

Abstract
NMR spectroscopy has been used to observe the effects of the amine ligand on the rate of reaction of platinum diamine and triamine complexes with DNA and protein residues. Whereas [Pt(dien)Cl]Cl and [Pt(dien)(D2O)]2+ have been known to react faster with thioether residues such as N-AcMet than with 5′-GMP, we found that [Pt(Me4en)(D2O)2]2+ appeared to react faster with 5′-GMP. To quantitatively assess the factors influencing the rates of reaction, rate constants at pH 4 were determined for the reactions of [Pt(en)(D2O)2]2+ [en = ethylenediamine] and [Pt(Me4en)(D2O)2]2+ with N-AcMet, N-AcHis, 5′-GMP, and Guo (guanosine). In each case the less bulky complex ([Pt(en)(D2O)2]2+) reacts more quickly than does the bulkier [Pt(Me4en)(D2O)2]2+, as expected. Both complexes reacted faster with 5′-GMP; however, analysis of the rate constants suggests that the [Pt(en)(D2O)2]2+ complex favors reaction with 5′-GMP due to hydrogen bonding with the 5′-phosphate, whereas [Pt(Me4en)(D2O)2]2+ disfavors reaction with N-AcMet due to steric clashes. Bulk had relatively little effect on the rate constant with N-AcHis, suggesting that peptides or proteins that coordinate via His residues would not have their reactivity affected by bulky diamine ligands.
Keywords: platinum complexes, NMR spectroscopy, ligand binding, guanine, methionine, histidine
1. Introduction
Cisplatin is a successful anticancer drug used worldwide, particularly among testicular and ovarian cancers. It is accepted that a 1,2-intrastrand cross-link formed by Pt at the N7 atoms of two guanine residues is responsible for the anticancer activity.[1, 2] However, protein interaction also has an important role in the activity of cisplatin; a significant number of protein adducts are formed due in part to the drug’s high affinity for S-containing ligands.[3] The formation of protein adducts has been suggested to be responsible for side effects and/or drug resistance.[4, 5] More recently, the copper transporter Ctr1 has been thought to assist with cisplatin uptake.[6] Reaction with serum albumin and inhibition of thioredoxin reductase[7] and acetylcholinesterase[8] by cisplatin and other platinum(II) complexes has also been noted.
When Guo (guanosine) or 5′-GMP (guanosine 5′-monophosphate) is added to cis-[PtA2(D2O)2]2+ (A2 = two unidentate ligands or one bidentate amine ligand) at pH 4, the guanine coordinates to platinum via the N7 atom.[1] At a 1:2 Pt:guanine ratio, cis-[PtA2(Guo-N7)2] or cis-[PtA2(5′-GMP-N7)2] is the predominant product formed even when the A2 ligand is bulky.[1, 9–13] Studies with cisplatin analogs in which one ammine ligand was replaced by a larger heterocyclic ligand found that the DNA binding mode was not altered by the heterocyclic ligand.[14] When platinum complexes with chiral and bulky 2,2′-bipiperidine ligands were reacted with an oligonucleotide, it was found that the duplex form was stabilized relative to the single stranded form.[15]
Due to cisplatin’s affinity for sulfur ligands, reaction with peptides and proteins at cysteine or methionine residues is also very common.[3] Thus, many studies have utilized the amino acid N-acetylmethionine (N-AcMet), which has an amide nitrogen and thus is representative of an internal methionine residue in a peptide or protein. When N-AcMet is added to platinum complexes with relatively small amine ligands in a 2:1 N-AcMet:Pt ratio, [PtA2(N-AcMet-S)2] is observed.[16, 17] When less N-AcMet is utilized, [PtA2(N-AcMet-S)(D2O)]+ and [PtA2(N-AcMet-S,N)]+ may be observed. Analogous S,N chelates can also be observed in small peptides containing internal methionine residues.[18–20]
Since the anticancer activity of cisplatin is due to the Pt-DNA crosslink, competing S-ligands could affect the efficiency of the drug. Intermolecular studies have shown that platinum reaction with methionine and related ligands is kinetically favored, while the reaction of platinum with guanine is thermodynamically favored.[3] Studies have found that within 1 day of administering cisplatin intravenously, 65–98% of the drug is bound to proteins.[21] It is therefore crucial to better understand the interaction of platinum complexes with DNA and proteins and to determine what factors affect the rates of reaction with each.
