

Abstract
To explore the possibility of producing a narrow distribution of mid- to long-chain hydrocarbons from ethylene as a chemical feedstock, co-oligomerization of ethylene and linear α-olefins (LAOs) was investigated, using a previously reported chromium complex, [CrCl3(PNPOMe)] (1, where PNPOMe = N,N-bis(bis(o-methoxyphenyl)phosphino)methylamine). Activation of 1 by treatment with modified methylaluminoxane (MMAO) in the presence of ethylene and 1-hexene afforded mostly C6 and C10 alkene products. The identities of the C10 isomers, assigned by detailed gas chromatographic and mass spectrometric analyses, strongly support a mechanism that involves five- and seven-membered metallacyclic intermediates comprising of ethylene and LAO units. Using 1-heptene as a mechanistic probe, it was established that 1-hexene formation from ethylene is competitive with formation of ethylene/LAO co-trimers, and that co-trimers derived from one ethylene and two LAO molecules are also generated. Complex 1/MMAO is also capable of converting 1-hexene to C12 dimers and C18 trimers, albeit with poor efficiency. The mechanistic implications of these studies are discussed and compared to previous reports of olefin co-trimerization.
INTRODUCTION
Ethylene, the most widely produced organic compound in the world,1,2 is attractive as a cheap and abundant feedstock for the manufacturing of value-added products including linear α-olefins (LAOs), a class of commodity chemicals that are used in industries such as synthetic polymers, detergents, plasticizers, and lubricants, and have a global demand of over four million tons per year.3 LAOs are produced primarily by catalytic oligomerization of ethylene via linear chain growth (Cossee-Arlman mechanism).4 Such reactions produce a non-selective Schulz-Flory distribution of oligomers, requiring additional procedures to separate the desired products. More recently, selective oligomerization of ethylene to 1-hexene and/or 1-octene has been reported,5–8 first using chromium/2-ethylhexanoate9,10 and subsequently with complexes containing a diverse array of transition metal ions and ligand architectures.5 These reactions most likely proceed through a metallacycle ring expansion mechanism, in which oxidative coupling of two ethylene molecules at an electrophilic metal center affords a metallacyclopentane intermediate, which undergoes ethylene insertion to give a metallacycloheptane that eliminates 1-hexene (Scheme 1).11–16 The elimination step may involve either a β-hydride elimination/reduction elimination sequence or a concerted 3,7-hydride shift; it is difficult to distinguish between these alternatives,13 but the latter is favored by recent theoretical studies.15–17 Whichever mechanism operates, the elimination is expected on geometric grounds to be much more favorable, relative to further insertion, for the seven-membered rather than for the five-membered metallacycle, accounting for the observed selectivity. With some catalysts, and especially at higher ethylene pressures, a second insertion becomes favored over elimination, yielding 1-octene. This chemistry has also been extended to homo-trimerization of LAOs18,19 and other olefinic substrates.20–22
Scheme 1.
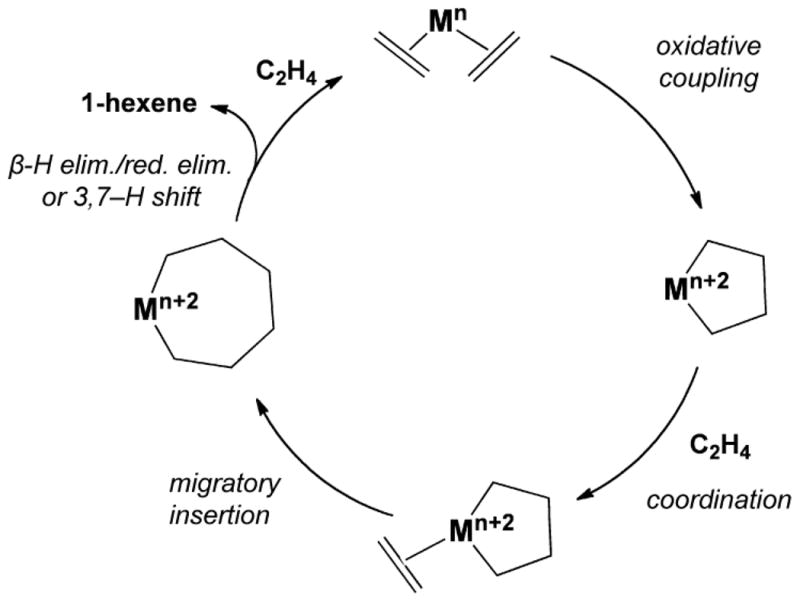
Proposed catalytic cycle for the selective trimerization of ethylene to 1-hexene
Significant effort has been devoted to understanding the chemistry of selective ethylene oligomerization,5–7 including issues such as catalyst initiation, effect of activators, metal oxidation states, and reaction mechanism. One interesting feature of ethylene trimerization is the formation of some C10 alkenes in addition to 1-hexene, suggested to be secondary products from the coupling of two ethylenes with 1-hexene. Co-oligomerization of ethylene and an α-olefin, such as propylene,13 1-octene,23 and styrene,20 by homogeneous chromium catalysts has been demonstrated previously. “Product recycling,” wherein co-oligomerization of ethylene with the primary α-olefin produced competes effectively with homo-oligomerization, could constitute a selective route to hydrocarbons larger than C6 or C8, using only ethylene as starting material.24 Here we explore the feasibility of such a process by examining the efficiency of 1-hexene incorporation in co-trimerization, using the previously reported chromium complex, [CrCl3(PNPOMe)] (1, where PNPOMe = N,N-bis(bis(o-methoxyphenyl)phosphino)methylamine, Chart 1) as the catalyst precursor.25,26
Chart 1.
