

In the past five years, the covalent mechanochemistry of polymeric1 and small molecule2 systems has generated a great deal of interest, in terms of both material science and synthetic chemistry. Most studies to date have focused on systems with a single mechanophore per polymer chain,3–7 an approach that has enabled the discovery of new mechanically induced chemistry and tools for stress monitoring. Single molecule architectures are likely to be limited, however, for applications in stress-responsive mechanical properties or scalable stoichiometric reactivity, due to low mechanophore concentration.8 gem-Dihalocyclopropane mechanophores have demonstrated a rich array of mechanochemical activity,9–12 and their post polymerization addition to polybutadiene based polymers has yielded the highest mechanophore content13 and highest single-chain toughness14 demonstrated in a synthetic polymer to date. While simple to synthesize, olefin containing polymers suffer from inherent instability due to ambient light, heat, and oxygen, often causing uncontrolled crosslinking. Furthermore, it is difficult to create complex architectures based on the post polymerization modification of polybutadiene.
In considering more robust and useful synthetic approaches, we start from the desire to engineer non-scissile mechanophores, which typically means that ring systems must be embedded along the polymer backbone.15 Backbone rings cannot easily be introduced by the traditional polymerization of vinyl monomers, and although ring-opening metathesis polymerization (ROMP) has been used,11 the synthesis of complex fused ring monomers can be cumbersome. Furthermore, the ROMP methodology produces olefin-containing backbones whose stability is limited by the factors discussed above. While a rich array of functional groups can easily be incorporated into condensation polymers, mild and controllable polycondensation procedures typically generate polymers of low molecular weight (MW). This limits both bulk material utility and mechanochemical activation by pulsed ultrasound, where high molecular weights (> 40 kDa) are typically required to experience sufficient shear forces along the polymer backbone. Here, we report that the synthesis of an ABA triblock copolymer harvests the advantages of polycondensation and radical polymerization, giving access to stable, mechanophore-rich polymers of desirable molecular weight.
Results and Discussion
Recently, Soucek16 and Oh17 have described the chain-extension of polyester based macroinitiators by controlled radical polymerization (CRP). From a design perspective, this approach appeared ripe for use in mechanochemical systems. Under ultrasound induced elongational flow, forces tend to accumulate about the center of a polymer chain,18,19 while in the bulk either physical entanglements or thermoplastic domains are required for the accumulation of sufficient stress for activation. In an ABA triblock copolymer, the B block would then represent a potential “sweet spot” in terms of mechanochemical activation. The general strategy was to build a mechanophore rich block by polycondensation and utilize chain extension by CRP to yield a high MW, mechanically active material featuring the known mechanophore gem-dichlorocyclopropane (gDCC),13 which undergoes a mechanically-accelerated transformation to a 2,3-dichloroalkene (Fig. 1). The synthesis (Scheme 1) began with a polyesterification procedure based on that of Moore and Stupp.20 A gDCC containing diol (Diol 1) was reacted with stoichiometric glutaric acid in the presence of diisopropylcarbodiimide (DIC) and dimethylaminopyridinium toluenesulfonate (DPTS) to suppress N-acylurea termination. After 24 hours, an excess of Diol 1 was added to ensure the formation of a predominantly α,ω-hydroxy ditelechelic polymer (Polymer 1). Esterification of the endgroups in the presence of α-bromoisobutyryl bromide generated the difunctional, mechanophore-laden macroinitiator (Polymer 2) with Mn = 26 kDa and PDI = 1.50 by SEC-MALS. Polymer 2 was then chain extended (Fig. 2) under standard single-electron transfer living radical polymerization (SET-LRP) conditions21 in DMSO with methyl acrylate (MA) to generate a 136 kDa, 1.17 PDI triblock copolymer (Polymer 3).
Fig. 1.

Mechanophore rich ABA triblock copolymers
Scheme 1.
