetics observed. Uninfected cells recover fluorescence of the entire nuclei by 79% ± 3.3; however IL-6 activated cells showed minimal recovery (29% ± 2.5) (Fig. 2D). Similarly, a reduction in recovery of fluorescence was detected in the nuclei of H. pylori-infected cells post bleaching (39% ± 2.6%), indicating that the majority of STAT3-GFP was localized in the nucleus of infected cells, thereby permitting minimal recovery from the cytoplasm post nuclear bleaching. Taken together, these results indicate that H. pylori infection triggers STAT3 nuclear translocation.
Figure 2. H. pylori induces STAT3 nuclear translocation and transcriptional activation in a CagA-dependent manner.

(A-C) HEp-2 cells were treated with culture media (A), IL-6 for 30mins (B) or infected with H. pylori strain 7.13 (C) for 4hrs. Fixed cells were stained with anti-STAT3 and donkey anti-mouse Cy3 conjugated secondary antibodies. Confocal images were taken of representative cells for each treatment.
(D) Whole nuclei of uninfected, IL-6-treated and H. pylori infected STAT3-GFP transfected cells were bleached and the percentage recovery of fluorescence was measured relative to the initial values. Six cells for each group were analyzed. (* p <0. 05 using the Student’s t test, n=4). HEp-2 cells were transfected with luciferase reporter constructs containing three repeat STAT3 DNA binding domains, p950m4 or control pSTAT3 plasmids. Transfected cells were treated with IL-6 or infected with H. pylori strain 60190 for 48hrs and luciferase activity was measured and normalised against renilla luciferase activity. (* p <0.05 using the Student’s t test, n=3).
H. pylori induces STAT3 transcriptional activity
To test the ability of H. pylori to activate STAT3 signaling at the level of transcription, a STAT3- luciferase reporter system was employed. As shown in Figure 2D, IL-6 treatment of p950M4-transfected cells enhanced luciferase activity relative to control cells. Similarly, p950M4-transfected cells infected with H. pylori strain 60190 demonstrated an increase in relative luciferase activity in comparison with transfected, uninfected control cells (Fig. 2D). H. pylori infection induces comparable levels of transcriptional activity of STAT3 as IL-6 treatment (P=NS), despite promoting a lower level of pSTAT3 (Figure 3A). Therefore, we compared phosphorylation of STAT3 serine 727, which is thought to regulate maximal transcriptional activity of STAT3 regulated genes, in IL-6 treated and H. pylori infected cells. No differences in levels of STAT3 serine 727 phosphorylation were seen in whole cell extracts from IL-6 treated or H. pylori-infected cells (Suppl. Fig. 4B). However, nuclear extracts collected from IL-6 treated cells showed greater serine phosphorylated STAT3 in comparison to H. pylori infected cells (Suppl. Fig. 4B).
Figure 3. H. pylori-mediated STAT3 activation is CagA dependent.

(A) HEp-2 cells were infected with either H. pylori strain 60190, its isogenic cagA mutant or the type IV secretion system mutant cagE (MOI of 100:1 for 4 hrs). Whole cell lysates were collected and probed for STAT3 activation. Untreated and IL-6 treated cells (50ng/ml) served as controls. Graph depicts the densitometric analysis for pSTAT3 bands relative to the native STAT3 protein (* p < 0.05 using the Student’s t test, n=4).
(B) (i) Untreated control (ii) H. pylori wildtype or (iii) cagA mutant infected HEp-2 cells were immunolablled for STAT3 and images obtained using confocal micrscopy.
(C) HEp-2 cells were transfected with untagged GFP or CagA-GFP for 20hrs, cell lysates collected and probed for phosphoSTAT3.
H. pylori-mediated STAT3 activation is CagA-dependent
Since infection with CagA is associated with an increased risk for the development of gastric cancer, we next assessed the role of this effector protein in H. pylori mediated STAT3 activation. Infection with cagA isogenic mutants of strain 61090 failed to activate STAT3 (Fig. 3A). Similarly, cagE mutants, possessing a disrupted type IV secretion system, were also unable to induce STAT3 phosphorylation during infection. In addition, infection with heat-killed bacteria, which destroys heat-labile proteins such as CagA but does not affect LPS, resulted in an inhibition of H. pylori mediated STAT3 activation (suppl. Fig 5). These findings were confirmed using an additional H. pylori Cag+ isolate, strain 251 and its type IV secretion system mutant lacking cagM (suppl Fig. 6). In addition, cells infected with the 7.13 cagA mutant (Fig. 3Biii) did not display nuclear STAT3 immunolabelling. Furthermore, HEp-2 cells transfected with CagAGFP for 20hrs displayed STAT3 activation as assessed by Western blotting for phospho-STAT3 (Fig. 3C) compared to cells transfected with GFP alone. Taken together these findings indicate that CagA translocation is essential for STAT3 activation.