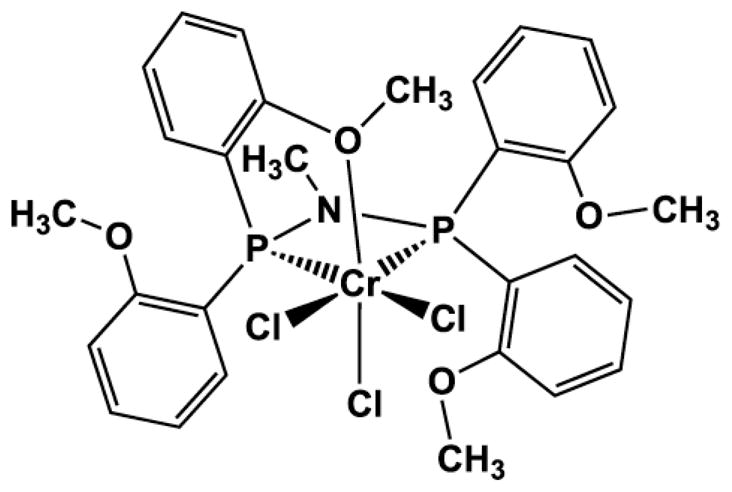
Structure of the chromium pre-catalyst [CrCl3(PNPOMe)] (1)
RESULTS
Co-oligomerization of ethylene and 1-hexene
To establish a benchmark for the present studies, compound 1 was treated with modified methylaluminoxane (MMAO) in chlorobenzene under an atmosphere of ethylene. Analysis of the reaction mixture after 1 h by gas chromatography (GC) (Figure S1A) revealed that C6 (92 wt %) and C10 (8 wt %) alkenes were the primary products (Table 1, entry 1), with a productivity of 571 ± 14 g/g Cr·h; 1-hexene was the only detectable C6 isomer and only trace amounts (<10 mg) of polyethylene were obtained.
Table 1.
Product distribution of olefin homo- and co-trimerizationa
| Entry | Monomer(s) | Productivitye (g/g Cr/h) | Products (wt %) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C6f | C10 | C11 | C12 | C13 | C14 | C15 | C16 | C18 | |||
| 1 | ethyleneb | 571 ± 14 | 92 | 8 | – | – | – | – | – | – | – |
| 2 | ethyleneb/1-hexenec | 926 ± 93a | nd | 94 | – | 1 | – | 4 | – | – | 0.2 |
| 3 | ethyleneb/1-heptene,d | 863 ± 97 | 51 | 2 | 43 | – | 0.5 | 0.6 | 0.8 | 1.2 | – |
| 4 | 1-hexenec | 7.6 ± 0.3 | – | – | – | 27 | – | – | – | – | 73 |
Reaction condition: complex 1 (12 μmol), monomer(s), and MMAO (250 mg, ~55 equiv, solution in isohexanes) diluted to 10 mL with chlorobenzene, room temperature.
Ethylene (1 atm).
1-Hexene (35 mmol).
1-Heptene (24 mmol).
The standard deviations were determined from reactions performed in triplicate.
The C6 fraction contains >99% 1-hexene.
To favor the production of C10 olefins and to obtain sufficient material for characterization, the same reaction was performed with an excess of added 1-hexene; the ratio of 1-hexene:ethylene in solution, under the reaction conditions employed, based on NMR spectroscopic measurement, was approximately 1:1. Quantification of the olefins heavier than C6 by GC showed that C10 olefins were the major products (94 wt %), followed by much smaller amounts of C12 (1 wt %), C14 (4 wt %), and C18 (0.2 wt %) olefins (Figure S1B). Because 1-hexene was used as a starting material, the amount that was formed during the reaction could not be determined; the productivity based on the other products was 926 ± 93 g/g Cr·h (Table 1, entry 2).
The C10 olefins were isolated by fractional distillation of the crude reaction mixture. GC analysis showed three major species with retention times of 36.98, 37.19, and 37.38 min, and several minor ones between 37.75–38.07 min (Figure 1, black trace). By comparison to authentic samples of the nine isomers expected from co-trimerization through a metallacycle mechanism (Scheme 2), it is apparent that 3-propyl-1-heptene (P, red trace) corresponds to the peak at 36.98 min, 4-ethyl-1-octene (O, orange trace) corresponds to the peak at 37.19 min, 5-methyl-1-nonene (N, yellow trace) corresponds to the peak at 37.38 min, 2-butyl-1-hexene (M, green trace) corresponds to the peak at 37.75 min, and the linear C10 olefins (H-L; light blue, brown, pink, purple, and blue traces) correspond to the peaks between 37.78–38.07 min. The relative amounts of C10 olefins were determined from the integrated areas of the peaks in the gas chromatogram: linear C10 olefins H–L, 3%; M, 5%; N, 47%; O, 18%; and P, 27% (Scheme 2).
Figure 1.