Previously we have shown that platinum complexes that have significant bulk at one or both amine nitrogens form only a 1:1 complex with methionine and N-acetylmethionine.[17, 22] Molecular mechanics studies have indicated that steric clashes would be present in cis-[PtA2(Met-S)2]2+ complexes when the A2 ligand is bulky.[17] Such bulk does not prevent coordination of a second guanine. More recently, we found that a bulky triamine complex, [Pt(Me5dien)(D2O)]2+ [Me5dien = N, N, N′, N′, N″-pentamethyldiethylenetriamine], reacted significantly faster with 5′-GMP than with N-AcMet,[23] a reversal from the long-known preference for thioethers by [Pt(dien)Cl]Cl.[3, 5, 24]
In this study, we have focused on [Pt(Me4en)(D2O)2]2+ (Me4en = N, N, N′, N′, tetramethylethylenediamine) as a bulky platinum diamine complex (Figure 1). [Pt(en)(D2O)2]2+ (en = ethylenediamine) and [Pt(dien)(D2O)]2+ (dien = diethylenetriamine) are used to show the effects of a less bulky ligand. We have utilized NMR spectroscopy to monitor “competition” reactions in which N-AcMet and 5′-GMP are added to a platinum complex to see which products are formed preferentially. We have also evaluated rate constants for the reactions of [Pt(en)(D2O)2]2+ and [Pt(Me4en)(D2O)2]2+ with N-AcMet, 5′-GMP, and Guo in order to see how hydrogen bonding, steric interactions, and other factors affect the rates of these reactions.
Figure 1.

Representations of platinum complexes and ligands used in this study. Top row (left to right): [Pt(en)(D2O)2]2+, [Pt(Me4en)(D2O)2]2+. Bottom row (left to right): Guo, 5′-GMP, N-AcMet, N-AcHis. The charge state that should be dominant at pH 4 is shown.
2. Materials and Methods
N-AcMet (Acros), N-AcHis (Acros), Guo (Acros), GMP (Acros), and Pt(en)Cl2 (Alfa Aeser) were used as supplied. Pt(Me4en)Cl2 was prepared using methods described previously.[25]
2.1 Reaction of [Pt(en)(D2O)2]2+ or [Pt(Me4en)(D2O)2]2+ with 5′GMP, Guo, N-AcMet, and N-AcHis
Samples of Pt(en)Cl2 and Pt(Me4en)Cl2 were each treated with 2 equivalents of AgNO3 in H2O and stirred for at least 24 hours in the dark. The samples were filtered to remove AgCl and evaporated to dryness to make Pt(en)(NO3)2 and Pt(Me4en)(NO3)2. The appropriate amount of PtA2(NO3)2 was then dissolved in D2O to yield the [PtA2(D2O)2]2+ complex.
No buffers were utilized, as phosphate buffer has been shown to coordinate to platinum complexes.[26, 27] For samples in which rate constants were determined, sodium nitrate was added to give a final concentration of 0.1 M; less than 2% of the platinum would be expected to have a coordinated NO3− ion assuming an equilibrium constant similar to that (0.17 M−1) reported for cis-[(NH3)2Pt(H2O)2]2+,[28] and we noted no significant difference between rate constants that were obtained with sodium triflate compared with sodium nitrate. N-AcMet, N-AcHis, Guo, and 5′GMP were individually dissolved in D2O. Stock solutions of the platinum complexes, amino acids, and nucleobases were prepared at typical concentrations of 25–40 mM except for Guo, which was prepared at 3.5 mM due to solubility. The pH (uncorrected) of each solution was adjusted to 4.0 and the appropriate volume of each reactant was added to a sample of D2O to give the desired final concentration (typically 0.5–5 mM, except for the N-AcHis reactions in which 30 mM concentrations of the platinum complexes were utilized). In the reaction of [Pt(en)(D2O)2]2+ and N-AcMet, <1 μL of methanol was added to give an internal reference peak. The solutions were maintained at 25 °C before and after mixing. Immediately after mixing, the samples were transferred to an NMR tube, and the progress of reaction was determined by 1H NMR spectroscopy. For reactions in which rate constants were determined, each reaction was repeated a minimum of three times.
2.2 NMR Measurements
The 1H NMR spectra were collected on a JEOL 500 MHz NMR instrument. NMR spectra were referenced to the residual HOD signal adjusted for temperature.[29]
2.3 Determination of rate constants
Rate constants were determined using the software DynaFit (BioKin, Ltd.). For reactions involving Guo or 5′-GMP, the amount of product and reactant was determined by integration of the H8 signals in the NMR spectra. The data was fit to a mechanism involving two steps that were first order in each reactant: reaction of platinum complex with the first guanine ligand, followed by reaction of the mono product with a second guanine ligand. Since a large excess of platinum complex was used for the N-AcHis reactions, only one step was included in the reaction mechanism; experimentally, the N-Ac CH3 group was integrated to determine the extent of reaction. For reactions involving [Pt(Me4en)(D2O)2]2+ and N-AcMet, the amount of product and reactant were determined by integration of the Hα signals in the NMR spectra. Only one step that was first order in platinum and N-AcMet was used since two N-AcMet ligands cannot coordinate to this platinum complex.[17] For reactions involving [Pt(en)(D2O)2]2+ and N-AcMet, the size of the unreacted N-AcMet peak was integrated and compared with the integration of the methanol reference signal; this method of integration allowed us to ignore the subsequent chelation of the nitrogen atom of the N-AcMet. The data was fit to an equation that was first order in both platinum complex and N-AcMet.
