

Abstract
Interest in the role of the microbiome in human health has burgeoned over the past decade with the advent of new technologies for interrogating complex microbial communities. The large-scale dynamics of the microbiome can be described by many of the tools and observations used in the study of population ecology. Deciphering the metagenome and its aggregate genetic information also can be used to understand the functional properties of the microbial community. Both the microbiome and metagenome probably have important functions in health and disease; their exploration is a frontier in human genetics.
Until recently, the properties of the microbiota of humans (formerly called ‘the normal flora’) were largely a black box. Cultivation in vitro, which has been the cornerstone of microbiology since the 19th century, cannot be applied to many of the most densely populated microbial communities1. However, DNA-based analyses have expanded our horizon, by generating enormous new data sets that can be mined for information on the composition and functional properties of vastly greater numbers of microbial communities. For example, the Human Microbiome Project (HMP) by the NIH has produced a 2.3 terabyte 16S rRNA metagenomic dataset of over 35 billion reads taken from 690 samples from 300 U.S. subjects, across 15 body sites. Large-scale endeavors (e.g. the HMP2 and also the European project, Metahit3) provide a preliminary understanding of the biology and medical significance of the human microbiome and its collective genes (the metagenome).
The aim of these projects, particularly the HMP, is to characterize the compositional range of the ‘normal’ microbiome of healthy individuals. Important questions concerning the commonalities and differences between healthy individuals in both microbial taxa and functional pathways are being addressed. The presence of major clustering patterns at body sites such as the vagina4 and the gastrointestinal tract5 provide new ways to classify individuals, and possibly, their disease risks. Substantial progress has been made in developing the tools for inquiry, and defining the overarching concepts that advance the field. However, the subject is vast, and the implications for human health and disease are wide-ranging. The study of humans and model animal systems with strong phenotypes is essential for making progress in this field of applied genetics. Although a focus on bacteria is important, inquiries aimed at archaea, viruses, and retroviruses, is also needed.
The purpose of this review is to develop the theoretical basis for investigating how microbiome composition and function affect human health. We provide examples of applying this knowledge to better understand human health, and discuss how microbiome changes could alter host–microbiome interactions to mitigate disease. We also consider the next steps in the development of this field, particularly on the need to focus on the inheritance of the microbiome, and on its involvement in modulating complex traits.
Characterizing the microbiome
Animals have had residential microbes performing metabolic functions for at least 500 million years, at a conservative estimate6,7. Extensive congruent phylogenies of animal hosts and their microbiota, involving both individual organisms and whole microbial populations1,8,9, suggest the existence of specific selection based on co-adaptation. Cooperative interactions between microbes and their hosts typically involve microbial participation in host functions such as defence, metabolism, and reproduction10. For example, comparing germ-free and conventional mice indicates that microbiota are responsible for most of the metabolites that are detected in plasma11.
Functional variation of host microbiota can be mediated by the introduction or extinction of particular microbial groups or by a change in population structure12–14. Such alterations can in turn be induced by selection by environmental factors10,15 such as dietary changes or exposure to antibiotics10,15. Below we describe the efforts to categorize the composition and complex dynamics of the microbiota.
Tools for studying the metagenome
The taxonomic diversity inherent in complex environmental communities and the task of identifying specific associations with host traits create unique challenges. One approach to metagenome analysis involves assigning unassembled sequences generated by shotgun high-throughput sequencing (HTS)16 to the NCBI non-redundant Clusters of Orthologous Groups (COG) or the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases17. This method facilitates the assessment of interactions that occur within the microbiome, and potentially between microbiome and host18. However, because a substantial fraction of the metagenome (~ 33%) is not well-represented by reference genomes, this strategy provides only a limited understanding of the functional potential of the microbiota. An alternative approach is to use catalogues of known genes to identify functional clusters within a sample; such clusters could correspond to the proposed taxonomic enterotypes5. A catalogue of the microbial genes present in the human gut, for example, is being generated using several approaches including sequencing, assembling and characterizing non-redundant microbial genes from fecal samples19, and whole genome sequencing of reference microbial species20.