Gas chromatogram of the C10 alkenes (black, bottom trace) obtained by fractional distillation from the reaction of 1/MMAO/ethylene/1-hexene; the products appear at 36.98, 37.19, 37.38, 37.75, 37.86, 38.07 min. The C10 olefin standards are shown directly above (from top to bottom, RT = retention time in min): cis-5-decene (H, light blue; 38.08), cis-4-decene (I, brown; 38.07), 1-decene (J, pink; 37.84), trans-4-decene (K, purple; 37.86), trans-5-decene (L, blue; 37.90), 2-butyl-1-hexene (M, green; 37.76), 5-methyl-1-nonene (N, yellow; 37.44), 4-ethyl-1-octene (O, orange; 37.22), 3-propyl-1-heptene (P, red; 37.02). The peak marked with an asterisk (*) is the trans-4-decene impurity present in the 3-propyl-1-heptene standard (see SI).
Scheme 2.

C6 and C10 alkenes formed via co-trimerization of ethylene and 1-hexene and their hydrogenation productsa
aThe percentages shown in green correspond to the relative amounts of C10 alkenes present in the reaction product. Similarly, the percentages shown in blue correpond to the relative amounts of C10 alkanes present after the alkenes were subjected to hydrogention. The discrepancies (up to ±5%) between the total amount of each group of alkene isomers observed and the amounts of hydrogenation products obtained are attributed to experimental error.
To obtain further support for the assignments, a sample of the C10 olefin mixture was hydrogenated and analyzed by GC. As shown in the black trace in Figure 2, the gas chromatogram of the saturated C10 products displays two prominent peaks at 36.79 and 36.90 min, and a significantly smaller one at 37.72 min. Comparing the retention times of the peaks in this trace to those of the expected products (Scheme 2) indicate that indeed, they correspond to 2-ethyloctane (S, red trace), 5-methylnonane (R, orange trace), and decane (Q, green trace), respectively, with a relative ratio of 40:56:4 (Figure 3). (Although the expected ratio based on the distribution of C10 alkenes is 45:52:3, this discrepancy is attributable to experimental error, such as non-ideal peak integration, slight differences in the GC response factors for the different isomers, or incomplete hydrogenation.) Consistent with the chemical formula C10H22, mass spectral analysis reveals that all three species in the hydrogenated sample display a peak with an m/z value of 142. In addition, each compound exhibits a unique fragmentation pattern in its mass spectrum that matches well with that of the species to which it was assigned (Figure 3).
Figure 2.
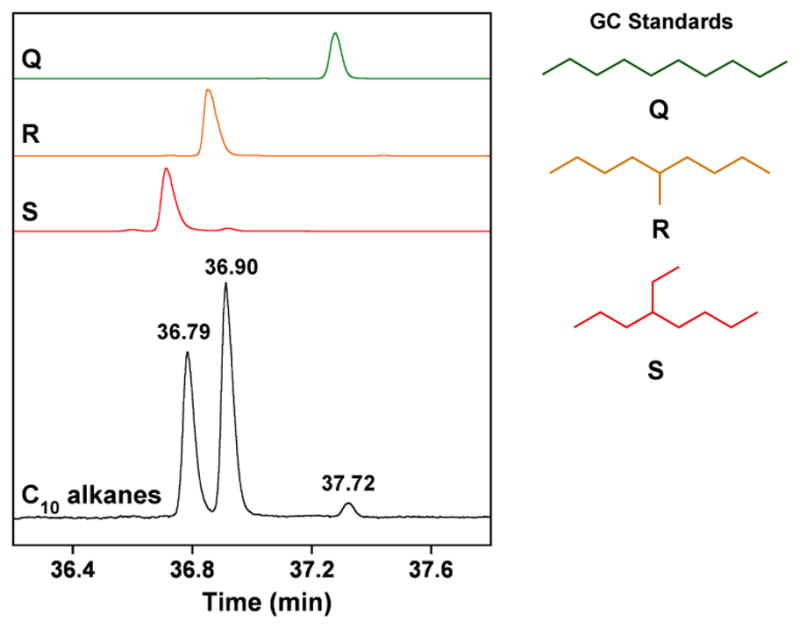
Gas chromatogram of the C10 alkanes (black, bottom trace) obtained from hydrogenation of the C10 alkene mixture (see Figure 1); the products appear at 36.79, 36.90, and 37.72 min. The C10 alkane standards are shown directly above (from top to bottom, RT = retention time in min): decane (Q, green; 37.28), 5-methylnonane (R, orange; 36.86), and 2-ethyloctane (S, red; 36.71).
Figure 3.

Electron-impact (EI) ionization mass spectra (black) of the peaks that appear in the C10 alkane sample (Figure 2) at A) 36.79 min, B) 36.90 min, and C) 37.72 min. For comparison, the mass spectra for authentic samples of 4-ethyloctane (pink), 5-methylnonane (blue), and decane (green) are shown directly above that for A, B, and C, respectively. RT = retention time.
Homo-oligomerization of 1-hexene
To test the feasibility of LAO homo-oligomerization by the Cr-PNPOMe system, reactions were conducted using 1/MMAO in the presence of 1-hexene without added ethylene. GC-MS analysis of this reaction mixture showed that both C12 and C18 olefins were formed, in a ratio of 27:73 wt % (Figure S2; Table 1, entry 4). Although several isomers of each chain length were obtained, no attempt was made to ascertain their identities. The overall productivity was quite low, 7.6 ± 0.3 g/g Cr·h.
