

Abstract
Lack of an adequate animal model of Plasmodium falciparum severe malarial anemia (SMA) has hampered the understanding of this highly lethal condition. We developed a model of SMA by infecting C57BL/6 mice with P. chabaudi followed after recovery by P. berghei infection. P. chabaudi/P. berghei-infected mice had an initial 9- to 10-day phase of relatively low parasitemia and severe anemia, followed by a second phase of hyperparasitemia, more profound anemia, reticulocytosis, and death 14 to 21 days after infection. P. chabaudi/P. berghei-infected animals had more intense splenic hematopoiesis, higher interleukin-10 (IL-10)/tumor necrosis factor alpha and IL-12/gamma interferon (IFN-γ) ratios, and higher antibody levels against P. berghei and P. chabaudi antigens than P. berghei-infected or P. chabaudi-recovered animals. Early treatment with chloroquine or artesunate did not prevent the anemia, suggesting that the bulk of red cell destruction was not due to the parasite. Red cells from P. chabaudi/P. berghei-infected animals had increased surface IgG and C3 by flow cytometry. However, C3−/− mice still developed anemia. Tracking of red cells labeled ex vivo and in vivo and analysis of frozen tissue sections by immunofluorescence microscopy showed that red cells from P. chabaudi/P. berghei-infected animals were removed at an accelerated rate in the liver by erythrophagocytosis. This model is practical and reproducible, and its similarities with P. falciparum SMA in humans makes it an appealing system with which to study the pathogenesis of this condition and explore potential immunomodulatory interventions.
INTRODUCTION
Plasmodium falciparum is an intracellular parasite of humans responsible for 1 million to 2 million deaths per year. Severe malarial anemia (SMA) due to Plasmodium falciparum claims the lives of thousands of children in sub-Saharan Africa every day. The pathogenesis of this anemia is complex and not well understood. There is evidence supporting a role for bone marrow suppression (1, 42) as well as evidence to suggest that uninfected red blood cells (URBCs) are destroyed at an accelerated rate in a manner independent of the level of parasitemia (26, 38). A mathematical model has shown that an average of 8.5 uninfected red cells are destroyed for every parasitized red cell (20). In a prospective study, the proportion of red cell mass lost attributable to the parasite was calculated to be 7.9% of the total lost (37). Additionally, patients treated for P. falciparum malaria continue to experience red cell destruction after treatment (4), indicating that there are alternative mechanisms for the destruction of red cells that are not directly related to the parasite.
The study of host and parasite factors that contribute to the pathogenesis of SMA has been hampered by the lack of an inexpensive and easy-to-reproduce animal model that is relevant to the clinical picture seen with P. falciparum infection. Although there are currently multiple rodent models available, all differ significantly from the clinical picture of severe anemia seen with P. falciparum. The rodent parasites most commonly used to study anemia are P. chabaudi, P. berghei, P. vinckei, and P. yoelii. Whereas SMA in individuals with P. falciparum infection is characterized by relatively low parasitemia, most mouse malaria species either lead to early death or result in severe anemia associated with high-level parasitemia (39). The most frequently used model, P. chabaudi AS, causes severe anemia with hyperparasitemia of 20 to 40% which differs in lethality depending upon the strain of mouse used (5). In addition to rodent models, there are nonhuman primate models of malarial infection. Semi-immune Aotus monkeys infected with P. falciparum have been used to study SMA (10, 23). While the use of nonhuman primates is advantageous due to their similarity to humans, their short supply and cost make this approach impractical.
Recently, Evans et al. (11) described a model of SMA caused by P. berghei ANKA infection in semi-immune BALB/c mice and naïve Wistar rats. These animals developed severe anemia in the presence of a low parasite burden, which is similar to what is seen in human P. falciparum infection. They also demonstrated an accelerated destruction of uninfected red cells, which has been reported in humans infected with P. falciparum (4, 27). While this model does represent an improvement over previous models, it has significant drawbacks. Its biggest limitation is the long preparative time (up to 6 months) required to establish partial immunity with repeated cycles of infection and drug treatment in mice. In addition, the timing of the anemia in mice is unpredictable, making it difficult to plan experiments. Further, subsequent work has shown that the anemia in rats is not as profound as originally reported (16).
On the basis of the factors described above, there is a critical need to develop a model of SMA that is simple, inexpensive, highly reproducible, and relevant to human malarial infections. Therefore, we sought to develop this model in C57BL/6 mice and report here its initial characterization.
MATERIALS AND METHODS
Mice and malaria infections.
Mice were used under protocols approved by the Institutional Animal Care and Use Committees (IACUC) of the Uniformed Services University of the Health Sciences and of the Pennsylvania State University College of Medicine. C57BL/6 mice were purchased from Jackson Laboratories. All mice used in the experiments were 6 to 12 weeks of age at the time of the initial P. chabaudi infection. Mice were kept in a pathogen-free barrier facility until initiation of the experiments.
All experiments were repeated 2 to 3 times. Plasmodium berghei ANKA parasites were a gift from Martha Sedegah at the Naval Medical Research Center. Plasmodium chabaudi chabaudi AS parasites were obtained from David Walliker at the University of Edinburgh. Infected RBCs (IRBCs; 106) were injected intraperitoneally (i.p.) into each mouse to start an experimental infection. On day 5 after P. chabaudi infection, a Giemsa-stained thin blood smear was prepared directly from tail blood and the parasitemia was determined to confirm that all animals were infected. Mice were then allowed to continue through the entire course of infection without any further handling. At approximately 6 to 8 weeks after P. chabaudi infection, tail blood was again obtained to ensure that the parasitemia was cleared and that blood count parameters had returned to normal. If so, mice were inoculated i.p. with either 106 P. berghei ANKA IRBCs or RPMI 1640 medium as a sham control. In some experiments, a group of naïve C57BL/6 mice was inoculated with P. berghei ANKA. For drug treatments, mice were injected intramuscularly with 50 μl of 10 mg/ml chloroquine in phosphate-buffered saline (PBS; pH 7.4) or 50 μl of 24 mg/ml artesunate (Sigma-Aldrich) in 5% sodium bicarbonate starting on day 5 postinfection and continuing once daily until euthanasia was carried out. At designated time points, 20 to 40 μl of tail vein blood was collected into EDTA capillary tubes (Heska Corp., Loveland, CO). RBC concentrations were determined using a hemacytometer or a Mindray BC-2800 vet hematology analyzer (Mindray Bio-Medical Electronics Co., People's Republic of China). Giemsa-stained thin blood smears were prepared directly from a droplet of tail blood. Reticulocyte smears were prepared with methylene blue (Sigma-Aldrich). A minimum of 500 total RBCs in consecutive high-power fields were counted to determine the percent parasitemia or reticulocytemia. For collection of serum or plasma, blood was obtained from the retro-orbital plexus and placed into an EDTA-coated or uncoated Pasteur pipette.