As sequencing and bioinformatics technologies continue to evolve (see ref 21 for a review of the state-of-the-art technologies), scientific priorities will include elucidating the ‘core’ metagenome that occupies a specific human niche, and discerning the differences between normal and diseased hosts.. As an example of the latter, Greenblum et al. applied new tools to understand interhost metagenomic variation in relation to phenotypes such as obesity and inflammatory bowel disease (IBD)22. By categorizing metagenomic sequences based on gene function, they constructed community-level metabolic networks varying in gene abundance, and examined the topological features of these networks in relation to host phenotype. Their analysis identified specific network topologies related to obesity and IBD; skewed topologies chiefly differ in genes related to host interactivity, particularly in relationship to metabolism. Such topological tools can be applied to explore differences in other host disease states.
Taxonomic and functional variation
The composition of the microbiome varies by anatomical site (Figure 1). The primary determinant of community composition is anatomical location: interpersonal variation is substantial23,24 and is higher than the temporal variability seen at most sites in a single individual25. Such relative temporal stability suggests that individuals can be grouped according to the major enterotypes present in the colon5 or the vagina4. However, dietary changes can rapidly cause substantial intestinal metagenomic changes, and enterotypes are known to cluster based on dietary abundance of animal protein vs carbohydrate26. Similarly, nasopharyngeal microbiota in young children varies seasonally24, and vaginal microbiota varies with menses4. In the absence of marked perturbations, the aggregate microbiota of an individual appears to vary relatively narrowly within host-specific boundaries; the basis of such boundaries have not been established, but may represent Nash equilibria13. Because minor microbial populations have the potential to bloom, the temporal variation observed in a host may be mirrored by the interindividual variation at a single time27; that the system is dynamic suggests that there are greater interpersonal similarities than a snap-shot view indicates. However, large perturbations such as antibiotic exposure28 or enteric infections (LA David, personal communication), can lead to transient disequilibrium29, or to the development of a new stable state.
Figure 1. Compositional differences in the microbiome by anatomic site.
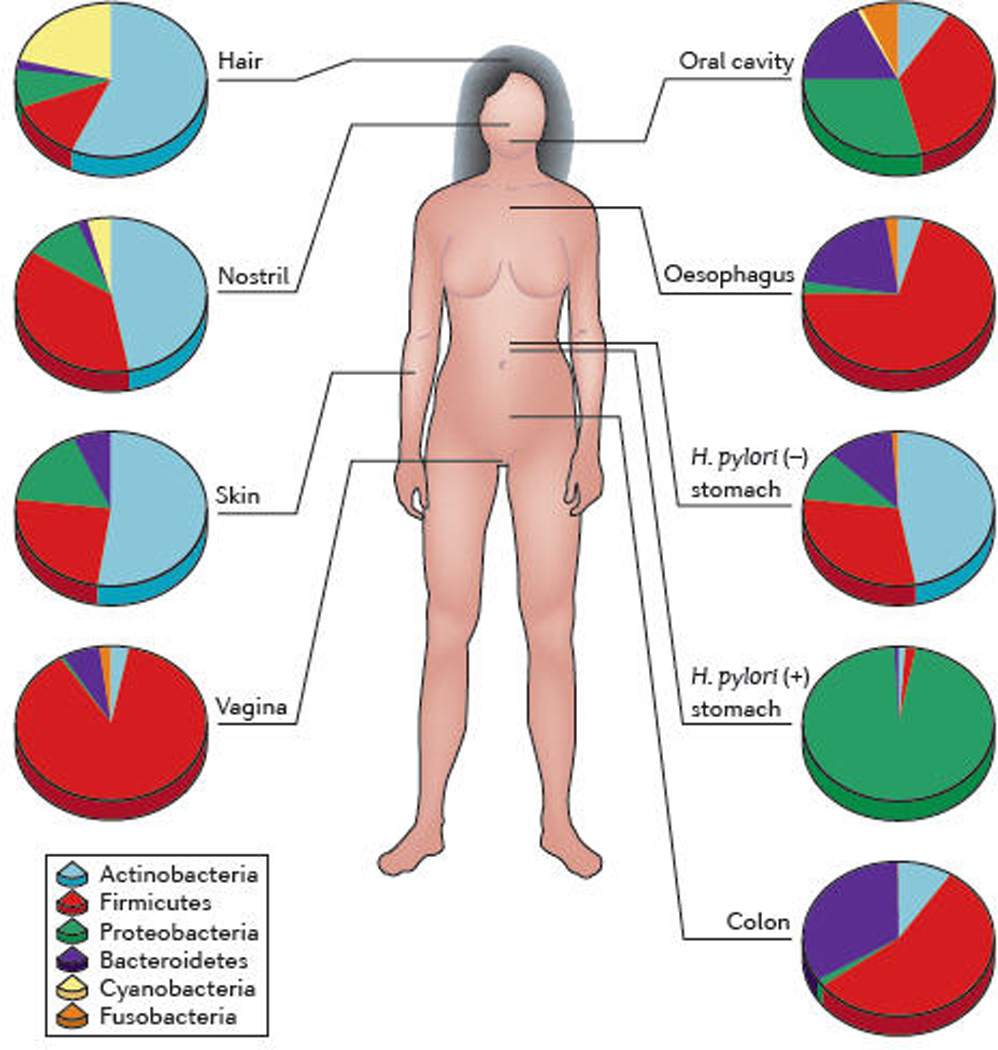