Co-oligomerization of ethylene and 1-heptene
An analogous reaction was carried out with ethylene and 1-heptene as co-monomers, in approximately 4:3 ratio (as determined by NMR spectroscopic measurements). GC analysis of the crude mixture showed that 1-hexene and C11 olefins constituted approximately 51 and 43 wt %, respectively, of the total products (Figure S1C; Table 1, entry 3). Minor amounts of C10 (2 wt %), C13 (0.5 wt %), C14 (0.6 wt %), C15 (0.8 wt %), and C16 (1.2 wt %) olefins were also observed. The overall productivity was 863±97 g/g Cr·h.
DISCUSSION
The chromium (III) complex CrCl3(PNPOMe) (1) was selected as the pre-catalyst for the present studies because of its high selectivity for ethylene trimerization.25–27 That selectivity has been attributed, in part, to the interaction of a pendant methoxy moiety of the PNPOMe ligand with the chromium center;7 when these methoxy groups are absent, as in the [CrCl3(PNP)]2 variant (where PNP = N,N-bis(diphenylphosphino)methylamine), both ethylene tri- and tetramerization occur.28–30 Although the methoxyaryl functionalities are not essential to obtaining a high ratio of 1-hexene versus 1-octene,31 only the PNPOMe ligand forms a catalyst that produces 1-hexene with >99% trimerization selectivity.
The ability of 1 to facilitate selective trimerization is indicated by the high yield of C10 olefins, ~94 wt % of the quantifiable products (i.e., those other than C6) obtained when ethylene/1-hexene were used as co-monomers (Table 1, entry 2). Because identification of the reaction products would provide insight into their possible mechanism of formation, studies were undertaken to characterize the C10 species. As shown in Scheme 2, there are three possible metallacycloheptanes from co-trimerization of two ethylenes with one 1-hexene, and two possible routes to each of them. Unsubstituted chromacyclopentane A (from oxidative coupling of two ethylene molecules with the Cr center) can undergo insertion of ethylene to give D (i.e. homo-trimerization), or either 2,1- or 1,2-insertion of 1-hexene, leading to chromacycloheptanes E and F respectively. Alternatively chromacyclopentanes B and C, formed by oxidative coupling of ethylene and 1-hexene, could undergo subsequent insertion of another ethylene to yield chromacycloheptanes E and G or F and G, respectively.
Elimination of olefin from the various chromacycloheptanes, by either the stepwise or concerted route (see above), would afford the products shown in Scheme 2. Only linear decenes can result from E, with the position of the double bond depending on the relative preference for hydride elimination (or transfer) from substituted vs. unsubstituted and endo- vs. exocyclic Cβ positions. Likewise, F should give rise to either 2-butyl-1-hexene (M) or 5-methyl-1-nonene (N), and G, to 4-ethyl-1-octene (O) or 3-propyl-1-heptene (P). As shown in Figure 1, GC analysis shows that all of the branched, and some or all of the unbranched, expected C10 olefins are indeed formed, supporting the proposed mechanism.
Detailed analysis of the C10 olefin distribution indicates several important characteristics of the [Cr-PNPOMe] intermediates. First, since only 3–4% of the C10 olefins are linear, their precursor intermediate E must be significantly disfavored. E can be generated either by 2,1-insertion of 1-hexene into chromacyclopentane A or ethylene insertion into the unsubstituted side of chromacyclopentane B; both sequences must therefore be of low probability. In each case there are several possible interpretations. A is certainly formed, since homo-trimerization of ethylene is taking place (see below); hence insertion of olefin into A exhibits a strong preference for ethylene over LAO and/or for 1,2-over 2,1-insertion. In previous studies using a Cr-PNPOMe/biphenyldiyl complex as a model for the chromacyclopentane intermediate,13 it was found that ethylene inserts into the five-membered metallacycle more than twenty times faster than LAOs, and the latter insert exclusively in 1,2-mode, consistent with the current observations. In contrast, in the co-trimerization of ethylene and styrene by 1/MAO the major products (~80%) were those derived from 2,1-insertion of styrene into A, and no 1,2-insertion products were observed.20 A preference for 2,1-insertion of styrene is common, both in the polymer literature32–34 and elsewhere,35–38 although 1,2-insertion can be achieved using sterically-hindered catalysts.39
It seems quite unlikely that insertion of ethylene into the substituted position of B, to give G, should be favored over insertion into the unsubstituted one, to give E. A much more reasonable explanation is that formation of B itself is relatively unfavorable, presumably for steric reasons, and that isomer C is strongly favored in the co-oxidative coupling of 1-hexene with ethylene.
The relative overall rates of homo- and co-trimerization may be inferred to be approximately the same, from the observation that the reaction of ethylene with 1-heptene (at approximately 4:3 ratio in solution) gives close to 50% each of 1-hexene and heavier products (Table 1). Since there is more than one available route to each intermediate, it is not possible to assess the relative contributions of all the pathways. However, as the above model studies show, insertion of LAO is significantly less favored than that of ethylene. Hence a reasonable interpretation would be that A and C are formed at about the same rate, with little or no B, and that both A and C react further almost entirely by insertion of ethylene rather than LAO. A smaller amount (~4%) of C14 and even less (~0.2%) C18 is formed in the reaction of ethylene with 1-hexene; these could result from sequential secondary reactions of C10 and C14 with two ethylenes. Another possible route to C14 would be insertion of C6 into C; that is, a C2/LAO/LAO co-trimerization; results from the ethylene/1-heptene co-oligomerization experiment suggest that route does contribute (see below). In a related study on co-oligomerization of ethylene with 1-octene using a bis(pyridine)carbene complex of Cr, insertion of LAO into the analog of intermediate C was indeed found to contribute to the products,23 although that system exhibits several other significant differences from the present one: there the C12 products (the result of co-trimerization of two ethylenes with one 1-octene) did not predominate in the product distribution, and the most abundant C12 species identified was 2-butyl-1-octene, the analog of M, which is not the major C10 product here.