Measurement of cytokines.
A mouse cytokine 10-plex panel was used according to the manufacturer's instructions (Invitrogen, Carlsbad, CA) to determine concentrations of interlukin-1β (IL-1β), IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 p40/70, granulocyte-macrophage colony-stimulating factor (GM-CSF), gamma interferon (IFN-γ), and tumor necrosis factor alpha (TNF-α) in plasma samples. Samples were run on a Luminex 100 system using Masterplex QT software (Luminex Corp., Austin, TX).
Preparation of parasite antigens and quantitation of parasite-specific antibodies.
For antigen extraction, the procedure was a modification of one previously reported (35). We obtained 500 μl of cardiac blood anticoagulated with citrate phosphate dextrose (CPD) and at ∼20% ring-stage parasitemia. One half of the sample was cultured overnight in 5 ml of RPMI 1640 (Sigma-Aldrich) containing 10% mouse serum plus 25 μg/ml gentamicin in a 5% O2, 5% CO2, and 90% N2 atmosphere at 37°C in order to obtain late trophozoites and schizonts. The remaining half was resuspended in PBS with 0.1% saponin (Sigma-Aldrich) and 1× protease inhibitor cocktail (Sigma-Aldrich) and incubated in ice for 5 min. The cell pellet was then washed twice in the same lysis buffer and resuspended in 500 μl of PBS containing 1× protease inhibitor (Sigma-Aldrich). A Giemsa-stained smear confirmed the presence of intact parasites and ghost red cells. The sample was frozen at −20°C overnight, and on the following day it was thawed and sonicated for 5 min. This was followed by another cycle of freeze-thawing and high-speed vortexing. Finally, the sample was cleared by pulsed centrifugation at 14,000 rpm, and the supernatant was removed and stored at −20°C. After overnight incubation, the cultured sample was processed exactly as described above. The total protein concentration was measured using the bicinchoninic acid method (Thermo Fisher Scientific, Rockford, IL). The presence of malaria parasite-specific antigens was confirmed by immunoblotting (see Fig. S1 in the supplemental material).
Parasite-specific IgG in mouse serum was measured by enzyme-linked immunosorbent assay (ELISA). Flat-bottom 96-well Immulon 2 HB plates (Dynex Technology Inc., Chantilly, VA) were coated overnight at 4°C with 100 ng per well of either P. chabaudi or P. berghei antigens in PBS. The plates were then blocked with 200 μl/well of 5% Blotto nonfat dry milk (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in Tris-buffered saline (TBS) containing 0.1% Tween 20 (Sigma-Aldrich) for 2 h at room temperature. Sera diluted 1:1,000 in dilution buffer (0.1% bovine serum albumin [BSA] in TBS containing 0.1% Tween 20) were added to duplicate wells and incubated at room temperature for 1 h. After extensive washing with TBS–0.1% Tween 20 (wash buffer), 0.1 μg/well of peroxidase-labeled goat anti-mouse IgG (H+L; KPL, Gaithersburg, MD) was added and the plate was incubated at room temperature for 1 h. The plates were washed extensively, 100 μl of ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] peroxidase substrate (KPL) was added to each well, and the plate was incubated at room temperature for 30 min. The reaction was stopped by addition of 100 μl/well of 1% sodium dodecyl sulfate (Sigma-Aldrich). The optical density at 405 nm (OD405) was read with an EMax microplate reader (Molecular Devices, Inc., Sunnyvale, CA) using SoftMax Pro (version 4.3) software. The ELISA data were converted to reactivity indexes by dividing the OD405 of each serum sample by the mean OD405 of the naïve serum sample.
Ex vivo and in vivo labeling of RBCs for red cell tracking and survival studies.
For ex vivo labeling, IRBCs were obtained from C57BL/6 mice infected with P. chabaudi followed after recovery by P. berghei infection (P. chabaudi/P. berghei-infected mice) with a P. berghei parasitemia of approximately 3%. URBCs were obtained from naïve uninfected animals. CPD-anticoagulated cardiac blood was collected, and RBCs were diluted to 10% hematocrit in PBS. Fifteen microliters of 2.5 mg/ml of the lipophilic fluorescent dye DiD (Invitrogen) in dimethyl sulfoxide (DMSO) was added to each ml of RBCs, followed by incubation at 37°C for 30 min in the dark. The RBCs were washed twice with PBS by low-speed centrifugation and resuspended at a concentration of 5 × 109 RBCs/ml in PBS. After sedation with ketamine-xylazine, 100 μl of labeled RBCs was injected into a retro-orbital plexus of infected or uninfected mice. For in vivo labeling, 10 mg/ml of Dylight 633 N-hydroxysuccinimide (NHS) ester (Thermo Fisher Scientific) in DMSO was diluted to 320 μg/ml with PBS and 100 μl was injected into a retro-orbital plexus of each animal on the day prior to infection. Following injection, 5 μl of EDTA-anticoagulated tail blood was collected after 30 min (baseline) and at predetermined intervals following infection. The blood was added to wells of a 96-well plate containing 2% paraformaldehyde in PBS. The samples were stored at 4°C in the dark until acquisition.
Acquisition was performed with an LSRII flow cytometer (Becton Dickinson, San Diego, CA) fitted with a 635-nm in-line laser. RBCs were gated on the basis of their forward and side scatter characteristics using logarithmic amplification. The percentage of positive cells in FL3 was determined in comparison to an unstained control.
Immunofluorescence microscopy of frozen tissues and quantitation of erythrophagocytosis.