CagA- mediated STAT3 activation is independent of tyrosine phosphorylation
To determine if tyrosine phosphorylation of the CagA EPIYA motif is required to induce STAT3 phosphorylation, we utilized an H. pylori cagA EPISA mutant, which cannot undergo tyrosine phosphorylation due to a tyrosine (Y) to serine (S) substitution. Infection of HEp-2 cells with either the wildtype parental strain G27 or the EPISA mutant induced comparable levels of STAT3 phosphorylation (Fig. 4A). To confirm that the cagA EPISA mutant did not undergo tyrosine phosphorylation, the same whole cell lysates already probed for pSTAT3 (shown in Fig 4A), were immunoblotted for the presence of a tyrosine phosphorylated protein at molecular weight of CagA (128- 144kDa) (3). As depicted in Fig. 4B, the cagA EPISA mutant injects CagA with a shift in the molecular weight as has been shown previously (19). However, unlike injected CagA from the wildtype G27 strain, tyrosine phosphorylation of the CagA EPISA mutant protein was not detected (Fig. 4B).
Figure 4. CagA-dependent STAT3 activation does not require tyrosine phosphorylation of CagA.

(A) Cells were infected with strain G27 or its cagA EPISA mutant. Cell lysates were probed for phosphoSTAT3. Graph depicts the density of pSTAT3 protein bands normalized to native STAT3 detected in each treatment (* p < 0.05 using the Student’s t test, n=6).
(B) Lysates initially probed for pSTAT3 were subsequently analyzed by immunoblotting using a tyrosine phosphorylation specific antibody to detect the presence of tyrosine phosphorylated CagA.
The IL-6 receptor and gp130 are pivotal for H. pylori STAT3 activation
To determine the requirement for IL-6 receptors in H. pylori-mediated STAT3 activation, Sant7, a highly specific receptor antagonist was employed (17). In cells pre-incubated with Sant7, IL-6 mediated pSTAT3 was significantly reduced (Fig. 5A). Similarly, Sant7 completely abrogated the pSTAT3 signal mediated by H. pylori infection. Next an anti-gp130 mAb (15), which prevents receptor dimerization and activation of the receptor was employed. As shown previously (17), IL-6 induced STAT3 activation was inhibited by the anti-gp130 mAb. Similarly, H. pylori-induced STAT3 phosphorylation was eliminated by the anti-gp130 mAb (Fig. 5B). Taken together, these findings indicate that both the IL-6 receptor and the gp130 receptor subunits were required for H. pylori mediated STAT3 phosphorylation.
Figure 5. H. pylori-mediated STAT3 activation requires the IL-6R and the gp130 receptor but is independent of the IL-6 cytokine.

(A) Cells were pre-incubated with the IL-6 receptor super-antagonist Sant7 prior to incubation with IL-6 (50ng/ml, 30mins) or infection with strain 60190. Lysates were probed for pSTAT3 and native STAT3 protein. Representative of three independent experiments.
(B) An anti-gp130 blocking Ab was incubated with cells prior to either H. pylori infection or incubation with IL-6, cell lysates obtained and immunoblotted for pSTAT3. Representative of three independent experiments.
(C) HEp-2 cells were treated with neutralizing IL-6 Ab followed by infection with strain 60190. Whole cell lysates were collected and probed for pSTAT3 via Western Blotting Densitometric analysis showing the ratio of pSTAT3 normalized to native STAT3 was performed and represented in the graph (* p <0.05 using Student’s t test, n=4).