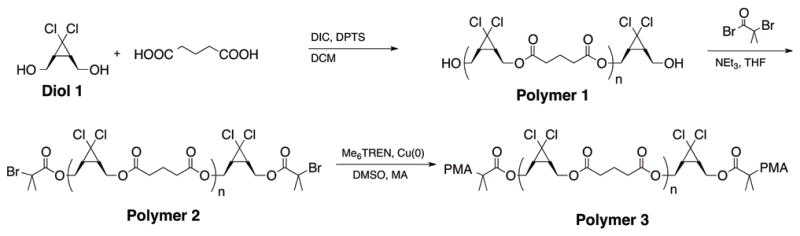
Synthesis of high mechanophore content ABA triblock copolymers.
Fig. 2.
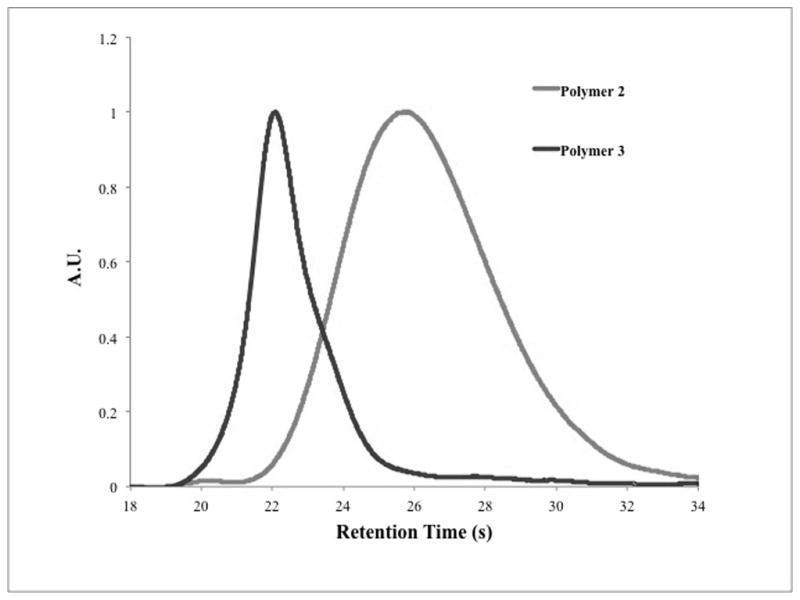
SEC-Refractive Index trace of chain extension of Polymer 2 (right) by SET-LRP to generate Polymer 3 (left)
The mechanochemical activity of Polymer 3 was then tested by subjecting it to pulsed ultrasound in acetonitrile at 6–9 °C under N2. These conditions mimic those previously employed by our group, and the expected conversion of gDCC units to 2,3-dichloroalkenes was indeed observed, with increased conversion as a function of sonication time (Fig. 3). The extent of ring opening was determined by 1H NMR, and the MWs were determined by SEC-MALS for various sonication times. After 1 scission cycle, where the molecular weight has been halved, the percent ring opening was determined to be 55%. Estimating a degree of polymerization (DPN) of 98 (See S.I.) for the mechanophore block, this corresponds to approximately 54 ring opening events per chain scission. This conversion, as a function of sonication time, is summarized in Figure 3, showing the gradual appearance of peaks at δ = 4.37, 4.70. 4.76, and 6.10 ppm corresponding to the 2,3-dichloroalkene products. For reference, previous studies on polybutadiene-gDCC copolymers showed a 35% ring opening per scission cycle;13 here we attribute the apparent increase in activity to the block architecture, where the gDCC functionalities are highly localized about the center of the chain, so that a higher fraction of them experience the force necessary for activation.
Fig. 3.