Following euthanasia of animals injected with RBCs labeled ex vivo or in vivo, livers and spleens were removed, weighed, embedded in OCT (Sakura Finetek, Torrence, CA), and snap-frozen in isopentane (Sigma-Aldrich) precooled in liquid nitrogen. Frozen tissues were stored at −80°C until used. Frozen sections of liver and spleen of 10-μm thickness were first fixed in 2% paraformaldehyde in PBS containing 2 μg/ml Hoechst 33342 dye (Invitrogen) for 30 min at room temperature. After they were rinsed twice with PBS for 5 min, the sections were blocked with 50 μg/ml of purified goat IgG (MP Biomedicals, Solon, OH) in PBS for 90 min. This was followed by incubation with a 1:25 dilution of rat anti-mouse F4/80 hybridoma supernatant for 30 min and by rinsing and incubation with a 1:200 dilution of Alexa Fluor 549-labeled goat anti-rat IgG (KPL) or Alexa Fluor 555-labeled goat anti-rat IgG preadsorbed with mouse IgG (Invitrogen). After a final rinse in PBS, the slides were air dried, mounted with Vectashield Hardset H-1400 mounting medium (Vector), and stored at 4°C in the dark. Slides were viewed using a Leica true confocal scanner (TCS) spectral photometry II (SP2) acousto-optical beam splitter (AOBS) confocal microscope. Four representative images from each mouse were obtained at the same magnification. An operator who was blinded to the experimental treatment of each animal counted the number of F4/80- and Hoechst-positive macrophages in each field that contained RBCs using Metamorph software (version 7.1.7; Molecular Devices, Sunnvale, CA) and obtained the average per field for each mouse.
Detection of RBC surface C3, IgG, and PS.
For detection of C3, we used rat anti-mouse C3 clone 11H9 (Santa Cruz Biotechnology, Inc.), and for detection of surface IgG, we used Dylight 488-labeled goat anti-mouse IgG (KPL). One to 2 μl of packed RBCs was added to wells of a 96-well plate containing 100 μl of 1% BSA in RPMI 1640 (wash buffer), centrifuged, and resuspended in the same buffer containing a 1:100 dilution of primary antibody. After a 30-min incubation at room temperature, the RBCs stained for IgG were washed once in washed buffer and resuspended in 2% paraformaldehyde in PBS. The RBCs stained for C3 were resuspended in wash buffer containing a 1:500 dilution of secondary Dylight 488-labeled goat anti-rat IgG (KPL) and incubated as before. After a final wash, they were resuspended in paraformaldehyde as described above. For detection of phosphatidylserine (PS), 1 to 2 μl of packed RBCs was added to 200 μl of annexin V binding buffer (10 mM HEPES, 0.14 M NaCl, 2.5 mM CaCl2, adjusted to pH 7.5 with NaOH), mixed, and resuspended in a 1:100 dilution of fluorescein isothiocyanate (FITC)-labeled annexin V (Sigma-Aldrich) in binding buffer. After a 30-min incubation at room temperature in the dark, the RBCs were resuspended in binding buffer containing 2% paraformaldehyde.
Statistical analysis.
All data, unless otherwise noted, are presented as means ± standard deviations (SDs). Statistical analysis was performed using the Sigma Plot (version 11) program (Systat Software Inc., Chicago, IL). Comparison of means between two groups was performed using a t test or the Mann-Whitney rank sum test for non-normally distributed data. To test for differences among more than two groups, we used the general analysis of variance (ANOVA) or Kruskal-Wallis ANOVA for nonparametric data. Post hoc tests were used to correct for multiple comparisons. All tests were two-tailed with α at ≤0.05.
RESULTS
P. chabaudi/P. berghei infection leads to severe anemia with low parasitemia.
Our approach was based on the premise that some level of preimmunity was required for the development of SMA. Therefore, we tested a number of strategies to confer partial immunity to mice, including immunizations with parasites killed by different methods such as freeze-thawing, heating, and gamma irradiation, followed by homologous challenge. All of these approaches resulted in solid homologous sterile immunity. We thus decided to add a heterologous challenge on the basis of the premise that there would be some level of cross-reactivity but not solid sterile immunity. Therefore, we decided to test P. chabaudi infection followed by P. berghei challenge. Infection of C57BL/6 mice with P. berghei ANKA following recovery from a P. chabaudi infection resulted in SMA with an accompanying low parasitemia for the first 10 days of infection (Fig. 1A). This was followed by a second phase of uncontrolled parasitemia, more profound anemia, and death by day 21 postinfection (Fig. 1B). On the other hand, naïve C57BL/6 mice infected with P. berghei developed a higher parasitemia without a dramatic decline in RBC count and by day 7 postinfection died from cerebral complications, as expected (Fig. 1A and B) (39). Mice that were previously infected with P. chabaudi and were sham-infected did not develop parasitemia or anemia. Interestingly, the rise in parasitemia always followed shortly after a rise in the reticulocyte count (Fig. 1C and E). Microscopic evaluation of Giemsa-stained thin smears during this phase of infection revealed that the majority of IRBCs were reticulocytes, reflecting the preference of P. berghei for reticulocytes (29). Both P. berghei-infected and P. chabaudi/P. berghei-infected mice developed thrombocytopenia (Fig. 1F). Subsequently, abbreviated infections were carried out where P. chabaudi/P. berghei-infected mice were euthanized approximately 9 to 10 days postinfection (Fig. 1C and D).
Fig 1.

P. chabaudi/P. berghei infection results in anemia with low parasitemia. C57BL/6 mice were infected with 106 P. berghei IRBCs when naïve (P. berghei infected) or following recovery from P. chabaudi infection (Pch-Pb). P. chabaudi- and sham-infected mice received RPMI 1640 i.p. Day 0 designates the day of infection with P. berghei (Pb). (A and B) Parasitemia and RBC density of an infection allowed to proceed to conclusion; (C to F) abbreviated infection. Data points are means with standard deviations. n = 4 to 5 mice per group.
Liver and spleen pathology in the P. chabaudi/P. berghei-infected mouse model.