Interaction between CagA and gp130 receptor
In order to determine if there was a direct interaction between CagA and the gp130 receptor, we attempted to perform co-immunoprecipitation experiments in cells transfected with CagA-GFP and gp130-CFP (See Methods in Supplementary materials). Although we were able to co-immunoprecipitate CagA and SHP-2 (suppl. Fig. 7), a known interacting partner of CagA, we were unable to co-immunoprecipitate gp130 with CagA indicating that CagA likely does not interact directly with gp130 (data not shown).
H. pylori-mediated STAT3 activation is IL-6, IL-11 and LIF independent
Since both IL-6 and IL-11 can induce STAT3 activation through the gp-130 receptor we next determined if autocrine activation by IL-6 or IL-11 was involved. IL-6 was measured in culture media from cells infected with either H. pylori strain 251 or the cagM mutant. IL-6 was undetectable in three separate groups of control HEp-2 cells and cagM infected cells. In H. pylori wildtype infected cells, minimal IL-6 was detected (19.22 pg/ml) (Table 2). Furthermore, this concentration of IL-6 was insufficient to activate STAT3 signaling (suppl. Fig. 8). To confirm that H. pylori induced STAT3 activation was independent of IL-6, we employed a neutralizing anti-IL-6 antibody. Upon pre- incubation with the anti-IL-6 antibody, the degree of IL-6-mediated pSTAT3 was significantly reduced (Fig. 5C). In contrast, H. pylori strains 60190 and 251 still induced STAT3 phosphorylation in the presence of the neutralizing antibody. These findings indicate that autocrine activation of STAT3 by IL-6 was not involved in H. pylori-mediated STAT3 phosphorylation. In addition, wild-type H. pylori infection of HEp-2 cells did not result in an increase in IL-11 in culture supernatants (Table 2). Furthermore, the sant7 inhibitor that blocks IL-6 and H. pylori mediated STAT3 activation had no effect on IL-11 mediated STAT3 activation (suppl. Fig. 9). Furthermore, LIF a known activator of the STAT3 signaling pathway was not increased in supernatants collected from H. pylori infected cells, (Table 2). These results suggest that H. pylori mediated STAT3 activation occurs independently of IL-6, IL-11 and LIF.
A gerbil-adapted carcinogenic CagA+ strain activates STAT3 signalling in vivo
Infection with H. pylori strain 7.13 induces dysplasia and gastric adenocarcinoma in Mongolian gerbils (16). To determine if H. pylori induced STAT3 activation in vivo, gerbils were infected with wild-type strain 7.13 or sham infected (Fig. 6) and sacrificed at 6 or 12 weeks. Gastric tissue lysates obtained from sham infected animals displayed minimal phospho-STAT3. In marked contrast, gastric tissue lysates obtained from gerbils infected with wildtype strain 7.13 displayed increased phospho-STAT3 following both 6 (suppl. Fig. 10) and 12 weeks of infection (Fig. 6). To determine if CagA was required for STAT3 activation in vivo, we compared STAT3 activation in gastric tissue lysates obtained from gerbils infected with the non-carcinogenic isogenic 7.13 cagA mutant (20). As shown previously, CagA+ infected gerbils demonstrated a higher degree of chronic antral inflammation in comparison to CagA- infected animals, however there was greater colonization detected in CagA- infected gerbil stomachs (Fig. 6Bi-ii, supplementary Fig. 11). In contrast to wild-type infected gerbils, following 12 weeks of infection, phospho-STAT3 activation was not detected in gerbils infected with the cagA mutant strain (Fig. 6A). These findings indicate that infection with the known carcinogenic H. pylori strain 7.13 (16) induces STAT3 activation in a CagA dependent manner. To determine the specific cell types exhibiting STAT3 activation, immunohistochemistry was performed on gastric tissue sections. STAT3 nuclear translocation was detected in gastric epithelial cells in a patchy distribution in the antrum of stomachs from CagA+ H. pylori- infected animals (Fig. 6C iv). In addition, nuclear STAT3 was detected in inflammatory cells within infiltrates (data not shown). In marked contast, nuclear STAT3 staining was not detected in gastric tissue obtained from sham infected or H. pylori CagA- infected gerbils (Fig. 6 C ii, vi)
Figure 6. H. pylori carcinogenic strain 7.13 induces STAT3 activation in vivo in a CagA dependent manner.