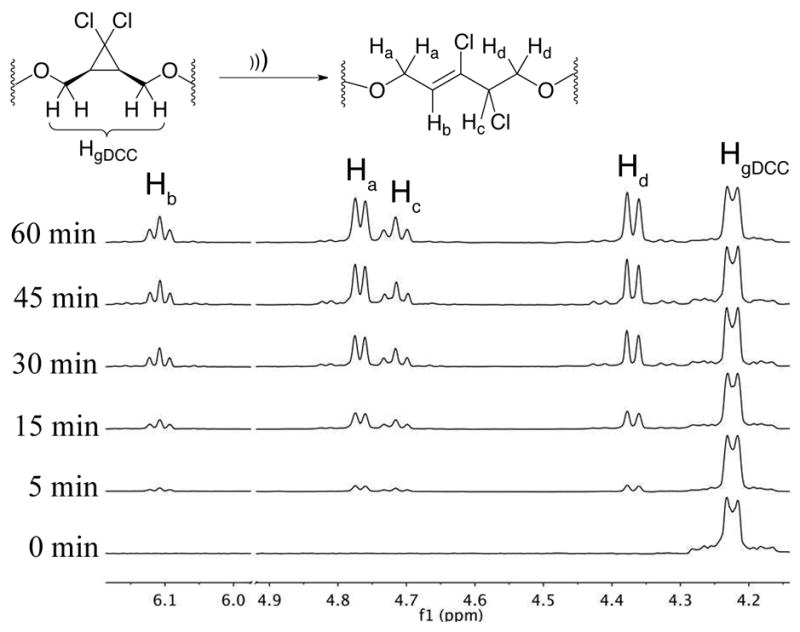
Mechanochemical activation of gDCC mechanophores by 1H NMR with increasing sonication times.
During the course of these studies, we noted a dramatic increase in the stability of Polymer 3 compared to analogous polybutadiene based systems. In our experience, casual handling of polybutadiene or ROMP derived gDHC polymers tends to result in a loss of solubility over time, often in less than one week, but we were able to dissolve and characterize Polymer 3 after one month at room temperature in the presence of light with no measurable change in MW or 1H NMR spectrum. The lack of olefin functionality can most likely be credited with this increase in stability, which in our experience is similar to that of simple acrylate polymers synthesized by copper catalyzed CRP.
Experimental
Materials and Methods
Diol 122 and 4-(Dimethylamino)pyridinium-4-toluenesulfonate (DPTS)20 were synthesized as previously described. Methyl acrylate was purchased from Sigma Aldrich and passed through a short column of basic alumina to remove inhibitor. Dry solvents (THF and DCM) were obtained from a Pure Solv™ solvent purification system. All other reagents were purchased from Sigma Aldrich and used without further purification.
Size Exclusion Chromatography (SEC) was performed on an in-line two column system (Agilent Technology PL gel, 103 and 104 Å) using inhibitor-free tetrahydrofuran (THF) as mobile phase. Molecular weights were calculated using an inline Wyatt Dawn EOS multi-angle light scattering (MALS) detector and a Wyatt Optilab DSP Interferometric Refractometer (RI). The dn/dc values were determined by on-line calculation using injections of known concentration and mass. 1H NMR was conducted on either a 400 MHz or 500 MHz Varian spectrophotometer and the residual solvent peak (CDCl3, 7.26 ppm) was used as the chemical shift reference. Fourier transform infrared spectroscopy was performed on a Nicolet 6700 FT-IR spectrophotometer.
Ultrasound experiments were performed in acetonitrile on a Vibracell Model VCX500 operating at 20 kHz with a 13.1 mm replaceable titanium tip probe from Sonics and Materials (http:www.sonics.biz/). Sonications were performed at polymer concentrations of 2.5 mg/mL in 16 mL of acetonitrile. Solutions were sparged with bubbling N2 for the thirty minutes prior to sonication. Sonications were performed at 6–9 °C in an ice-water bath at 35% amplitude (~12.5 W*cm−2) with a pulse sequence of 1 s on/1 s off.
Synthesis of Polymer 1
Using a modified procedure by Moore and Stupp:20 Diol 1 (1.00 g, 5.85 mmol), glutaric acid (772 mg, 5.85 mmol), and DPTS (687 mg, 2.34 mmol) were added to a 25 mL 2-neck round bottom flask and flushed with argon. Dry CH2Cl2 (10 mL) was added by syringe and the solution was heated to 37 °C while stirring until solution became homogenous. After cooling to room temperature, DIC (2.73 mL, 17.6 mmol) was added dropwise by syringe and the solution was allowed to stir under argon for 24 hours. An additional portion of Diol 1 (200 mg, 1.16 mmol) and DIC (0.91 mL, 5.85 mmol) were added to ensure hydroxy end-functionalization. The solution was allowed to stir for an additional 24 hours then precipitated twice from CH2Cl2 into MeOH to yield 698 mg (44% based on initial monomer mass minus 11.7 mmol (187 mg) H20) of a white gummy polymer.