3. Results
3.1 Competition of N-AcMet and 5′-GMP with [Pt(Me4en)(D2O)2]2+
We first considered a competition reaction between N-AcMet and 5′-GMP for [Pt(Me4en)(D2O)2]2+; this reaction is similar to that studied previously [23] for [Pt(Me5dien)(D2O)]2+ except that two coordination sites are available. We observed the reactions occurring in a solution containing 2 mM each of [Pt(Me4en)(D2O)2]2+, 5′-GMP, and N-AcMet; this platinum complex can react with only one N-AcMet, and while a [Pt(Me4en)(5′-GMP-N7)2] complex is possible, the second 5′-GMP addition is slower than the first and thus we saw no significant amount of this complex in the initial stages of reaction. At ~10 min (Figure 2), a product signal at 8.8 ppm is observed, corresponding to [Pt(Me4en)(5′-GMP)(D2O)]+.[17] The S-CH3 signal of [Pt(Me4en)(N-AcMet-S)(D2O)]+ occurs ~2.5 ppm. As shown in Figure 2, more product is initially formed with 5′-GMP than with N-AcMet. Throughout the reaction, the amount of 5′-GMP reacted exceeds the amount of N-AcMet reacted. Within ~45 min, signals due to [Pt(Me4en)(5′-GMP)2] were observed; two H8 resonances are observed due to restricted rotation around the Pt-N7 bond.[17, 30] Compared with the previous [Pt(Me5dien)(D2O)]2+ reactions, we note that the reactions with [Pt(Me4en)(D2O)2]2+ occurred more quickly and that the difference in reactivity of N-AcMet and 5′-GMP was much smaller.
Figure 2.

Partial 1H NMR spectra from the reaction of 2 mM [Pt(Me4en)(D2O)2]2+, 2 mM 5′-GMP, and 2 mM N-AcMet at pH 4.0. Mono products formed from reaction with 5′-GMP and N-AcMet have key resonances at ~ 8.8 ppm and ~ 2.5 ppm, respectively. Each side of the NMR spectrum has the y-axis scaled to the largest peak to make the signals easier to observe.
3.2 Rate constant determination and evaluation
In order to quantitatively assess the rates of reaction of the en and Me4en complexes with Guo, 5′-GMP, N-AcHis, and N-AcMet, we determined rate constants. We focused on the rate constants of the reactions of the ligands with the diaqua complexes in order to avoid the complications due to low solubility of the Pt(Me4en)Cl2 compound; this method also avoided differences that would result from differing rates of aquation of the chloride complexes. Likewise, we utilized a pH of 4 to avoid deprotonation of the coordinated D2O ligands, which could lead to dimerization products.[31, 32]
Although two coordination sites are available in the platinum complexes utilized, we focused on the rate constants for the coordination of the first ligand since in a larger DNA or protein molecule, chelation with the same biomolecule would be more likely than cross-linking of two different biomolecules. Thus, we tended to use excess of the platinum complexes especially in cases in which both mono- and bis-products were possible. The rate constants that were determined are summarized in Table 1.
Table 1.
Rate constants of the reactions of [Pt(en)(D2O)2]2+ and [Pt(Me4en)(D2O)2]2+ with N-AcMet, 5′-GMP, and Guo. All rates measured at an initial pH of 4.0 and a constant temperature of 25 °C with an ionic strength of 0.1 M Rates in M−1s−1
| Ligand | ken | kMe4en | ken/kMe4en |
|---|---|---|---|
| N-AcMet | 3.8 ± 0.7 | 0.237 ± 0.007 | 16 |
| N-AcHis | 7.2 ± 0.7 × 10−3 | 8.5 ± 0.6 × 10−3 | 1 |
| 5′-GMP | 5.0 ± 0.5 | 0.39 ± 0.06 | 13 |
| Guo | 0.56 ± .06 | 0.117 ± .003 | 5 |
In the reaction of [Pt(en)(D2O)2]2+ with N-AcMet, several products are possible. In addition to the [Pt(en)(N-AcMet-S)(D2O)]+ product, a [Pt(en)(N-AcMet-S)2] product could be formed in small amounts; however, under our conditions, only a very small amount (<5%) was observed by the end of the reaction. Furthermore, several different sets of signals due to chelation of the amide nitrogen in the mono product lead to several signals that make integration of the products difficult. Thus, we chose to introduce a small amount of methanol to use as an integration reference. With this method, the conversion of [Pt(en)(N-AcMet-S+(D2O)]+ to [Pt(en)(N-AcMet-S,N)]+ will have no effect on the integration of the signals that are being used. Thus, the mechanism used in DynaFit consisted of one step, consisting of reaction of the [Pt(en)(D2O)2]2+ with N-AcMet with the rate first-order in each reactant.