To characterize the pathology in the liver and spleen, we harvested these organs from the P. chabaudi/P. berghei-infected mice at day 9 or 10 after P. berghei infection or just prior to death for naïve P. berghei-infected animals. Both the livers and spleens of P. chabaudi/P. berghei-infected mice were larger than those of naïve, P. berghei-infected, and P. chabaudi- and sham-infected mice (Fig. 2). Livers from P. berghei-infected mice were also larger than those of naïve animals and P. chabaudi- and sham-infected animals (Fig. 2B). Compared to naïve animals, histological evaluation of the spleens of P. berghei-infected, P. chabaudi- and sham-infected, and P. chabaudi/P. berghei-infected animals showed expansion of the lymphoid follicles (see Fig. S2A in the supplemental material). In addition, the red pulp of P. chabaudi- and sham-infected mice and P. chabaudi/P. berghei-infected mice showed extensive infiltration by erythropoietic precursors which was more marked in the latter (see Fig. S2A in the supplemental material). This was confirmed by the observation of a greater proportion of lysis-resistant TER-119-positive/Ki67-positive cells in the spleens of P. chabaudi/P. berghei-infected mice by flow cytometry (see Fig. S3 in the supplemental material). Microscopic examination of the liver showed that hemozoin pigment was present within macrophages in all infected animals but was more markedly present in P. chabaudi/P. berghei-infected animals (see Fig. S2B in the supplemental material). In addition, livers of P. chabaudi/P. berghei-infected mice showed islands of erythropoietic precursors that were absent in all other groups (see Fig. S2B in the supplemental material). These findings are consistent with a strong extramedullary erythropoietic drive in P. chabaudi/P. berghei-infected animals as a result of SMA.
Fig 2.

P. chabaudi/P. berghei-infected animals have enlarged spleens and livers. Bars represent mean weights expressed as percentage of total body weight. Organs were harvested on day 6 or 7 postinfection for P. berghei-infected animals and day 9 for P. chabaudi- and sham-infected animals and P. chabaudi/P. berghei-infected animals. Error bars represent SDs. P values are based on analysis of variance with adjustment for multiple comparisons.
IL-12/IFN-γ and IL-10/TNF-α ratios were elevated in P. chabaudi/P. berghei-infected animals.
Plasma obtained at the time of euthanasia was used to measure the levels of 10 different cytokines. There were no detectable levels of IL-1β, IL-2, IL-4, IL-5, and GM-CSF among the experimental groups (data not shown). On the other hand, P. chabaudi/P. berghei-infected mice had higher levels of IL-12 and IL-10 than the other groups, although the difference did not reach statistical significance for the latter when corrected for multiple comparisons (Fig. 3A). P. berghei-infected mice had higher levels of IFN-γ and IL-6 than other mice, and TNF-α was similarly elevated in P. chabaudi/P. berghei-infected and P. berghei-infected mice. Because of antagonistic and stimulatory relationships between IL-10 and TNF-α and between IL-12 and IFN-γ, respectively, we expressed these cytokines as ratios. The IL-10/TNF-α and IL-12/IFN-γ ratios were higher in P. chabaudi/P. berghei-infected animals than in P. berghei-infected animals (Fig. 3B and C). These ratios were not calculated for P. chabaudi- and sham-infected mice because many of the mice had values of 0 for denominators, making this analysis meaningless.
Fig 3.

P. chabaudi/P. berghei-infected animals have increased IL-10/TNF-α and IL-12/IFN-γ ratios. Plasma samples from 3 separate experiments, obtained 9 or 10 days postinfection for P. chabaudi- and sham-infected animals and P. chabaudi/P. berghei-infected animals or 6 or 7 days postinfection for P. berghei-infected animals, were analyzed with a multiplex cytokine assay. (A) Mean cytokine concentrations across groups. The P value was obtained by analysis of variance with a post hoc test for multiple comparisons. *, significant difference when comparing that group to each of the other groups except when a horizontal bar is present, in which case the difference for a single comparison was significant. (B and C) IL-10/TNF-α and IL-12/IFN-γ ratios. Only ratios where the denominator was >0 were used for this analysis. The P value was obtained using the Mann-Whitney test. All error bars represent SDs.
P. chabaudi/P. berghei-infected animals have high levels of antibodies against P. berghei and P. chabaudi antigens.
To further characterize the model, parasite-specific total IgG antibodies were measured in sera obtained at the time of euthanasia, at about day 7 for P. berghei-infected mice and day 9 for P. chabaudi/P. berghei-infected mice. In P. chabaudi/P. berghei-infected mice, the response to P. berghei antigen was always more robust, even, surprisingly, with naïve serum. Therefore, we elected to present the antibody levels as reactivity indexes by dividing all values by the mean OD405 of the naïve serum (Fig. 4). There were no differences in the indexes of reactivity to P. chabaudi and P. berghei antigens within each group. However, P. chabaudi/P. berghei-infected animals had higher levels of antibody to either antigen than P. berghei-infected, P. chabaudi- and sham-infected, and naïve animals, but the difference was statistically significant only for P. berghei-infected and naïve animals due to the stringency of the multiple-comparison post hoc test.
Fig 4.
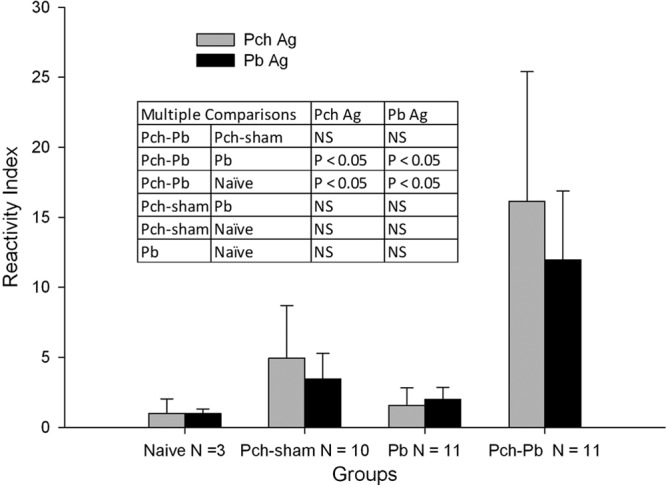
P. chabaudi/P. berghei-infected animals have increased parasite-specific IgG responses. Serum samples from 3 separate experiments, obtained at the time of euthanasia, were measured by ELISA. IgG responses to P. chabaudi (Pch) and P. berghei (Pb) antigens (Ag) are presented as mean reactivity indexes, the ratio of the OD405 for each sample to the mean OD405 for naïve sera. Error bars are standard deviations. Comparisons between two groups were by unpaired t test. There were no significant differences in reactivity to P. berghei or P. chabaudi antigen within groups. The table shows the results of the multiple-comparison testing between groups. NS, not significant.
Antimalarial treatment does not prevent the development of severe anemia in P. chabaudi/P. berghei-infected mice.