1H NMR (400 MHz, CDCl3): δ = 4.20–4.30 (br d, 4H), 2.43–2.49 (br t, 4H), 2.10–2.17 (br t, 2H), 1.95–2.04 (br m, 2H); 13C NMR (101 MHz, CDCl3): δ = 172.20, 61.46, 59.98, 32.72, 30.79, 19.75; SEC-MALS: Mn = 21,500, PDI = 1.70, dn/dc = 0.072
Synthesis of Polymer 2
Polymer 1 (626 mg, 0.029 mmol) was dissolved in 5 mL dry THF in a 10 mL round bottom flask with stirbar. After purging with argon, triethylamine (0.061 mL, 0.44 mmol) was added and the solution cooled to 0 °C. Bromoisobutyryl bromide (0.036 mL, 29 mmol) was added dropwise and the solution was allowed to warm to room temperature and stir overnight. The solution was precipitated into MeOH, redissolved in CH2Cl2 and reprecipitated into MeOH to yield 426 mg (68%) of a white gummy polymer.
1H NMR (500 MHz, CDCl3): δ = 4.20–4.30 (br d, 4H), 2.43–2.49 (br t, 4H), 2.10–2.17 (br t, 2H), 1.95–2.04 (br m, 2H);13C NMR (101 MHz, CDCl3): δ = 172.39, 61.56, 60.17, 32.91, 30.97, 19.91; SEC-MALS: Mn = 26,200, PDI = 1.50, dn/dc = 0.072
Synthesis of Polymer 3
Polymer 2 (100 mg, 0.0047 mmol) was dissolved in 2 mL DMSO in a 10 mL Schlenk flask with a stirbar wrapped in copper wire (~2 cm, 20 gauge). Methyl acrylate (0.980 mL, 10.8 mmol) was added and the solution was subjected to three freeze-pump-thaw cycles. After the final cycle, the flask was placed in a water bath thermostated at 25 °C and Me6TREN (1.4 μL, 0.0094 mmol) was added by microsyringe and the solution was allowed to stir under argon for 1 hour. The polymerization was stopped by exposing to air and the solution was diluted with DCM and twice precipitated into cold MeOH to yield 401 mg (77% based on conversion) of a white gummy polymer.
1H NMR (400 MHz, CDCl3): δ = 4.20–4.30 (br d, 4H), 3.59–3.72 (br s, 41H, PMA) 2.43–2.49 (br t, 4H), 2.25–2.40 (br, 14H, PMA), 2.10–2.17 (br t, 2H), 1.95–2.04 (br m, 2H), 1.85–2.00 (br, 7 H, PMA), 1.60–1.75 (br, 14 H, PMA), 1.35–1.60 (br, 7 H, PMA); 13C NMR (101 MHz, CDCl3): δ = 174.85, 172.44, 61.57, 60.22, 51.72, 41.25, 34.92, 32.95, 31.01, 19.95; SEC-MALS: Mn = 136,000, PDI = 1.17, dn/dc = 0.069.
For 1H, 13C, and FT-IR spectra, characterization of Polymer 3 after sonication, calculation of gDCC monomers per chain (SEC-MALS), and calculation of percent ring-opening per scission cycle see supporting information.
Conclusion
In conclusion, we have developed a versatile method for the abundant incorporation of cyclic mechanophores into high MW block copolymers. The ABA architecture ensures a mechanophore rich region about the center of the chain, allowing for large amounts of sonochemical activation. The polymer system described here also exhibits excellent long-term stability, potentially enabling extended quantitative studies, a previously tedious task, with a wider variety of systems. Additionally, CRP approaches to block copolymer synthesis potentially allow for a wide variety of phase-segregated microstructures and bulk mechanical properties to be generated. The general nature of the carbodiimide polyesterification procedure as well as the near ubiquitous use of SET-LRP conditions in the synthesis of mechanophore linked polymers will provide the opportunity to apply this approach to a variety of mechanochemical systems and enable the generation of functional, stress-responsive materials. Most intriguing in this regard are non-scissile cyclic mechanophores, which are the basis for mechanochemical remodelling in polymeric systems.8
Supplementary Material
Acknowledgments
This work was supported by the U.S. Army Research Laboratory and the Army Research Office under Grant W911NF-07-1-0409. ZSK thanks the NIH for a NIGMS Biotechnology Predoctoral Training Grant (T32GM8555).