Insertion of ethylene into C can take place either on the side near to or away from the β-substituent to give G and F respectively. Since the sums of the products (M + N) and (O + P) are not too different, it appears that there is relatively little preference in that step. On the other hand, it is notable that N (47%) is obtained in a significantly higher yield than M (5%), suggesting that hydrogen transfer, whether it occurs in a stepwise or concerted manner,15–17 is less favored from the tertiary substituted β position in F than from the secondary, unsubstituted one. Again, this pattern does not appear to be general for all ethylene trimerization catalysts. For example, ethylene trimerization by a titanium-phenoxyimine complex, believed to involve a similar metallacycle mechanism, afforded M as the major (~90%) C10 olefin product.40 The more similar yields of O and P (18% and 27%, respectively) imply that proximity to a substituent in the γ position has a much smaller effect on hydride transfer preference.
Although the formation of C10 olefins from the co-trimerization of one 1-hexene and two ethylenes is unambiguous, routes to the observed C12, C14, and C18 products (Table 1, entry 2) are less obvious. The various possible combinations are shown in Table 2; the C14 and C18 olefins are most simply explained as secondary and tertiary co-trimerization products of two ethylene molecules with the C10 and C14 olefins generated during the reaction. Co-trimerization of one ethylene with two 1-hexenes is an alternate route to C14, as noted above and discussed further below.
Table 2.
Possible combinations of ethylene (C2), 1-hexene (C6), and 1-heptene (C7) dimers and trimers that make up the observed product distributionsa
| Ethylene | Ethylene/1-Hexene | Ethylene/1-Hepteneb |
|---|---|---|
|
| ||
| C2 + C2 + C2 = C6 | C2 + C2 + C2 = C6 | C2 + C2 + C2 = C6 |
| C2 + C2 + C6 = C10 | C2 + C2 + C6 = C10 | C2 + C2 + C6 = C10 |
| C6 + C6 = C12 | C2 + C2 + C7 = C11 | |
| C2 + C2 + C10 = C14 | C6 + C7 = C13 | |
| C2 + C6 + C6 = C14 | C7 + C7 = C14 | |
| C2 + C2 + C14 = C18 | C2 + C2 + C10 = C14 | |
| C6 + C6 + C6 = C18 | C2 + C6 + C6 = C14 | |
| C2 + C6 + C7 = C15 | ||
| C2 + C7 + C7 = C16 | ||
Each combination must satisfy the following criteria: 1) homo-oligomerization of ethylene form trimers, 2) co-oligomerization of ethylene/LAO forms trimers, 3) homo-oligomerization of LAO forms dimers and trimers, 4) dimers do not form between LAO and ethylene, and 5) tetramers of alkenes do not form.
Use of 1-heptene allowed the determination of trimers formed from two molecules of LAOs and one molecule of ethylene.
Because co-dimerization of ethylene/LAO does not occur, as evidenced by the absence of any C8 products from ethylene and 1-hexene, C12 cannot be the coupling product of ethylene and C10. A more reasonable route is through homo-dimerization of 1-hexene; 1/MMAO was found to homo-oligomerize 1-hexene, at a productivity about 1% of that of reactions involving ethylene (Table 1, Entry 4; Figure S2). The yield of C12 in the ethylene/1-hexene co-trimerization, about 1% of all products heavier than C6, is roughly consistent with that relative reactivity; some of the C18 products might also arise by that route. In comparison, chromium complexes supported by triazacyclohexane ligands (Cr-NNN) can oligomerize LAO with high efficiency, up to ~98% conversion;18,19 the bis(pyridine)carbene Cr complex is also effective for homo-dimerization of LAOs.23
Some additional inferences may be obtained from the reaction of ethylene/1-heptene with 1/MMAO, which affords C6, C10, C11, C13, C14, C15, and C16 species (Table 1, entry 3). Clearly C6 and C11 arise via homo-trimerization of ethylene and co-trimerization of two ethylenes and one 1-heptene respectively; because they are formed in comparable amounts (51 and 43 wt %), the reaction rates for homo-trimerization and co-trimerization by 1 must be similar. (Although no attempts were made to measure actual reaction kinetics in the present work, it has been the subject of other reports in the literature.41,42) The remaining identified products comprise only a small percentage of the total. The only obvious pathway to C13 is by co-dimerization of C6 and C7; a small amount of LAO dimerization was indicated by appearance of C12 in the ethylene/1-hexene reaction, but since much more C7 than C6 is present at any time here, it is surprising that the amounts of C14 and C13 are nearly the same —especially given that there are additional possible routes to C14 isomers, co-trimerization of C2/C2/C10 (not likely to be very significant, as the amount of C10 is relatively low) or C2/C6/C6. In contrast to the situation for ethylene/1-hexene, where the two sequences C2/C2/C10 and C2/C6/C6 both give C14 and hence their relative contributions cannot be determined, the corresponding sequences here would be C2/C2/C11 and C2/C7/C7, giving C15 and C16 respectively. Again, comparable amounts of the two are obtained, implying that some co-trimerization of the form C2/LAO/LAO does take place.