(A) Stomach sections from Mongolian Gerbils sham infected (n=4) or infected with H. pylori strain 7.13 (n=4) or its isogenic cagA- mutant (n=4) for 12 weeks were collected and homogenized to obtain protein lysates. Lysates were probed for pSTAT3, STAT3 and actin. Densitomtric analysis was performed and depicted in graph (* p<0.05 using Student’s t test)
(Bi) Severity of Inflammation within the gastric antrum of animals infected with carcinogenic H. pylori strain 7.13. Chronic antral gastritis was scored from numbers 1 to 3 (14) in control, CagA+ and cagA- infected gerbil stomachs and mean values represented on scatter plots. (* p <0.05 for infected gerbils compared to control sham infected animals, **p< 0.05 for H. pylori CagA+ infected gerbils compared with H. pylori cagA- animals).
(Bii) Colonization density was quantitated using a standardized grading system (14) and data presented in scatter plot (p< 0.05 for control animals compared to infected gerbils (* p <0.05 for infected gerbils compared to control sham infected animals)
(C) Immunohistochemistry showing DAPI and STAT3 staining of cells within the antral mucosa. Panels on the left show DAPI stained sections from (i) control (iii) H. pylori CagA+ and (v) H. pylori cagA- infected stomachs (x 10 magnification). White boxes highlight the corresponding regions magnified in panels (ii), (iv) and (vi) (X 63 magnification) showing anti-pSTAT3 staining (red), and DAPI (blue) co-staining. Spinning Disk Microscopy was utilized to obtain images represented.
Discussion
Although current evidence strongly supports a role for STAT3 activation in the development of gastric cancer, the exact mechanism of STAT3 activation is still unknown. Here we demonstrate that infection with H. pylori, the single most important risk factor for the development of gastric cancer, induces STAT3 activation both in vitro and in vivo. In addition, our studies define a role for the cancer-associated effector CagA, independent of tyrosine phosphorylation, in STAT3 activation.
We have shown that H. pylori-mediated STAT3 activation occurs at the IL-6 receptor level but is independent of the known activation ligands IL-6, IL-11, and LIF indicating a novel mechanism involving the interaction of both host receptors and CagA. Previous studies indicate that forced dimerization of the gp130 receptor promotes STAT3 activation independent of the IL-6 cytokine (21). Therefore, we hypothesize that CagA modulates STAT3 activation by promoting receptor dimerization either directly or indirectly. In support of this contention, recent evidence indicates that CagA may interact with several membrane associated proteins and signalling molecules. For example, Churin and colleagues (5) demonstrated that CagA in a tyrosine phosphorylation independent manner interacted directly with c-Met in AGS gastric epithelial cells. Mimuro and colleagues (4) demonstrated that CagA, independent of its EPIYA tyrosine phosphorylation motif, associated with the intracellular signalling molecule Grb2. In addition, Amieva et al. (6) demonstrated that in addition to altering the location ZO-1 and JAM, CagA, independent of phosphorylation, also altered localization of apical and basolateral membrane proteins in MDCK cells (22). Based on our findings, we hypothesize a similar mechanism whereby CagA triggers clustering of the IL-6 and gp130 membrane receptors, favouring STAT3 activation observed during H. pylori infection. Based on the results of our coimmunoprecipitation studies, there does not appear to be a direct interaction between CagA and gp-130 to promote gp-130 clustering. Further studies are now required to elucidate the exact mechanism involved.
Other bacterial pathogens can modulate the STAT3 intracellular signalling pathway, likely to promote infection. In contrast to our findings, the majority of these bacterial pathogens modulate STAT3 activation via autocrine activation by IL-6. For example, Listeria monocytogenes induces STAT3 activation in an IL-6-dependent manner in Kupffer cells during the early stages of infection (23). Similarly, Escherichia coli-induced pneumonia is accompanied by an increase in IL-6, leading to the activation of STAT3 in the lungs of infected mice, which facilitates improved bacterial clearance (24). Lin and colleagues demonstrated that Salmonella enterica serovar Typhimurium activates STAT3 in leukocytes and macrophages early during infection and hypothesized that STAT3 activation would reduce the initial destructive inflammatory response (25). However, the mechanism responsible for Salmonella-mediated STAT3 activation in these immune cells was independent of IL-6 but was not defined.