High-throughput sequencing has revealed substantial intra-individual microbiome variation at different anatomical sites, and inter-individually for the same anatomical sites 4,5,25,52,89,93. However, higher level (e.g. phylum) taxonomic features display temporal (longitudinal) stability in individuals at specific anatomical sites. Such site-specific differences as well as observed conservation between human hosts provide an important framework to determine the biological and pathological significance of a particular microbiome composition. The figure indicates percentages of sequences at the taxonomic phylum level from selected references. Certain features, such as the presence or absence of Helicobacter pylori, can lead to permanent and marked perturbations in community composition93.
Among all mammals, the microbiota is extensively conserved at high taxonomic levels7, but variation increases at progressively lower taxonomic levels30. Consequently, 85% of the sequences obtained from the distal mouse gut represent genera that are not detected in humans31. Furthermore, intra-species variability of the microbiota within the human population is also substantial5, This degree of variation was unanticipated a priori. However, in retrospect, extensive taxonomic variation is unsurprising: a human harbours a climax population of about 1014bacterial cells, can host 105 – 106 bacterial generations per human generation, is omniverous, and has accumulated genetic and epigenetic diversity as a host species for >1 million years. Indicator organisms such as Helicobacter pylori32 and Streptococcus mutans33 highlight some differences across the microbiota34 and metagenome35 among human ethnic groups; however, the extent of ethnic variation in overall metagenomic composition is unknown. The microbiomes of monozygotic twins are more closely related to one another than those of unrelated individuals36,37 but not strikingly so, indicating important post-natal influences on composition.
The extensive lower-level taxonomic variation and large compositional differences observed even among highly related host organisms (e.g. mice and humans) is counterbalanced by the substantial conservation of metagenomic core functions (Figure 2). This reflects the conservation of core bacterial properties involved in nucleic acid and protein synthesis, and in metabolic and structural requirements. Of the >50 known phyla, most of the human microbiota is composed of <10 (and mostly 6) phyla. Bacteria from other phyla, usually of plant origin, that may be present in skin, nasopharyngeal, or gut samples24,25,38; are generally infrequent (<0.01% of the sequences) and probably represent transient carriage from food- and air-borne exposures. Why did the particular restriction of diversity within a few phyla evolve, not only in humans, but in perhaps all other vertebrates? One possibility is that within the relatively conserved boundaries for the microbiome permitted by the human genome, there exists a large array of contingency organisms and contingency genes. According to this hypothesis, the genes may only be active at some moment in the host’s lifespan, or perhaps at a frequency of less than once per lifespan.
Figure 2. Conservation of bacterial genes despite taxonomic variation.