The reaction of [Pt(Me4en)(D2O)2]2+ with N-AcMet yields production of the [Pt(Me4en)(N-AcMet-S,O)]+ chelate; the intermediate [Pt(Me4en)(N-AcMet-S)(D2O)]+ product is not observed separately, and [Pt(Me4en)(N-AcMet-S)2] does not form even when excess N-AcMet is present.[17] Therefore, the mechanism used consisted of the reaction of [Pt(Me4en)(D2O)2]2+ with N-AcMet, with the rate first-order with respect to each reactant.
Reactions of either platinum complex with N-AcHis proceeded very slowly at pH 4 because of the protonation of the histidine ligand. However, use of 30 mM platinum complex and 5 mM N-AcHis led to formation of significant product within ~1 hour. Coordination of the platinum(II) to the imidazole ring was established by raising the pH after the reaction; no significant shift of the imidazole signals occurred with a pKa of ~6 as would be expected due to deprotonation of the imidazole ring.
For the reactions of [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ with either 5′-GMP or Guo, two steps are possible: reaction of the diaqua complexes with the first guanine ligand, followed by reaction of the mono product with a second guanine ligand. As shown in Figures 2 and 3, complexes with one and two guanines attached are easily distinguished with the former having the more downfield shift. Because we wanted to focus on rate constants for the coordination of the first guanine ligand, the reaction conditions utilized for rate constant determination had significant excess of the platinum complex since two guanines can coordinate even with equimolar amounts of platinum and GMP (Figure 2). Only a small amount of bis product was observed during the course of the rate constant reactions under the conditions utilized; nevertheless, a mechanism to account for further reaction to the bis-products was utilized.
Figure 3.
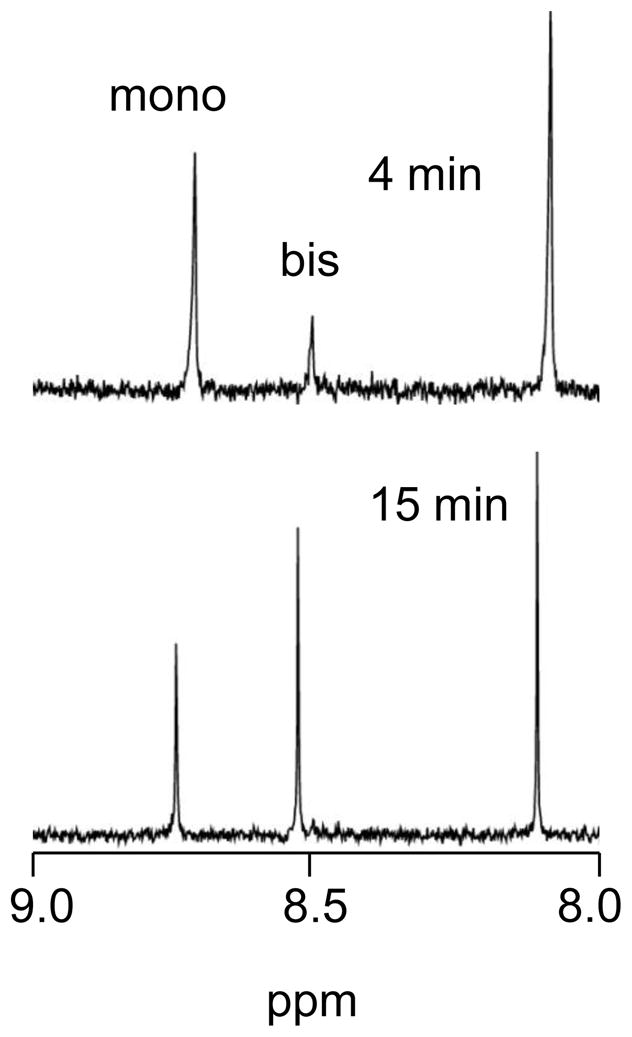
Partial 1H NMR spectra from the reaction of 2 mM [Pt(en)(D2O)2]2+ and 4 mM 5′-GMP at pH 4.0. Under these conditions, the H8 resonances both [Pt(en)(5′-GMP)(D2O)]+ (mono) and [Pt(en)(5′-GMP)2] (bis) are clearly visible and are labeled. The unlabeled peak is due to unreacted 5′-GMP.