To determine the contribution of parasitemia to the anemia, we treated animals with chloroquine on day 5 postinfection. Chloroquine treatment of P. berghei-infected mice led to a rapid decline in parasitemia (Fig. 5A) and a small decline in RBC density of infected animals but not in uninfected animals (Fig. 5B). On the other hand, treatment of P. chabaudi/P. berghei-infected mice resulted in a degree of anemia similar to that in untreated animals (Fig. 5D and E), except that chloroquine-treated animals did show a faster recovery by day 9 in this experiment. Treatment of P. chabaudi- and sham-infected mice with chloroquine had no significant effect on the RBC density.
Fig 5.

Chloroquine (CQ) treatment does not prevent the development of anemia, and IRBCs are removed at an accelerated rate compared to URBCs. (A) P. berghei-infected mice were treated with chloroquine on day 5 postinfection, which resulted in rapid clearance of parasitemia and mild anemia (B). (C) DiD-labeled RBCs from P. chabaudi/P. berghei-infected animals at 3% parasitemia (IRBCs) or from uninfected mice (URBCs) were injected on day 5 postinfection into mice whose results are shown in panel A, and the clearance was tracked by flow cytometry. The data present the decline in the percentage of labeled RBCs from baseline. At 48 h, IRBCs (filled symbols) disappeared from the circulation faster than URBCs. (D) P. chabaudi/P. berghei-infected and P. chabaudi- and sham-infected mice were infected with P. berghei, and some groups of animals were treated with chloroquine on day 5 postinfection. P. chabaudi/P. berghei-infected animals developed severe anemia, despite treatment with chloroquine (E). (F) DiD-labeled RBCs, as for panel C, were injected on day 5 postinfection into P. chabaudi/P. berghei-infected animals or P. chabaudi- and sham-infected animals whose results are shown in panel D. IRBCs injected into P. chabaudi/P. berghei-infected animals disappeared from the circulation than faster URBCs. Data points are means ± SDs. P values were obtained using unpaired t test.
IRBCs are removed from circulation at an accelerated rate compared to URBCs.
In order to compare the rate of removal of IRBCs and URBCs in our model, we labeled RBCs obtained from P. chabaudi/P. berghei-infected mice with a parasitemia of 3% (IRBCs) or from uninfected animals (URBCs) ex vivo with the lipophilic membrane dye DiD (Invitrogen) and injected them into the retro-orbital plexuses of infected and uninfected animals under cover of chloroquine. Figures 5C and F depict the proportion of circulating labeled RBCs normalized to the baseline. IRBCs, whether injected into P. berghei-infected or naïve animals treated with chloroquine, disappeared faster than URBCs during the first 48 h (Fig. 5C). Likewise, when labeled IRBCs were injected into P. chabaudi/P. berghei-infected animals, they were removed from the circulation faster than URBCs (Fig. 5F). Surprisingly, URBCs injected into P. chabaudi- and sham-infected animals disappeared faster in the first 2 days following injection than those injected into P. chabaudi/P. berghei-infected animals (Fig. 5F). Frozen sections of spleen and liver tissue from these animals showed that most of the label accumulated in the liver (see Fig. S4 in the supplemental material). Paradoxically, untreated animals had more fluorescence in the liver than chloroquine-treated animals. This may have been due to dilution of the labeled RBCs in untreated animals by trapped unlabeled RBCs. This is supported by the larger liver size in untreated than treated animals. In addition, analysis of global fluorescence in P. chabaudi- and sham-infected animals showed increased spleen fluorescence (see Fig. S4 in the supplemental material), consistent with their ability to trap RBCs.
RBCs of P. chabaudi/P. berghei-infected mice have increased surface C3 and IgG.
Since the above-described experiments showed that IRBCs have decreased survival compared to URBCs, we set out to detect changes on the RBC surface that could mark them for destruction. Translocation of PS from the inner leaflet of the cell membrane to the outer leaflet is a marker of apoptosis. PS can bind to scavenger receptors on macrophages, leading to phagocytosis (24). Surface PS can be detected with annexin V (14). In addition, IgG and C3b are well-known opsonic agents that synergize to lead to erythrophagocytosis (13). Figure 6 shows that RBCs from P. chabaudi/P. berghei-infected mice had a progressive increase in surface PS, IgG, and C3. However, only the last two were significantly increased on day 9 postinfection. To test the contribution of C3 to the anemia, we infected mice with a C3−/− background with P. chabaudi-P. berghei. Figure 6D shows that the absence of C3 failed to prevent the development of anemia, although parasitemia was somewhat higher in C3−/− animals (Fig. 6E).
Fig 6.

RBCs of P. chabaudi/P. berghei-infected animals develop increased surface IgG and C3, but C3−/− mice develop anemia. RBCs from naïve, P. berghei-infected, or P. chabaudi/P. berghei-infected animals were obtained at various time points after infection and stained with annexin V–FITC (A), FITC-labeled goat anti-mouse IgG (B), or rat anti-mouse C3 monoclonal, followed by FITC-labeled anti-rat IgG (C). C3−/− mice developed severe anemia like wild-type mice (D). (E) The parasitemia of C3−/− mice is slightly higher than that of wild-type mice in the early phases of infection. MFI, mean fluorescence intensity.
Tracking of RBCs labeled in vivo and quantitation of in vivo erythrophagocytosis.
To track the fate of autologous RBCs, mice were subjected to in vivo RBC labeling by retro-orbital injection of the fluorescent dye Dylight 633 NHS ester (Thermo Fisher Scientific). Preliminary experiments in uninfected animals confirmed that most of the RBCs were promptly labeled following injection, with no decrease in survival of labeled RBCs. However, in infected animals we observed a gradual decline in the percentage of the positive population that was due to a decline in the median fluorescence intensity (data not shown). Immunofluorescence microscopy revealed significant accumulation of labeled RBCs within F4/80 macrophages in the liver that were more abundant in P. chabaudi/P. berghei-infected mice than in P. berghei-infected animals on days 3 and 7 postinfection (Fig. 7). Importantly, P. berghei-infected animals had higher parasitemia and less anemia than P. chabaudi/P. berghei-infected animals.
Fig 7.