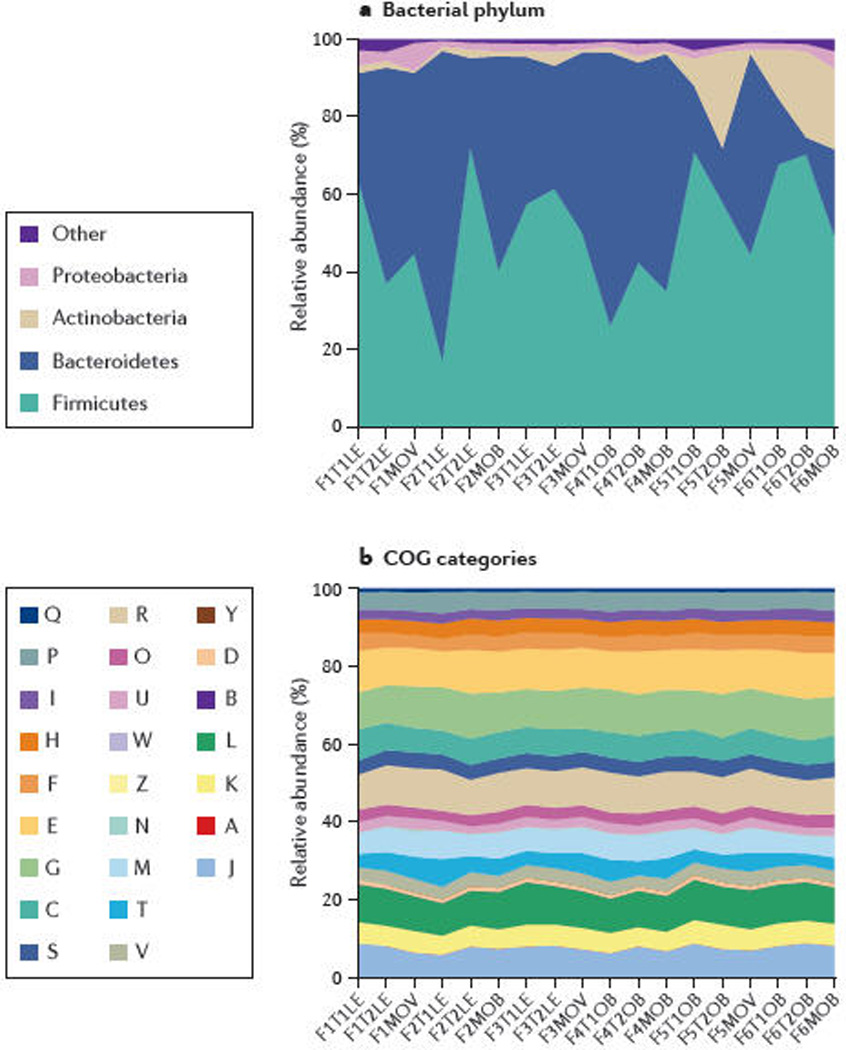
A) Turnbaugh et al. studied the distal gut microbiome in lean and obese twins and their mothers37. There were substantial and significant taxonomic variations amongst the individuals, although Firmicutes and Bacteroidetes still constituted the majority of the taxa. B) Through metagenomic analyses, the functional characteristics of the microbiomeas identified by COG pathways are largely conserved, despite the taxonomic variation37. COG pathways are denoted by: S – Unknown; R – General function; L – DNA; G – Carbohydrates; E – Amino acids; M – Envelope; K – Transcription; J – Translation; C – Energy; T – Signal transduction; P – Inorganic; V – Defense; H – Coenzymes; O – Protein turnover; F – Nucleotides; U – Secretion; I – Lipids; D – Cell cycle; B – Chromatin; Q – Second metabolites; N – Cell motility; W – Extracellular; Z – Cytoskeleton; A – RNA. Reproduced with permission from Turnbaugh et al37, Nature and the authors © Macmillan Publishers Ltd
The parallel needs of individual bacteria lead to both competition for key substrates and to functional redundancy in the microbiota. Nevertheless, the enormous bacterial biomass also provides many unique or minimally redundant bacterial genes19.
Resilience and community disturbance
Resilience, the ability to withstand disturbance, is a central concept in ecology. The resilience of the human colonic microbiome is beautifully illustrated by recent studies of twins examined before, during, and after 7-weeks of ingesting a fermented milk product containing a sample composed of ~108 Bifidobacterium, Lactobacillus, Lactococcus, and Streptococccus species36. Despite the daily oral inoculations, the composition of the microbiota at the 16S rDNA level and the metagenome were essentially unaffected. While the microbiome of human adults appears highly resilient, no comparable studies have been performed in children, but because population structures appear more dynamic and developmental39, resilience may be lower. An important natural experiment has been occurring over the past 70 years in which most of the world’s population has been exposed to pharmacological doses of antimicrobial agents. Such usage has been based on the implicit belief that the human microbiome is completely resilient, and returns to the status quo ante after antibiotic-induced perturbation. However, study of indicator organisms such as H. pylori, indicates that extirpations of a bacterial species can occur in individual hosts40. There also may be medium- and long-term selection of resistant organisms, and destabilization of the microbiome with new species compositions, in the absence of further antibiotic exposure28,41. Thus, despite the extensive resilience inherent in a complex eco-system, there may be loss of recovery from continued perturbations29, with important implications for human health42.