4. Discussion
Platinum complexes such as [Pt(dien)Cl]Cl have been shown to react with thioether complexes such as N-acetylmethionine and S-methylglutathione faster than they react with 5′-GMP.[3, 5, 24] The thioether ligands can eventually be displaced by guanine nucleotides,[33–36] leading to speculation that peptide or protein adducts could be intermediate products that will eventually be replaced by DNA adducts.[3, 35, 36] However, ammine loss is a competing reaction in cisplatin,[37, 38] and it was found that little displacement of a methionine ligand from a complex containing both methionine and guanine ligands was observed.[16]
Our previous study found that [Pt(Me5dien)(D2O)]2+ reacted much faster with 5′-GMP than with N-AcMet.[23] Thus, in the present study we first performed a similar competition with [Pt(Me4en)(D2O)2]2+. Whereas a preference for 5′-GMP was noted for [Pt(Me4en)(D2O)2]2+ (Figure 2, Table 1), the difference in reactivity between the 5′-GMP and N-AcMet was considerably less than was noted for [Pt(Me5dien)(D2O)]2+.[23] Previously, a tridentate DNSH-dien ligand[39] was found to lessen the kinetic preference for methionine over 5′-GMP due to the bulk of the DNSH; reaction occurred with 5′-GMP and with methionine at approximately equal rates. The available coordination site is cis to one bulky site (DNSH) and to one nonbulky site (NH2). The Me4en complexes used in the current study likewise have coordination sites with one bulky ligand (the tertiary amine ligand) and one nonbulky ligand (D2O) that are cis to the coordination site. We note that the rates of reaction of [Pt(Me4en)(D2O)2]2+ are also similar with 5′-GMP and with methionine (Table 1).
Since the [Pt(Me5dien)(D2O)]2+ complex has bulk on two amine nitrogens that are cis to the coordination site, the effect on N-AcMet reactivity is much greater.[23] This observation is consistent with observations that N-AcMet will coordinate in one but not both coordination sites of [Pt(Me4en)(D2O)2]2+;[17] the steric hindrance of the Me4en ligand combined with the steric hindrance of one coordinated N-AcMet renders the [Pt(Me4en)(N-AcMet-S,O)]+ complex stable to further reaction with N-AcMet.
An analysis of the rate constants for the reactions of [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ with N-AcMet, 5′GMP, and Guo give us insights into the factors affecting the rates of reaction of methionine versus guanine (Table 1). First, we note that [Pt(en)(D2O)2]2+ reacts ~5–15 times faster than [Pt(Me4en)(D2O)2]2+ with these ligands. This could be due to several factors, including the steric bulk of the Me4en ligand, hydrogen bonding to the NH2 groups in [Pt(en)(D2O)2]2+, and possible trans effect differences due to reaction trans to an NH2 vs. an N(CH3)2 group.
In order to probe the effects of hydrogen bonding with the phosphate of 5′-GMP, we compared rate constants of Guo and 5′-GMP reacting with [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ complexes. Guo reacts more slowly than 5′-GMP for both platinum complexes (Table 1), which could be due to hydrogen bonding with the phosphate of 5′-GMP and to increased electrostatic attraction of the anionic 5′-GMP with the cationic platinum complexes. Previously, [cis-Pt(NH3)2(H2O)2]2+ and [Pt(dien)(H2O)]2+ were shown to react faster with 5′-GMP than with 3′-GMP,[40] indicating that at least some hydrogen bonding with the 5′-GMP phosphate is important in similar complexes.
Comparing the reactivity of the en complex with that of the Me4en complex with each ligand, the rate is 5 times faster with Guo but 13 times faster with 5′-GMP (Table 1). Steric clashes with the phosphate would be unlikely due to the conformational flexibility; thus, the data suggest that the hydrogen bonding with 5′-GMP is more significant for [Pt(en)(D2O)2]2+ than for [Pt(Me4en)(D2O)2]2+. Since both platinum complexes can hydrogen bond via attached D2O molecules but only the en complex can hydrogen bond through NH2 groups (Figure 4), these results suggest that additional hydrogen bonding with the NH2 groups is an important factor in the rate of reaction with the en complex. The presence of a 5′-phosphate on nucleotides is known to have a stabilizing effect on palladium complexes in which hydrogen bonding between the phosphate and amine hydrogens are possible.[41, 42] However, an alternative explanation is that the methyl groups of Me4en donate electron density to the platinum atom and thus diminish the acidity (and hence the hydrogen bonding ability) of the D2O.