P. chabaudi/P. berghei-infected mice have increased erythrophagocytosis in the liver. Animals were injected into a retro-orbital plexus with Dylight 633 NHS ester, which bound to RBCs. Animals were euthanized at predefined intervals. After euthanasia, livers and spleens were snap-frozen and sections were stained with Hoechst 33342 and monoclonal antibody F4/80, followed by FITC-labeled goat anti-rat IgG. The pictures show liver Kupffer cells in green with intracellular RBCs in red. Little or no erythrophagocytosis was observed in livers from uninfected animals (data not shown). PI, postinfection.
DISCUSSION
We aimed to develop and characterize a relevant murine model of SMA that is highly reproducible. Infection of C57BL/6 mice with P. berghei ANKA is uniformly fatal, whereas infection with P. chabaudi leads to severe anemia with high parasitemia, followed by full recovery (39). We determined that infection with P. chabaudi followed after recovery by infection with P. berghei ANKA results in anemia with relatively low level parasitemia in a highly reproducible manner. The major advantages of this model over a recently published rodent model (11, 18) are the occurrence of synchronized infections, the predictable timing of the anemia, and a short preparative time of only 6 to 8 weeks. Another important advantage of our model is the strain of mouse that we used. C57BL/6 is the most commonly used background for genetically modified mice, allowing the exploration of the role of different genes in the pathogenesis of SMA.
Our model has a number of features that are very relevant to human P. falciparum infections. SMA is more often found in areas of intense malaria transmission where children suffer repeated infections (17, 41). This observation suggests that some level of immunity or at least immune stimulation is required for SMA to occur. Thus, our model is in agreement with this observation. Other features of our model that have been documented in humans include a level of anemia that is out of proportion to the parasite burden (20, 37) and the fact that uninfected RBCs continue to be destroyed after treatment (4, 28).
The carefully orchestrated balance between proinflammatory and regulatory cytokines produced by the host is important in determining the outcome of malaria infection (2, 9). Therefore, we measured proinflammatory and anti-inflammatory cytokines. We found that P. berghei-infected mice had relatively high levels of the proinflammatory cytokines IL-6, IFN-γ, and TNF-α, whereas P. chabaudi/P. berghei-infected mice had relatively high levels of IL-10 and IL-12, although the higher IL-10 levels were not statistically significant. The unregulated production of proinflammatory cytokines is central to the development of cerebral malaria (CM) (7, 48). Normally, IL-12, produced by macrophages and dendritic cells (43), induces the production of IFN-γ by NK cells and CD8+ T cells, but in P. berghei-infected mice, IFN-γ is produced in an IL-12-independent manner (12), which explains the high IL-12/IFN-γ ratios in these animals. IL-12 has been shown to play a critical role in controlling parasitemia (3, 30, 31, 40, 44), which may explain the low parasitemia in P. chabaudi/P. berghei-infected mice. IL-12 also plays a role in the stimulation of erythropoiesis (32, 33), and this may account for the excessive extramedullary hematopoiesis and brisk erythropoietic response in the same animals. The other important regulatory cytokine is IL-10, which normally counters the action of TNF-α and IL-12 by downregulating their production (6, 8, 19). P. berghei-infected mice produced inappropriately low levels of this cytokine. Thus, sequential P. chabaudi-P. berghei infection restored the normal regulatory relationship between IL-12, IL-10, IFN-γ, and TNF-α. These data suggest that the P. chabaudi-P. berghei infection model can be an important tool to study the role of these cytokines in the pathogenesis of CM and SMA. In addition, the model can serve to explore how innate immune responses change with repeated infections, an important but poorly understood phenomenon.
P. berghei infection of P. chabaudi-experienced mice elicited strong cross-reactive antibody responses against P. chabaudi and P. berghei antigens. The raw antibody data showed higher recognition of P. berghei antigens than P. chabaudi antigens by all groups, even naïve animals, suggesting that P. berghei antigens are more promiscuous in their interactions with antibodies. Similar cross-reactive responses were previously observed (22). When this innate promiscuity was corrected by expressing the data as reactivity indexes (Fig. 4), we observed no significant difference in the antibody responses to P. berghei or P. chabaudi antigens. The increased antibody responses in P. chabaudi/P. berghei-infected animals may be in part due to their ability to produce IL-12, which has been found to be critical for production of cytotoxic antibodies (45).
In an attempt to determine the contribution of red cell destruction by the parasite to development of anemia, we treated mice with antimalarials on day 5 postinfection and infused labeled RBCs by retro-orbital injection to track their disappearance from the circulation. Treatment with neither chloroquine nor artesunate (data not shown) prevented the development of SMA in P. chabaudi/P. berghei-infected mice. Further, P. berghei-infected mice treated with chloroquine did not develop SMA. These data provide additional evidence that the mechanism of SMA in our model is not due to the destruction of the RBCs by the parasite, requires previous exposure to malaria, as it does in humans, and it is set in motion early during infection. Furthermore, the failure of antimalarial treatment to prevent anemia in our model has one very clinically relevant implication which should be investigated in clinical studies: that RBC destruction may continue in children with SMA, despite initiation of antimalarial therapy.
Infusion of labeled IRBCs and URBCs in naïve, P. berghei-infected, and P. chabaudi/P. berghei-infected mice showed that IRBCs were removed at an accelerated rate compared to URBCs, at least during the first 48 h postinfusion. However, these experiments also demonstrated the contribution of the host, since the survival of both URBCs and IRBCs was shorter in P. chabaudi/P. berghei-infected animals than in P. berghei-infected or naïve animals. Paradoxically, the survival of URBCs in the first 2 days postinjection was shorter in P. chabaudi- and sham-infected mice than in P. chabaudi/P. berghei-infected mice, suggesting that the filtering capacity of the animals evolves during infection. Analysis of the global fluorescence of frozen sections of livers and spleens clearly showed that the liver is the major site where RBCs are removed, regardless of the setting (see Fig. S4 in the supplemental material). This observation is consistent with the finding of hepatomegaly in P. chabaudi/P. berghei-infected mice and in children with SMA (15, 37). The spleen seemed to play a minor role in our model, perhaps due to the extensive infiltration with erythropoietic cells, but its role increased in P. chabaudi- and sham-infected mice, judging from the increased global fluorescence of their spleens, which was confirmed microscopically. Extramedullary hematopoiesis has not been reported in the spleens of patients with severe P. falciparum malaria (46), and it is possible that in this setting it may play a larger role in trapping URBCs. Our results are remarkably consistent with published results from animal and human studies showing premature removal of uninfected RBCs in mice (38) and destruction of RBCs, despite appropriate treatment of malaria infection in humans (4, 28, 37).