To promote their survival, many pathogens employ techniques to commandeer host cell signal transduction responses. This is particularly important for organisms which promote chronic infection such as H. pylori. We have previously shown that H. pylori alters other STAT signalling pathways thereby modulating host immune responses. For example, H. pylori infection does not by itself effect STAT1 activation but disrupts IFNγ-induced STAT1 activation in epithelial cells which could lead to a reduction in the Th1 immune response (26). Of interest, reciprocal interactions between STAT1 and STAT3 by several proposed mechanisms have been described (27). Therefore it remains possible that as yet unidentified effects on STAT1 may enhance H. pylori-mediated STAT3 transcriptional activity providing a possible explanation for the comparable activation in H. pylori and IL-6 treated cells.
H. pylori also disrupts IL-4-mediated STAT6 signalling in epithelial cells, inhibiting a Th2 immune response, which is important for eliminating the pathogen (28). H. pylori-mediated disruption of both STAT1 and STAT6 occurs in a cagA, cagE, and vacA independent manner. In contrast, in the present study, the injection of CagA is essential to facilitate STAT3 activation. In addition to STAT3’s direct role in tumorigenesis, STAT3 is well recognized to facilitate immune evasion of tumors at many levels including suppressing pro-inflammatory responses by inhibiting dendritic cell maturation and promoting development of T regulatory cells (8). Of interest, recent evidence indicates that the insufficient immune response observed during H. pylori infection may be mediated, in part, by both abnormal T regulatory (Treg) cells (29, 30) and inhibition of dendritic cell activation (31). In contrast to playing a role in immune surveillance, seemingly paradoxically STAT3 has also been implicated in modulating certain inflammatory responses, which promote cancer including both inhibition of IL-12 and upregulation of IL-23 (8). In this study, we detected nuclear STAT3 both in epithelial cells and in inflammatory infiltrates of CagA+ infected gerbils. Thus, H. pylori-mediated STAT3 activation may facilitate both immune evasion and pro-carcinogenic inflammatory responses in vivo.
Our findings of CagA- mediated activation of STAT3 in gastric epithelial cells in H. pylori infected gerbils, which develop gastric adenocarcinoma in a CagA- dependent manner (20), raises the hypothesis that STAT3 activation plays a role in oncogenesis in this model. In support of our in vivo findings, a recent study from Jackson and colleagues indicates that STAT3 activation is also detected in gastric tissue obtained from H. pylori-infected human subjects (32). In this study, similar to our results, STAT3 activation was augmented in subjects infected with a CagA positive strain. A variety of functional effects are attributed to the oncogenic potential of STAT3 including: proliferation, apoptosis, angiogenesis, invasion, migration and disruption of immune surveillance (7). In contrast, inhibiting STAT3 activation in tumor cells promotes cell death, inhibits angiogenesis, and triggers anti- tumor immune responses (7). Furthermore, tumor cells appear to be more sensitive to these effects compared with normal cells (7, 8). Thus, STAT3 may represent an ideal target for cancer therapy. Our studies indicate that the Mongolian gerbil model may prove useful to more precisely define the role of STAT3 in the multi-step process of gastric carcinogenesis and the effect of STAT3 inhibition on lesion progression.
In conclusion, this study identifies the H. pylori effector protein CagA as potentially enhancing the risk for gastric cancer by increasing STAT3 signaling in epithelial cells. The prognosis for gastric cancer is poor with a limited five year survival rate (1). Currently, there is no conclusive evidence that eradication of H. pylori infection prevents the development of gastric cancer (33). Our results indicate that inhibition of STAT3 activation may be a viable therapy for H. pylori-mediated carcinogenesis. Thus, the findings from our studies set the stage for ongoing investigations addressing the efficacy of STAT3 inhibition for chemoprevention during H. pylori-mediated gastric carcinogenesis.
Supplementary Material
Acknowledgements
This work was supported by funds from an operating grant from the Canadian Institute for Health and Research and the Ontario Ministry of Research and Innovation Early Researcher Award (Nicola L. Jones) and NIH (DK 58587), NIH (CA 77955) and NIH (DK 73902) to Richard M. Peek Jr.
Support: CIHR (Nicola L. Jones)
Glossary
Abbreviations used in this paper
- H. pylori
- STAT3
- pSTAT3
- IL-6R
- Ab
Footnotes
No conflicts of interest exist
Website for accessing Image J software: (http://rsb.info.nih.gov/ij/).