References
- 1.Caruso MM, Davis DA, Shen Q, Odom SA, Sottos NR, White SR, Moore JS. Chem Rev. 2009;109:5755–5798. doi: 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]
- 2.Yang QZ, Huang Z, Kucharski TJ, Khvostichenko D, Chen J, Boulatov R. Nature Nanotechnology. 2009;4:302–306. doi: 10.1038/nnano.2009.55. [DOI] [PubMed] [Google Scholar]
- 3.Brantley JN, Wiggins KM, Bielawski CW. Science. 2011;333:1606–1609. doi: 10.1126/science.1207934. [DOI] [PubMed] [Google Scholar]
- 4.Wiggins KM, Syrett JA, Haddleton DM, Bielawski CW. J Am Chem Soc. 2011;133:7180–7189. doi: 10.1021/ja201135y. [DOI] [PubMed] [Google Scholar]
- 5.Berkowski KL, Potisek SL, Hickenboth CR, Moore JS. Macromolecules. 2005;38:8975–8978. [Google Scholar]
- 6.Hickenboth CR, Moore JS, White SR, Sottos NR, Baudry J, Wilson SR. Nature. 2007;446:423–427. doi: 10.1038/nature05681. [DOI] [PubMed] [Google Scholar]
- 7.Potisek SL, Davis DA, Sottos NR, White SR, Moore JS. J Am Chem Soc. 2007;129:13808–13809. doi: 10.1021/ja076189x. [DOI] [PubMed] [Google Scholar]
- 8.Kean ZS, Craig SL. Polymer. 2012;53:1035–1048. [Google Scholar]
- 9.Black AL, Lenhardt JM, Craig SL. J Mater Chem. 2011;21:1655–1663. [Google Scholar]
- 10.Lenhardt JM, Black AL, Beiermann BA, Steinberg BD, Rahman F, Samborski T, Elsakr J, Moore JS, Sottos NR, Craig SL. J Mater Chem. 2011;21:8454–8459. [Google Scholar]
- 11.Lenhardt JM, Ogle JW, Ong MT, Choe R, Martinez TJ, Craig SL. J Am Chem Soc. 2011;133:3222–3225. doi: 10.1021/ja107645c. [DOI] [PubMed] [Google Scholar]
- 12.Lenhardt JM, Ong MT, Choe R, Evenhuis CR, Martinez TJ, Craig SL. Science. 2010;329:1057–1060. doi: 10.1126/science.1193412. [DOI] [PubMed] [Google Scholar]
- 13.Lenhardt JM, Black AL, Craig SL. J Am Chem Soc. 2009;131:10818–10819. doi: 10.1021/ja9036548. [DOI] [PubMed] [Google Scholar]
- 14.Wu D, Lenhardt JM, Black AL, Akhremitchev BB, Craig SL. J Am Chem Soc. 2010;132:15936–15938. doi: 10.1021/ja108429h. [DOI] [PubMed] [Google Scholar]
- 15.Hermes M, Boulatov R. J Am Chem Soc. 2011;133:20044–20047. doi: 10.1021/ja207421v. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee U, Wang X, Jewrajka SK, Soucek MD. Macromol Chem Phys. 2011;212:1879–1890. [Google Scholar]
- 17.Nelson-Mendez A, Aleksanian S, Oh M, Lim HS, Oh JK. Soft Matter. 2011;7:7441–7452. [Google Scholar]
- 18.Frenkel J. Acta Physicochim USSR. 1944;19:51–76. [Google Scholar]
- 19.Koda S, Mori H, Matsumoto K, Nomura H. Polymer. 1994;35:30–33. [Google Scholar]
- 20.Moore JS, Stupp SI. Macromolecules. 1990;23:65–70. [Google Scholar]
- 21.Lligadas G, Rosen BM, Monteiro MJ, Percec V. Macromolecules. 2008;41:8360–8364. [Google Scholar]
- 22.Pustovit YM, Ogojko PI, Nazaretian VP, Rozhenko AB. J Fluorine Chem. 1994;69:231–236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.