CONCLUSIONS
The following generalizations appear to best account for the observations in co-oligomerization of ethylene and LAO by the chromium-PNPOMe complex: 1) the initial intermediates are chromacyclopentanes formed by oxidative coupling of either two ethylenes, or one ethylene and one LAO, at the Cr center; 2) the rate constants for formation of those two species are similar; 3) the isomer of the mixed chromacyclopentane with the alkyl chain in the β position is strongly favored; 4) insertion of ethylene into the 5-membered ring is strongly favored over insertion of LAO; 5) insertion occurs preferentially on the side of the ring away from the substituent; 6) hydride transfer occurs preferentially from a secondary over a tertiary carbon site; 7) homo-di- (and -tri-) merization of LAOs does take place, but at a rate much lower than that of any reactions involving ethylene; and 8) co-dimerization of ethylene with LAO is not observed. While points 7) and 8) seem at first to be at odds, they need not be: it may be that the disubstituted chromacyclopentane is sufficiently inhibited towards a further insertion that formation of dimer (presumably by slow β-hydrogen elimination/reductive elimination) competes effectively with further growth, whereas for un- or monosubstituted rings (intermediates A–C in Scheme 2) ethylene insertion dominates over elimination. Only small amounts of the products that would result from insertion of ethylene into a disubstituted ring (C16 from ethylene/1-heptene; probably some of the C14 from ethylene/1-hexene) are observed.
Comparison of 1 to other olefin trimerization catalysts in the literature (see Discussion section) shows clearly that these generalizations do not universally apply to reactions proceeding via the metallacycle-based mechanism. There is a wide range of reactivity behaviors and consequent product distribution profiles, including preferences for ethylene vs. LAO in both oxidative coupling and insertion steps, relative rates of growth by insertion vs. elimination by hydride transfer, and regiochemistry of insertion and hydride transfer. Ongoing work is aimed at gaining the understanding the factors influencing those preferences, at a quantitative as well as qualitative level, that will be needed to make this chemistry a viable alternative to traditional non-selective oligomerization as a route from ethylene to C10+ hydrocarbons.
EXPERIMENTAL SECTION
General Considerations
Reagents obtained from commercial suppliers were used as received. The chromium pre-catalyst [CrCl3(PNPOMe)] (1) was synthesized according to a literature procedure.12 The following gas chromatography standards were obtained from the sources shown in parentheses: 4-ethyl-1-octene (Novel Chemical Solutions), 2-butyl-1-hexene (ChemSampCo), trans-5-decene (Aldrich), cis-5-decene, (TCI), trans-4-decene (ChemSampCo), 1-decene (Aldrich), and decane (Aldrich). The compounds 5-methylnonane and 4-ethyloctane were prepared by hydrogenation of 2-butyl-1-hexene and 4-ethyl-1-octene, respectively, using H2 over Pd/C. Syntheses of 3-propyl-1-heptene, 5-methyl-1-nonene, and cis-4-decene are provided in the Supporting Information. The modified methylaluminoxane activator (MMAO-C4 solution in isohexanes, 7 wt % Al; referred to as MMAO in the text) was obtained from Albemarle. MMAO-C4 was the activator of choice because it is available as a homogeneous solution that could be transferred quantitatively in repeated trials and gave the most reproducible data. Chlorobenzene was purged with argon and dried over calcium hydride before use. All ethylene oligomerization trials were performed using a high vacuum Schlenk manifold. Polymer-grade ethylene gas (>99.9% purity) was purified by passage through columns containing activated molecular sieves and Ridox, an oxygen-scavenger.
Gas Chromatographic Analyses
Gas chromatography-mass spectrometry (GC-MS) was performed using an Agilent 6890N system with an HP-5MS capillary column (30 m length, 0.25 mm diameter, and 0.5 μm film) that is equipped with an Agilent 5973N mass selective detector. Gas chromatography (GC) was performed using an Agilent 6890N instrument with a flame ionization detector (FID). Routine runs were performed using a DB-1 capillary column (10 m length, 0.10 mm diameter, 0.40 μm film) with the following heating program: hold at 40°C for 3 min, ramp temperature at 50°C/min to 290°C and then hold for 3 min (total run time = 13 min). For detailed C10 isomer analyses, an Agilent GS-GasPro capillary column (30 m, 0.32 mm diameter) was used, with the following heating program: hold at 90°C for 30 min, ramp temperature at 50°C/min to 260°C, and then hold for 10 min (total run time = 43 min).
The amount of products in each oligomerization trial was determined from the integrated areas of the peaks observed in the gas chromatogram, using the integrated area of a biphenyl internal standard as a reference. The integrated area for each peak was corrected using the appropriate response factor, which was determined experimentally. The response factors for all isomers containing the same number of carbon atoms were assumed to be the same.
To determine the identities of the C10 alkene isomers, the C10 fraction was separated from the crude reaction mixture by fractional distillation under atmospheric pressure. The GC retention times of the C10 products were compared to those of authentic samples that were either prepared independently or obtained from commercial sources. These assignments were further confirmed by GC analysis of the C10 sample after it had been hydrogenated.