Reference List
- 1.Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 2.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;3:449–90. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–94. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 4.Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2000;10:745–55. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 5.Churin Y, Al Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann MJ. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. Cell Biol. 2003;161:249–55. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–4. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–5. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 9.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 10.Kanda N, Seno H, Konda Y, Marusawa H, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921–9. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 11.Judd LM, Alderman BM, Howlett M, et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 12.Tebbutt NC, Giraud AS, Inglese M, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–97. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 13.Judd LM, Bredin K, Kalantzis A, Jenkins BJ, Ernst M, Giraud AS. STAT3 activation regulates growth, inflammation, and vascularization in a mouse model of gastric tumorigenesis. Gastroenterology. 2006;131:1073–85. doi: 10.1053/j.gastro.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark G. Mutant cells that do not respond to Interleukin-1 (IL-1) reveal a noel role for IL-1 receptor-associated kinase. Mol and Cell Biol. 1999;19:4643–52. doi: 10.1128/mcb.19.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-κB and IRF3 diverges at Toll-IL-1 receptor domain- containing adapter inducing IFN-β. Proc Natl Acad Sci USA. 2004;101:3533–8. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franco AT, Israel DA, Washington MK, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–51. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee M, Honemann D, Lentzsch S, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2006;100:3311–8. doi: 10.1182/blood-2002-01-0102. [DOI] [PubMed] [Google Scholar]
- 18.Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–8. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]
- 19.Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl W, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Micro. 2002;43:971–80. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 20.Franco A, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–87. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuhlmann-Laeisz C, Lang S, Chalaris A, et al. Forced dimerization of gp130 leads to constitutive STAT3 activation, cytokine-independent growth, and blockade of differentiation of embryonic stem cells. Mol Biol Cell. 2006;17:2986–95. doi: 10.1091/mbc.E05-12-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci USA. 2005;102:16339–44. doi: 10.1073/pnas.0502598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregory SH, Wing EJ, Danowski KL, van Rooijen N, Dyer KF, Tweardy DJ. IL-6 produced by Kupffer cells induces STAT protein activation in hepatocytes early during the course of systemic listerial infection. J Immunol. 1998;160:6056–61. [PubMed] [Google Scholar]
- 24.Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis. 2006;193:360–9. doi: 10.1086/499312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin T, Bost KL. STAT3 activation in macrophages following infection with Salmonella. Biochem Biophys Res Commun. 2004;321:828–34. doi: 10.1016/j.bbrc.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell DJ, Huynh HQ, Ceponis PJ, Jones NL, Sherman PM. Helicobacter pylori disrupts STAT1-mediated gamma interferon-induced signal transduction in epithelial cells. Infect Immun. 2004;72:537–45. doi: 10.1128/IAI.72.1.537-545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Regis G, Pensa S, Boselli D, Novelli F, Poll V.Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signaling Sem Cell Biol Devel 2008; epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 28.Ceponis PJ, McKay DM, Menaker RJ, Galindo-Mata E, Jones NL. Helicobacter pylori infection interferes with epithelial Stat6-mediated interleukin-4 signal transduction independent of cagA, cagE, or VacA. J Immunol. 2003;171:2035–41. doi: 10.4049/jimmunol.171.4.2035. [DOI] [PubMed] [Google Scholar]
- 29.Lundgren A, Stromberg E, Sjoling A, et al. Mucosal FOXP3-expressing CD4+ CD25 high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73:523–31. doi: 10.1128/IAI.73.1.523-531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rad R, Brenner L, Bauer S, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–37. doi: 10.1053/j.gastro.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Kao JY, Rathinavelu S, Eaton KA, et al. Helicobacter-pylori secreted factors inhibit dendritic cell IL-12: a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol. 2006;291:G73–81. doi: 10.1152/ajpgi.00139.2005. [DOI] [PubMed] [Google Scholar]
- 32.Jackson CB, Judd LM, Menheniott TR, et al. Augmented gp130-mediated cytokine signaling accompanies human gastric cancer progression. J. Pathol. 2007;213:140–51. doi: 10.1002/path.2218. [DOI] [PubMed] [Google Scholar]
- 33.Wong BC, Lam SK, Wong WM, et al. China Gastric Cancer Study Group. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–94. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.