Medical scientists are familiar with Koch’s postulates, which are used as criteria to determine whether a microbe causes disease43. However, pathogenetic considerations about the microbiome may better focus on community characteristics, which are largely governed by richness, composition, and interactions among the constituent members7,16,44. Substantial perturbation (community disturbance45) tests the resilience of the community, e.g., it ability to resist invasion by exogenous microbes; stable diverse communities resist pathogens46. At present, 16S rRNA analyses focus on taxonomic differences at or above the species level. However, examinations below the subspecies level, relating to strain, or even allele, ultimately may be more significant, although the technology, importantly the informatics tools, are not yet developed enough.
Extinctions
The human microbiome represents one or more complete ecosystems. The trophic organization of species-rich communities is similar to other complex network topologies, in that it shows extreme heterogeneity, with a relatively small number of highly connected nodes dominating47. Such communities may resist random perturbations, but if keystone species48 are lost, effects may cascade, with secondary extinctions; high biodiversity diminishes this risk12. The substantial non-linear interactions in complex co-evolved systems ensure that ecological networks are robust against random removals49. However, with repeated perturbations, the effects of gene loss can be amplified by downstream effects on co-colonizing microbes (secondary extinctions) and on the host. Because of allelopathy, the effects of extinctions may magnify50. In the short-term, functional redundancy may mask extinction effects, but in the longer-run, extinctions lead to loss of contingency responses that can cause ecological crashes49. Considering the importance of guilds of bacteria exploiting parallel and sequential metabolic pathways, these concepts are germane to the human metagenome. With modernity, horizontal microbial transmission has been diminishing, and there has been unprecedented selection against existing, long-present microbes40. As has occurred in the human stomach with the loss of a dominant species51,52, body sites then can harbor alternative stable states.
In summary, as with other complex ecosystems, the microbiomes that populate specific human anatomical niches are species-rich, but possess particular overall community characteristics at higher organizational levels. All are subject to perturbation in the course of normal development and aging, and especially with disease. As our knowledge of the fundamental characteristics and biology of the human microbiome grows, so will our ability to understand disease-related variation.
Influences on the microbiota during the life cycle
As described in the previous section, differences in microbiota composition exist across body sites and between individuals. However, changes are also evident across the human lifespan. Important questions in this field involve determining if such temporal changes are life-stage specific, and whether they are predetermined by host genetic characteristics, or by environmental factor.
Inheritance of microbiota
The congruent phylogenies of mammals and their microbiota8 provide strong evidence for the inheritance of the microbiota7 Although inheritance of the microbiota from the father is presently little studied, increasing evidence supports inheritance from the mother34,53. Until the amniotic sac ruptures, a fetus is considered to be sterile, or essentially sterile. Immediately after vaginal delivery, founding microbial populations in the baby closely resemble that of their mother’s vagina54, with lactobacilli predominating. Since lactic acid-producing bacteria dominate in both vagina and in mother’s milk, the initial gastro-intestinal tract bloom of lactobacilli in the baby cannot be considered accidental. Lactobacilli represent the pioneer community in mice55 as well as humans39, preparing the gastrointestinal tract for subsequent microbial successions, until microbial maturity is reached.
The multiple opportunities for the microbiota to be transferred from a mother to her baby may be disrupted by modern lifestyles. Caesarian section instead of vaginal delivery is an obvious example of the potential impact of medical practice on microbiota composition, with substantial differences in founding population54, that may persist for months56 (Figure 3). In many host species, paternal contributions to offspring traits have been well documented57,58; these observations have been extended to the microbiome, where paternal contributions to offspring H. pylori allele composition have been shown59, In any event, there is evidence for extensive horizontal gene transfer (HGT) within human populations, functional classes, and ecological niches60, indicating the site-specificity and dynamism of selection on the human microbiome. Even so, microbial inheritance can provide important confirmation of human ancestry61.
Figure 3. Acquisition of the microbiome in early life by vertical transmission and factors modifying mother-to-child microbial transmission.