Procedure for Olefin Oligomerization Trials
Homo-trimerization of ethylene
Inside a nitrogen-filled glovebox, [CrCl3(PNPOMe)] (8.0 mg, 12 μmol) was suspended in 10 mL of chlorobenzene, along with a stir bar, in a 250 mL round bottom flask equipped with a 180° needle valve. The flask with sealed, brought outside of the glovebox, and then attached to a high vacuum manifold. The reaction flask was cooled to −78°C in a dry ice/acetone bath, degassed, and then warmed to room temperature. The solution was stirred under an atmosphere of ethylene and a solution of MMAO (250 mg diluted to 1 mL in chlorobenzene) was added using an airtight syringe through a septum that was placed over the needle valve adapter, resulting in the formation of a pale yellow-green homogeneous mixture. Ethylene consumption was monitored using a U-tube mercury monometer during the course of the reaction, and the ethylene pressure was maintained between 700–790 Torr. (It should be noted that gradual catalyst decomposition was observed during the course of the reaction; the underlying cause for such behavior has not yet been determined.) After 1 h, the reaction with quenched with HCl(aq) and stirred for 1 min, resulting in a colorless solution. Solid biphenyl was added (50 mg, 325 μmol) as an internal standard. A 2.0 mL aliquot of the organic layer was filtered through a short silica plug and then analyzed by GC.
Co-oligomerization of ethylene/1-hexene
The same procedure was used as described for homotrimerization of ethylene except that 1-hexene (2.98 g, 35.4 mmol) was added to the reaction flask in addition to the chromium pre-catalyst before diluting to 10 mL with chlorobenzene.
Co-oligomerization of ethylene/1-heptene
The same procedure was used as described for homotrimerization of ethylene except that 1-heptene (2.32 g, 24 mmol) was added to the reaction flask in addition to the chromium pre-catalyst before diluting to 10 mL with chlorobenzene. Less 1-heptene was added to the reaction mixture, compared to the amount of 1-hexene used in the ethylene/1-hexene trials, because its increased lipophilicity was found to cause clumping of the chromium complex at high concentration when activated with MMAO-C4.
Homo-oligomerization of 1-hexene
Inside a nitrogen-filled glovebox, [CrCl3(PNPOMe)] (8.0 mg, 12 μmol) and 1-hexene (2.98 g, 35 mmol) were diluted to 10 mL with chlorobenzene. The mixture was treated with MMAO-C4 (250 mg) and then stirred at room temperature for 1 h. Aqueous HCl was added to quench the reaction and then biphenyl (50 mg, 325 μmol) was added to the organic layer as an internal GC standard. A 2.0 mL aliquot of the organic phase was filtered through a short silica plug and then analyzed by GC.
Supplementary Material
Acknowledgments
This work was supported by BP. L.H.D. acknowledges the National Institute of General Medical Sciences for a postdoctoral fellowship (F32 GM099189-02). The authors thank Dr. Emmanuelle Despagnet-Ayoub, Matt Winston, and Rachel Klet for helpful comments on this manuscript.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Syntheses of gas chromatography standards and GC traces. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Facts & Figures of the Chemical Industry. Chem Eng News. 2010;88:33–67. [Google Scholar]
- 2.Nexant. Process Evaluation Research Planning (PERP) Report–Ethylene 08/09–5. 2009. [Google Scholar]
- 3.Nexant. Process Evaluation Research Planning (PERP) Report–Alpha Olefins 06/07–5. 2008. [Google Scholar]
- 4.Vogt D. Oligomerization of Ethylene to Higher Linear α-Olefins. In: Cornils B, Herrmann WA, editors. Applied Homogeneous Catalysis with Organometallic Compounds. Vol. 1. VCH Publishers; New York: 1996. pp. 245–258. [Google Scholar]
- 5.Dixon JT, Green MJ, Hess FM, Morgan DH. J Organomet Chem. 2004;689:3641–3668. [Google Scholar]
- 6.McGuinness DS. Chem Rev. 2011;111:2321–2341. doi: 10.1021/cr100217q. [DOI] [PubMed] [Google Scholar]
- 7.Agapie T. Coord Chem Rev. 2011;255:861–880. [Google Scholar]
- 8.Wass DF. Dalton Trans. 2007:816–819. doi: 10.1039/b616291f. [DOI] [PubMed] [Google Scholar]
- 9.Manyik RM, Walker WE, Wilson TP. US Pat. 3300458 1967
- 10.Manyik RM, Walker WE, Wilson TP. J Catal. 1977;47:197–209. [Google Scholar]
- 11.Briggs JR. Chem Commun. 1989:674–675. [Google Scholar]