The main mechanism of RBC destruction seems to be phagocytosis, as evidenced by the increased erythrophagocytosis in the livers of P. chabaudi/P. berghei-infected and, less so, of P. berghei-infected animals (Fig. 7). Immunofluorescence microscopy of the spleen was a lot more difficult to interpret due to the overwhelming hypercellularity. Our experiments suggest that RBCs suffer a number of surface alterations during rodent malaria infection to include translocation of PS to the outer membrane, increased deposition of IgG, and increased C3 deposition, any one or combination of which could lead to erythrophagocytosis. Similarly, RBCs of children with SMA have been shown to have increased surface IgG and C3 deposition (36, 47). The fact that P. chabaudi/P. berghei-infected C3−/− mice developed SMA as well suggests that C3 deposition alone cannot account for the RBC destruction and that damage from multiple mechanisms may be required. Alternatively, the presence of higher parasitemia in C3−/− mice may have cancelled any beneficial effect from C3 deficiency (Fig. 6E). Possible mechanisms of antibody-mediated uninfected red cell opsonization include targeting of self-antigens (21, 34) or malaria antigens on the RBC surface (25). In addition, antibodies against malaria antigens could form immune complexes that deposit on the RBC surface, leading to complement activation and deposition via a bystander phenomenon.
In summary, we have presented a practical and novel mouse model of SMA that has many immunological and clinical features that are relevant to P. falciparum infection in humans. This model provides an excellent platform on which to study the role of protective immune responses against CM and SMA in the pathogenesis of these conditions, the changes in the innate response to malaria with repeated infections, and the fate of uninfected RBCs during SMA and begin to test adjunctive immunomodulatory strategies for the treatment of SMA.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant HL 71502 (principal investigator, José A. Stoute), funds from the Uniformed Services University of the Health Sciences, the U.S. Department of Defense Infectious Diseases Research Program, and the Pennsylvania State University College of Medicine. Core facility services and instruments used in this project were funded, in part, under a grant from the Pennsylvania Department of Health using Tobacco Settlement Funds.
The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
We are grateful to Wade Edris for assistance with the use of the confocal core facilities at the Pennsylvania State University College of Medicine. We are also thankful to Christopher Norbury for the kind gift of F4/80 hybridoma supernatant.
Footnotes
Published ahead of print 11 June 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abdalla SH, Weatherall DJ, Wickramasinghe SN, Hughes M. 1980. The anaemia of P. falciparum malaria. Br. J. Haematol. 46:171–183 [DOI] [PubMed] [Google Scholar]
- 2. Akanmori BD, et al. 2000. Distinct patterns of cytokine regulation in discrete clinical forms of Plasmodium falciparum malaria. Eur. Cytokine Netw. 11:113–118 [PubMed] [Google Scholar]
- 3. Boutlis CS, et al. 2003. Plasma interleukin-12 in malaria-tolerant Papua New Guineans: inverse correlation with Plasmodium falciparum parasitemia and peripheral blood mononuclear cell nitric oxide synthase activity. Infect. Immun. 71:6354–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camacho LH, et al. 1998. The course of anaemia after the treatment of acute, falciparum malaria. Ann. Trop. Med. Parasitol. 92:525–537 [DOI] [PubMed] [Google Scholar]
- 5. Chang KH, Tam M, Stevenson MM. 2004. Modulation of the course and outcome of blood-stage malaria by erythropoietin-induced reticulocytosis. J. Infect. Dis. 189:735–743 [DOI] [PubMed] [Google Scholar]
- 6. D'Andrea A, et al. 1993. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 178:1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Kossodo S, Grau GE. 1993. Profiles of cytokine production in relation with susceptibility to cerebral malaria. J. Immunol. 151:4811–4820 [PubMed] [Google Scholar]
- 8. de Waal MR, Abrams J, Bennett B, Figdor CG, de Vries JE. 1991. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174:1209–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dodoo D, et al. 2002. Absolute levels and ratios of proinflammatory and anti-inflammatory cytokine production in vitro predict clinical immunity to Plasmodium falciparum malaria. J. Infect. Dis. 185:971–979 [DOI] [PubMed] [Google Scholar]
- 10. Egan AF, Fabucci ME, Saul A, Kaslow DC, Miller LH. 2002. Aotus New World monkeys: model for studying malaria-induced anemia. Blood 99:3863–3866 [DOI] [PubMed] [Google Scholar]
- 11. Evans KJ, Hansen DS, van Roolijen N, Buckingham LA, Schofield L. 2006. Severe malarial anemia of low parasite burden in rodent models results from accelerated clearance of uninfected erythrocytes. Blood 107:1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng C, et al. 1999. An alternate pathway for type 1 T cell differentiation. Int. Immunol. 11:1185–1194 [DOI] [PubMed] [Google Scholar]
- 13. Fries LF, Siwik SA, Malbran A, Frank MM. 1987. Phagocytosis of target particles bearing C3b-IgG covalent complexes by human monocytes and polymorphonuclear leucocytes. Immunology 62:45–51 [PMC free article] [PubMed] [Google Scholar]
- 14. Geldwerth D, et al. 1999. Detection of phosphatidylserine surface exposure on human erythrocytes using annexin V-ferrofluid. Biochem. Biophys. Res. Commun. 258:199–203 [DOI] [PubMed] [Google Scholar]
- 15. Giha HA, Elghazali G, Elgadir TM, Elbasit IE, Elbashir MI. 2009. Severe malaria in an unstable setting: clinical and laboratory correlates of cerebral malaria and severe malarial anemia and a paradigm for a simplified severity scoring. Eur. J. Clin. Microbiol. Infect. Dis. 28:661–665 [DOI] [PubMed] [Google Scholar]
- 16. Gomez ND, et al. 2011. Deletion of a malaria invasion gene reduces death and anemia, in model hosts. PLoS One 6:e25477 doi:10.1371/journal.pone.0025477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greenwood BM. 1997. The epidemiology of malaria. Ann. Trop. Med. Parasitol. 91:763–769 [DOI] [PubMed] [Google Scholar]