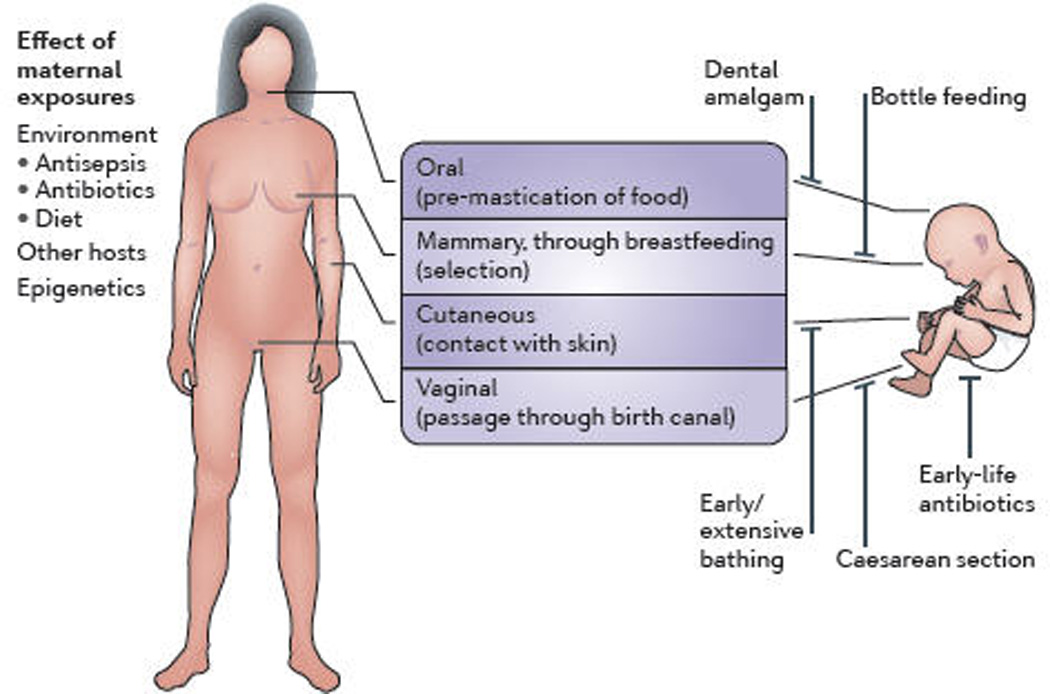
Through live-birth, mammals have important opportunities for mother → child microbial transmission, via direct-surface contact. However, many modern practices can reduce the organism and gene flow; several examples are illustrated. After initial introductions, there is strong selection by hosts for microbes with specific phenotypes, consistent with the extensive conservation shown in Figure 1. Acquisition is modified by offspring genetic and epigenetic differences (with respect to both maternal and paternal genes) that inform the competition for host resources by the vertically transmitted and environmentally acquired microbes. Ancestral organisms that have particular tissue- and niche-specific adaptations facilitate tissue tropisms and are selected, explaining the conserved niche-specificity compositions.
In Drosophila melanogaster, microbial influences have an effect on mating preference for >30 generations62; can microbiome composition affect mating in humans? Odor is one means to affect mating preference, since human axillary and oral odors are largely influenced by microbial products, especially mercaptans63. In general, the greater the force of mating preference, the more likely those populations become sexually isolated64,65; this could affect tribal differentiation and other ethnic differences in humans. We speculate that metagenome composition may have affected mating preference in humans, representing another phenotype under strong selective pressure.
Postnatal influences on the microbiota
Over a lifetime, each human develops a densely populated microbiome that is recapitulated in every individual and in every generation. The eruption of teeth is responsible for major successions in the oral microbiota66,67, suggesting that succession may be a general property of microbiome dynamics in humans. In mice, this clearly occurs in the GI tract68. Exposure (or not) to environmental microbes is another important but highly variable reservoir for the resident microbiota. Early life antibiotic use produces major shifts in both microbiota characteristics and in host developmental phenotypes, both on the farm69 and in experimental animals70,71. Whether such precedents are applicable to human children is unknown, but seems likely. If so, then both the timing of microbiome succession and the specific organisms present may affect development. The concept of time-dependent compositional variation affecting host immunological, metabolic, cognitive, and reproductive development is a potentially important and testable hypothesis. We further speculate that nature orchestrates microbiome development so to optimize fecundity, reaching a climax state at or near parturition to maximize success for the next generation. The noted heterozygote advantage for fecundity72 may be an analogue to a genetically diverse microbiota.
Microbiome dynamics in adults
Our knowledge of microbiome dynamics, especially age-related changes, during human adulthood is limited. The older literature (predating the use of HTS), clearly shows that the post-menopause vaginal microbiota differs substantially from that during the reproductive period73,74. Similarly, in the stomach,, the age-related progressive development of gastric atrophy (enhanced by H. pylori presence75,76) selects for gastric microbiota that are substantially different from the norm 77. Analogous changes may be occurring in other body sites as senescence advances. In the gut, the ratio of Bacteroidetes to Firmicutes changes with age78.