Figure 4.

Representation of the [Pt(en)(5′-GMP)(H2O)]+ complex showing possible hydrogen bonding between the phosphate group and either the attached water molecule or the NH2 of the amine ligand.
When the ratio of rate constants for [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ reactions with N-AcMet are compared, it is seen that [Pt(en)(D2O)2]2+ reacts ~16 times faster than [Pt(Me4en)(D2O)2]2+. Thus, the presence of the methyl groups on the Me4en ligand affects reaction with N-AcMet more than it affects reaction with Guo and slightly more than it affects 5′-GMP. One possible explanation would be that hydrogen bonding between the NH2 groups of [Pt(en)(D2O)2]2+ and N-AcMet is comparable to, if not superior to, that of the NH2 groups and the phosphate of 5′-GMP; however, if that were true then we would expect that [Pt(en)(D2O)2]2+ would react faster with N-AcMet than with 5′-GMP since faster reactivity with the thioether is typical.[3, 24, 39] Thus, the observation that the [Pt(en)(D2O)2]2+ reacts ~16 times faster than [Pt(Me4en)(D2O)2]2+ suggests that the reaction between N-AcMet and [Pt(Me4en)(D2O)2]2+ is unusually slow. Support for the effects of steric clashes comes from the DNSH-dien ligand studies;[39] the preference for methionine was diminished despite the presence of a cis NH2 ligand that could hydrogen bond. We conclude that the presence of the bulky Me4en ligand hinders the reaction with N-AcMet more than it hinders the reaction with 5′-GMP.
Previously, we studied the reaction of N-AcMet with the platinum complex Pt(Et2en)(D2O)22+ [Et2en = N,N-diethylethylenediamine].[22] The Et2en ligand had one nitrogen atom that had an en-like NH2, while the other nitrogen atom had two ethyl groups attached, similar to (though even more bulky than) the nitrogens of Me4en. Only one geometric isomer of the [Pt(Et2en)(N-AcMet-S,O)]+ chelate was observed,[22] namely the one in which the sulfur atom was cis to the nonbulky NH2 nitrogen atom, despite the molecular mechanics calculations which suggested that both geometric isomers would be somewhat similar in energy. The observation of only one geometric isomer was concluded to be due to a strong kinetic preference for the nonbulky site. The lack of formation of any significant amount of the other geometric isomer is consistent with our observations that N-AcMet reacts substantially faster with [Pt(en)(D2O)2]2+, which has NH2 groups cis to the D2O ligands, than with [Pt(Me4en)(D2O)2]2+, which has N(CH3)2 groups cis to the D2O ligands.
When the reaction of [Pt(Me4en)(D2O)2]2+ with a mixture of N-AcMet and 5′-GMP was continued for several hours, it was observed that [Pt(Me4en)(5′-GMP)2] was observed beginning around 45 minutes, whereas [Pt(Me4en)(N-AcMet)(5′-GMP)] was not significant even at 3.5 hours. [Pt(Me4en)(N-AcMet)(5′-GMP)] could be formed by two routes (Figure 5): reaction of N-AcMet with [Pt(Me4en)(5′-GMP)(D2O)]+, or reaction of 5′-GMP with [Pt(Me4en)(N-AcMet-S,O)]+, a reaction that was previously observed to occur slowly.[17] [Pt(Me4en)(5′-GMP)2] can be formed when 5′-GMP reacts with the [Pt(Me4en)(5′-GMP)(D2O)]+ complex and could also form if 5′-GMP displaced N-AcMet from [Pt(Me4en)(N-AcMet)(5′-GMP)]. However, such displacement was not previously observed for this complex[17] and has been observed to occur very slowly for [Pt(en)(N-AcMet-S)(5′-GMP)][16]; furthermore, [Pt(Me4en)(5′-GMP)2] was observed much earlier than [Pt(Me4en)(N-AcMet)(5′-GMP)]. Thus, [Pt(Me4en)(5′-GMP)(D2O)]+ clearly reacts faster with 5′-GMP than with N-AcMet.
Figure 5.

Scheme of the possible reactions of [Pt(Me4en)(D2O)2]2+ with N-AcMet and 5′-GMP. For simplicity, the sugar and phosphate are not shown for 5′-GMP.