- 18. Helegbe GK, et al. 2009. Rate of red blood cell destruction varies in different strains of mice infected with Plasmodium berghei-ANKA after chronic exposure. Malar. J. 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho M, et al. 1998. Endogenous interleukin-10 modulates proinflammatory response in Plasmodium falciparum malaria. J. Infect. Dis. 178:520–525 [DOI] [PubMed] [Google Scholar]
- 20. Jakeman GN, Saul A, Hogarth WL, Collins WE. 1999. Anaemia of acute malaria infections in non-immune patients primarily results from destruction of uninfected erythrocytes. Parasitology 119(Pt 2):127–133 [DOI] [PubMed] [Google Scholar]
- 21. Jarra W. 1983. Protective immunity to malaria and anti-erythrocyte autoimmunity. Ciba Found. Symp. 94:137–158 [DOI] [PubMed] [Google Scholar]
- 22. Jarra W, Hills LA, March JC, Brown KN. 1986. Protective immunity to malaria. Studies with cloned lines of Plasmodium chabaudi chabaudi and P. berghei in CBA/Ca mice. II. The effectiveness and inter- or intra-species specificity of the passive transfer of immunity with serum. Parasite Immunol. 8:239–254 [DOI] [PubMed] [Google Scholar]
- 23. Jones TR, et al. 2002. Anemia in parasite- and recombinant protein-immunized Aotus monkeys infected with Plasmodium falciparum. Am. J. Trop. Med. Hyg. 66:672–679 [DOI] [PubMed] [Google Scholar]
- 24. Kuypers FA, de Jong K. 2004. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell. Mol. Biol. (Noisy-le-grand) 50:147–158 [PubMed] [Google Scholar]
- 25. Layez C, et al. 2005. Plasmodium falciparum rhoptry protein RSP2 triggers destruction of the erythroid lineage. Blood 106:3632–3638 [DOI] [PubMed] [Google Scholar]
- 26. Lee SH, et al. 1989. Antibody-dependent red cell removal during P. falciparum malaria: the clearance of red cells sensitized with an IgG anti-D. Br. J. Haematol. 73:396–402 [DOI] [PubMed] [Google Scholar]
- 27. Looareesuwan S, et al. 1987. Dynamic alteration in splenic function during acute falciparum malaria. N. Engl. J. Med. 317:675–679 [DOI] [PubMed] [Google Scholar]
- 28. Looareesuwan S, et al. 1987. Reduced erythrocyte survival following clearance of malarial parasitaemia in Thai patients. Br. J. Haematol. 67:473–478 [DOI] [PubMed] [Google Scholar]
- 29. McNally J, O'Donovan SM, Dalton JP. 1992. Plasmodium berghei and Plasmodium chabaudi chabaudi: development of simple in vitro erythrocyte invasion assays. Parasitology 105(Pt 3):355–362 [DOI] [PubMed] [Google Scholar]
- 30. Mohan K, Moulin P, Stevenson MM. 1997. Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. J. Immunol. 159:4990–4998 [PubMed] [Google Scholar]
- 31. Mohan K, Sam H, Stevenson MM. 1999. Therapy with a combination of low doses of interleukin 12 and chloroquine completely cures blood-stage malaria, prevents severe anemia, and induces immunity to reinfection. Infect. Immun. 67:513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mohan K, Stevenson MM. 1998. Dyserythropoiesis and severe anaemia associated with malaria correlate with deficient interleukin-12 production. Br. J. Haematol. 103:942–949 [DOI] [PubMed] [Google Scholar]
- 33. Mohan K, Stevenson MM. 1998. Interleukin-12 corrects severe anemia during blood-stage Plasmodium chabaudi AS in susceptible A/J. mice. Exp. Hematol. 26:45–52 [PubMed] [Google Scholar]
- 34. Mota MM, Brown KN, Holder AA, Jarra W. 1998. Acute Plasmodium chabaudi chabaudi malaria infection induces antibodies which bind to the surfaces of parasitized erythrocytes and promote their phagocytosis by macrophages in vitro. Infect. Immun. 66:4080–4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MR4/ATCC 2008. Methods in malaria research. MR4/ATCC, Manassas, VA [Google Scholar]
- 36. Owuor BO, et al. 2008. Reduced immune complex binding capacity and increased complement susceptibility of red cells from children with severe malaria-associated anemia. Mol. Med. 14:89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Price RN, et al. 2001. Factors contributing to anemia after uncomplicated falciparum malaria. Am. J. Trop. Med. Hyg. 65:614–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salmon MG, De Souza JB, Butcher GA, Playfair JH. 1997. Premature removal of uninfected erythrocytes during malarial infection of normal and immunodeficient mice. Clin. Exp. Immunol. 108:471–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanni LA, Fonseca LF, Langhorne J. 2002. Mouse models for erythrocytic-stage malaria. Methods Mol. Med. 72:57–76 [DOI] [PubMed] [Google Scholar]
- 40. Sedegah M, Finkelman F, Hoffman SL. 1994. Interleukin 12 induction of interferon gamma-dependent protection against malaria. Proc. Natl. Acad. Sci. U. S. A. 91:10700–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Snow RW, et al. 1994. Severe childhood malaria in two areas of markedly different falciparum transmission in east Africa. Acta Trop. 57:289–300 [DOI] [PubMed] [Google Scholar]
- 42. Srichaikul T, Panikbutr N, Jeumtrakul P. 1967. Bone-marrow changes in human malaria. Ann. Trop. Med. Parasitol. 61:40–51 [DOI] [PubMed] [Google Scholar]
- 43. Stevenson MM, Riley EM. 2004. Innate immunity to malaria. Nat. Rev. Immunol. 4:169–180 [DOI] [PubMed] [Google Scholar]
- 44. Stevenson MM, Tam MF, Wolf SF, Sher A. 1995. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J. Immunol. 155:2545–2556 [PubMed] [Google Scholar]
- 45. Su Z, Stevenson MM. 2002. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J. Immunol. 168:1348–1355 [DOI] [PubMed] [Google Scholar]
- 46. Urban BC, et al. 2005. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect. Immun. 73:1986–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waitumbi JN, Opollo MO, Muga RO, Misore AO, Stoute JA. 2000. Red cell surface changes and erythrophagocytosis in children with severe Plasmodium falciparum anemia. Blood 95:1481–1486 [PubMed] [Google Scholar]
- 48. Wu JJ, et al. 2010. Natural regulatory T cells mediate the development of cerebral malaria by modifying the proinflammatory response. Parasitol. Int. 59:232–241 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.