- 12.Agapie T, Schofer SJ, Labinger JA, Bercaw JE. J Am Chem Soc. 2004;126:1304–1305. doi: 10.1021/ja038968t. [DOI] [PubMed] [Google Scholar]
- 13.Agapie T, Labinger JA, Bercaw JE. J Am Chem Soc. 2007;129:14281–14295. doi: 10.1021/ja073493h. [DOI] [PubMed] [Google Scholar]
- 14.Emrich R, Heinemann O, Jolly PW, Krüger C, Verhovnik GPJ. Organometallics. 1997;16:1511–1513. [Google Scholar]
- 15.Janse van Rensburg W, Grové C, Steynberg JP, Stark KB, Huyser JJ, Steynberg PJ. Organometallics. 2004;23:1207–1222. [Google Scholar]
- 16.Qi Y, Dong Q, Zhong L, Liu Z, Qiu P, Cheng R, He X, Vanderbilt J, Liu B. Organometallics. 2010;29:1588–1602. [Google Scholar]
- 17.Klemps C, Payet E, Magna L, Saussine L, Le Goff XF, Le Floch P. Chem–Eur J. 2009;15:8259–8268. doi: 10.1002/chem.200900986. [DOI] [PubMed] [Google Scholar]
- 18.Köhn RD, Haufe M, Kociok-Köhn G, Grimm S, Wasserscheid P, Keim W. Angew Chem, Intd Ed Engl. 2000;39:4337–4339. doi: 10.1002/1521-3773(20001201)39:23<4337::AID-ANIE4337>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Wasserscheid P, Grimm S, Köhn Randolf D, Haufe M. Adv Synth Catal. 2001;343:814–818. [Google Scholar]
- 20.Bowen LE, Wass DF. Organometallics. 2006;25:555–557. [Google Scholar]
- 21.Bowen LE, Charernsuk M, Wass DF. Chem Commun. 2007:2835–2837. doi: 10.1039/b702331f. [DOI] [PubMed] [Google Scholar]
- 22.Bowen LE, Charernsuk M, Hey TW, McMullin CL, Orpen AG, Wass DF. Dalton Trans. 2010;39:560–567. doi: 10.1039/b913302j. [DOI] [PubMed] [Google Scholar]
- 23.McGuinness DS. Organometallics. 2009;28:244–248. [Google Scholar]
- 24.Tomov AK, Chirinos JJ, Long RJ, Gibson VC, Elsegood MRJ. J Am Chem Soc. 2006;128:7704–7705. doi: 10.1021/ja0615369. [DOI] [PubMed] [Google Scholar]
- 25.Carter A, Cohen SA, Cooley NA, Murphy A, Scutt J, Wass DF. Chem Commun. 2002:858–859. doi: 10.1039/b201335e. [DOI] [PubMed] [Google Scholar]
- 26.Agapie T, Day MW, Henling LM, Labinger JA, Bercaw JE. Organometallics. 2006;25:2733–2742. [Google Scholar]
- 27.Schofer SJ, Day MW, Henling LM, Labinger JA, Bercaw JE. Organometallics. 2006;25:2743–2749. [Google Scholar]
- 28.Bollmann A, Blann K, Dixon JT, Hess FM, Killian E, Maumela H, McGuinness DS, Morgan DH, Neveling A, Otto S, Overett M, Slawin AMZ, Wasserscheid P, Kuhlmann S. J Am Chem Soc. 2004;126:14712–14713. doi: 10.1021/ja045602n. [DOI] [PubMed] [Google Scholar]
- 29.Elowe PR, McCann C, Pringle PG, Spitzmesser SK, Bercaw JE. Organometallics. 2006;25:5255–5260. [Google Scholar]
- 30.Blann K, Bollmann A, de Bod H, Dixon JT, Killian E, Nongodlwana P, Maumela MC, Maumela H, McConnell AE, Morgan DH, Overett MJ, Prétorius M, Kuhlmann S, Wasserscheid P. J Catal. 2007;249:244–249. [Google Scholar]
- 31.Blann K, Bollmann A, Dixon JT, Hess FM, Killian E, Maumela H, Morgan DH, Neveling A, Otto S, Overett MJ. Chem Commun. 2005:620–621. doi: 10.1039/b412431f. [DOI] [PubMed] [Google Scholar]
- 32.Pellecchia C, Pappalardo D, D’Arc M, Zambelli A. Macromolecules. 1996;29:1158–1162. [Google Scholar]
- 33.Correa A, Galdi N, Izzo L, Cavallo L, Oliva L. Organometallics. 2008;27:1028–1029. [Google Scholar]
- 34.Luo Y, Luo Y, Qu J, Hou Z. Organometallics. 2011;30:2908–2919. [Google Scholar]
- 35.Nelson JE, Bercaw JE, Labinger JA. Organometallics. 1989;8:2484–2486. [Google Scholar]
- 36.Terao J, Begum SA, Oda A, Kambe N. Synlett. 2005:1783–1786. [Google Scholar]
- 37.Kaisare AA, Owens SB, Jr, Valente EJ, Gray GM. J Organomet Chem. 2010;695:1472–1479. [Google Scholar]
- 38.Maura R, Steele J, Vendier L, Arquier D, Bastin S, Urrutigoïty M, Kalck P, Igau A. J Organomet Chem. 2011;696:897–904. [Google Scholar]
- 39.Nozaki K, Komaki H, Kawashima Y, Hiyama T, Matsubara T. J Am Chem Soc. 2001;123:534–544. doi: 10.1021/ja001395p. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki Y, Kinoshita S, Shibahara A, Ishii S, Kawamura K, Inoue Y, Fujita T. Organometallics. 2010;29:2394–2396. [Google Scholar]
- 41.Wöhl A, Müller W, Peulecke N, Müller BH, Peitz S, Heller D, Rosenthal U. J Mol Catal A. 2009;297:1–8. [Google Scholar]
- 42.Wöhl A, Müller W, Peitz S, Peulecke N, Aluri BR, Müller BH, Heller D, Rosenthal U, Al-Hazmi MH, Mosa FM. Chem–Eur J. 2010;16:7833–7842. doi: 10.1002/chem.201000533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.