Both [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ complexes favor reaction with 5′-GMP over N-AcMet, but for different reasons (steric hindrance in N-AcMet reactions and especially favorable hydrogen bonding with 5′-GMP, respectively). Since the phosphate groups in oligonucleotides are more conformationally restricted than the phosphate of 5′-GMP, hydrogen bonding with the phosphate group of oligonucleotides may have less effect on the reactivities; previous kinetic studies with short oligonucleotides suggested that hydrogen bonding to the 5′-phosphate did not have a substantial affect on the rate.[43] However, since peptides and proteins would likely have more steric clashes possible, these clashes could have more effect on the reactivity in peptides and proteins.
Since reactivity of platinum complexes with histidine residues is also possible,[36, 44, 45] we have also examined the effect of bulk on histidine residues. N-AcHis reacted very slowly with both [Pt(Me4en)(D2O)2]2+ and [Pt(en)(D2O)2]2+ complexes under the conditions utilized. Nevertheless, we can compare the effects of bulk by examining the rates of reaction of N-AcMet with each platinum complex. Of all of the ligands studied, N-AcHis was the least affected by the bulk of the Me4en ligand, as rates of reactivity were indistinguishable for [Pt(en)(D2O)2]2+ and [Pt(Me4en)(D2O)2]2+.
Thus, proteins that react with platinum via a methionine residue would likely have reduced reactivity with a platinum complex having a bulky diamine ligand whereas proteins that react via histidine could be significantly less affected by the bulky ligand. .
Research Highlights.
[Pt(Me4en)(D2O)2]2+ favors 5′-GMP over N-acetylmethionine due to steric clashes
[Pt(en)(D2O)2]2+ favors 5′-GMP over N-acetylmethionine due to hydrogen bonding
Bulk of diamine ligand does not affect reaction rate with N-acetylhistidine
Acknowledgments
This research was supported by awards from the NIH (1R15 GM074663-01), Research Corporation (Cottrell College Science Award CC6464), and Western Kentucky University (Summer Faculty Scholarship, Research and Creative Activities Program RCAP 11-8042, Materials Characterization Center Faculty Grants Program).
Abbreviations
- N-AcHis
N-acetylhistidine
- N-AcMet
N-acetylmethionine
- Me4en
N,N,N′,N′-tetramethylethylenediamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ano SO, Kuklenyik Z, Marzilli LG. In: Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Lippert B, editor. Wiley-VCH; Weinheim: 1999. pp. 247–292. [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Reedijk J. Chem Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- 4.Kartalou M, Essigmann JM. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 5.Reedijk J, Teuben JM. In: Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Lippert B, editor. Wiley-VCH; Weinheim: 1999. pp. 339–362. [Google Scholar]
- 6.Howell SB, Safaei R, Larson CA, Sailor MJ. Mol Pharmacol. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Free Radical Biol Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 8.Aljafari AA. Int J Biochem Cell Biol. 1995;27:965–970. doi: 10.1016/1357-2725(95)00044-p. [DOI] [PubMed] [Google Scholar]
- 9.Ano SO, Intini FP, Natile G, Marzilli LG. J Am Chem Soc. 1997;119:8570–8571. [Google Scholar]
- 10.Kiser D, Intini FP, Xu Y, Natile G, Marzilli LG. Inorg Chem. 1994;33:4149–4158. [Google Scholar]
- 11.Marzilli LG, Ano SO, Intini FP, Natile G. J Am Chem Soc. 1999;121:9133–9142. [Google Scholar]
- 12.Marzilli LG, Intini FP, Kiser D, Wong HC, Ano SO, Marzilli PA, Natile G. Inorg Chem. 1998;37:6898–6905. doi: 10.1021/ic980843f. [DOI] [PubMed] [Google Scholar]
- 13.Williams KM, Cerasino L, Intini FP, Natile G, Marzilli LG. Inorg Chem. 1998;37:5260–5268. doi: 10.1021/ic970704i. [DOI] [PubMed] [Google Scholar]
- 14.Kasparkova J, Marini V, Najajreh Y, Gibson D, Brabec V. Biochemistry. 2003;42:6321–6332. doi: 10.1021/bi0342315. [DOI] [PubMed] [Google Scholar]
- 15.Beljanski V, Villanueva JM, Doetsch PW, Natile G, Marzilli LG. J Am Chem Soc. 2005;127:15833–15842. doi: 10.1021/ja053089n. [DOI] [PubMed] [Google Scholar]
- 16.Barnham KJ, Guo ZJ, Sadler PJ. J Chem Soc, Dalton Trans. 1996:2867–2876. [Google Scholar]
- 17.Williams KM, Rowan C, Mitchell J. Inorg Chem. 2004;43:1190–1196. doi: 10.1021/ic035212m. [DOI] [PubMed] [Google Scholar]
- 18.Hahn M, Kleine M, Sheldrick WS. J Biol Inorg Chem. 2001:556–566. doi: 10.1007/s007750100232. [DOI] [PubMed] [Google Scholar]
- 19.Siebert AFM, Sheldrick WS. J Chem Soc, Dalton Trans. 1997:385–393. [Google Scholar]
- 20.Fröhling CDW, Sheldrick WS. J Chem Soc, Dalton Trans. 1997:4411–4420. [Google Scholar]
- 21.DeConti RC, Toftness BR, Lange RC, Creasey WA. Cancer Res. 1973;33:1310–1315. [PubMed] [Google Scholar]
- 22.Williams KM, Chapman DJ, Massey SR, Haare C. J Inorg Biochem. 2005;99:2119–2126. doi: 10.1016/j.jinorgbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Sandlin RD, Starling MP, Williams KM. J Inorg Biochem. 2010;104:214–216. doi: 10.1016/j.jinorgbio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Djuran MI, Lempers ELM, Reedijk J. Inorg Chem. 1991;30:2648–2652. [Google Scholar]
- 25.Cerasino L, Williams KM, Intini FP, Cini R, Marzilli LG, Natile G. Inorg Chem. 1997;36:6070–6079. doi: 10.1021/ic970704i. [DOI] [PubMed] [Google Scholar]
- 26.Frey U, Ranford JD, Sadler PJ. Inorg Chem. 1993;32:1333–1340. [Google Scholar]
- 27.Lempers ELM, Bloemink MJ, Reedijk J. Inorg Chem. 1991;30:201–206. [Google Scholar]
- 28.Appleton TG, Berry RD, Davis CA, Hall JR, Kimlin HA. Inorg Chem. 1984;23:3514–3521. [Google Scholar]
- 29.Hoffman RE, Davies DB. Magn Reson Chem. 1988;26:523–525. [Google Scholar]
- 30.Cramer RE, Dahlstrom PL. J Am Chem Soc. 1979;101:3679–3681. [Google Scholar]
- 31.Wimmer S, Castan P, Wimmer FL, Johnson NP. J Chem Soc, Dalton Trans. 1989:403–412. [Google Scholar]
- 32.Faggiani R, Lippert B, Lock CJL, Rosenberg B. J Am Chem Soc. 1977;99:777–781. [Google Scholar]
- 33.Teuben JM, Reedijk J. J Biol Inorg Chem. 2000;5:463–468. doi: 10.1007/pl00021447. [DOI] [PubMed] [Google Scholar]
- 34.van Boom SSGE, Chen BW, Teuben JM, Reedijk J. Inorg Chem. 1999;38:1450–1455. [Google Scholar]
- 35.Kleine M, Wolters D, Sheldrick WS, Inorg J. Biochem. 2003;2003:354–363. doi: 10.1016/s0162-0134(03)00289-7. [DOI] [PubMed] [Google Scholar]
- 36.Barnham KJ, Djuran MI, Murdoch PD, Sadler PJ. J Chem Soc Chem Comm. 1994:721–722. [Google Scholar]
- 37.Lau JK-C, Deubel DV. Chem-Eur J. 2005;11:2849–2855. doi: 10.1002/chem.200401053. [DOI] [PubMed] [Google Scholar]
- 38.Barnham KJ, Djuran MI, Murdoch PdS, Ranford JD, Sadler PJ. J Chem Soc, Dalton Trans. 1995;1995:3721–3726. [Google Scholar]
- 39.Christoforou AM, Marzilli PA, Marzilli LG. Inorg Chem. 2006;45:6771–6781. doi: 10.1021/ic0606375. [DOI] [PubMed] [Google Scholar]
- 40.Marcelis ATM, Erkelens C, Reedijk J. Inorg Chim Acta. 1984;91:129–135. [Google Scholar]
- 41.Kim SH, Martin RB. Inorg Chim Acta. 1984;91:11–18. [Google Scholar]
- 42.Vestues PI, Martin RB. J Am Chem Soc. 1981;103:806–809. [Google Scholar]
- 43.Gonnet F, Reeder F, Kozelka J, Chottard JC. Inorg Chem. 1996;35:1653–1658. doi: 10.1021/ic951136e. [DOI] [PubMed] [Google Scholar]
- 44.Hong J, Jiao Y, He WJ, Guo ZJ, Yu Z, Zhang JF, Zhu LG. Inorg Chem. 2010;49:8148–8154. doi: 10.1021/ic101191m. [DOI] [PubMed] [Google Scholar]
- 45.Wolters D, Sheldrick WS. J Chem Soc, Dalton Trans. 1999:1121–1129. [Google Scholar]