These concepts are particularly relevant to oncogenesis, which is generally age-related. In the multi-step Nordling hypothesis of oncogenesis79, 4–6 somatic cell mutations are needed for cancer development. We propose that age-related microbiota shifts contribute to this multi-step process. Residential microbes can contribute to somatic mutagenesis by causing genotoxicity secondary to inflammation, increased cell proliferation, and production of pro-mutagenic metabolites (e.g. butyrate)80. Genes may have alternative effects at different life stages, illustrating the idea of antagonistic pleiotropy81. We hypothesize that specific human microbiota and their genes that are beneficial early in life may be harmful later in life. The dominant gastric bacterium, H. pylori provides an example:early in life, inflammatory responses to the organism improve control of infection82,83 and allergy84, but later in life promote atrophy and oncogenesis85. A related hypothesis is that co-evolved microbiota are adaptive for the human species both by supporting early-in-life host functions and by leading to later-in-life host demise 86.
Disease links and health implications
How, then, does the microbiome affect human health? Current studies focus on describing the variant microbe populations that appear in specific disease states or the temporal microbial changes over the course of a disease. For many conditions, the challenge is to discover whether there is a causal link between microbiome variation and significant pathology. Limitations in the definitions and stratification of clinical syndromes, including irritable bowel syndrome (IBS) and non-ulcer dyspepsia (NUD), unfortunately reduce the potential of microbiome studies. The diseases listed below review some current recent investigations (Table 1). Investigations are preliminary, but some observations are promising.
Table 1.
Examples of association of human conditions with particular microbiota characteristics
| Disease | Relevant finding | Reference |
|---|---|---|
| Psoriasis | Increased ratio of Firmicutes to Actinobacteria | 88 |
| Reflux esophagitis | Esophageal microbiota dominated by gram-negative anaerobes Gastric microbiota with low or absent H. pylori |
75,134 |
| Obesity | Reduced ratio of Bacteroidetes to Firmicutes | 17,31 |
| Childhood-onset asthma | Absent gastric Helicobacter. pylori (especially cytotoxin-associated gene (cagA) genotype) |
96,135 |
| IBD (colitis) | Increased Enterobacteriaceae | 113 |
| Functional bowel diseases | Increased Veillonella and Lactobacillus | 136 |
| Colorectal carcinoma | Increased Fusobacterium spp. | 101,102 |
| Cardiovascular disease | Gut microbiota-dependent metabolism of phosphatidylcholine |
137 |
Cutaneous microbiome
The cutaneous microbiome is an obvious target in specific diseases, such as psoriasis, a chronic, idiopathic inflammatory dermatologic condition87. In studies predating HTS, the use of PCR and cloning led to observations that Firmicutes were significantly over-represented and Actinobacteria were significantly under-represented in psoriatic lesions compared to both unaffected skin in psoriasis patients and in normal controls88. Studies to explore these findings using HTS are currently underway89. Atopic dermatitis, another chronic inflammatory condition, has increased in incidence approximately three-fold over the last 30 years in industrialized countries, suggesting a potential role for microbiome alterations. Classic atopic dermatitis occurs in areas, such as the antecubital and popliteal fossae, with similar microbial populations89, suggesting a microbiome role. Similarly, Propionibacterium acnes has been implicated in the common dermatologic condition, acne. Although P. acnes thrives in the cutaneous pilosebaceous units and secretes enzymes that cause local injury and inflammation, and is widely accepted to have a function in acne development90, continuing investigations also are examining other microbes in the development of acne. Chronic skin ulcers, often secondary to venous stasis or diabetes, lead to substantial morbidity. Cutaneous microbiome shifts, such as an increased abundance of Pseudomonadaceae in patients with chronic ulcers treated with antibiotics and an increased abundance of Streptococcaceae in diabetic ulcers have been noted91. Such shifts may interact with aberrantly expressed host cutaneous defense response genes92, thereby increasing disease risk.
Gastric microbiome
The discovery that H. pylori was adapted to survive in the acidic gastric environment overturned the dogma that the stomach is sterile. In H. pylori-negative individuals, gastric microbiota diversity is high; most of the prominent gastric phylotypes (Streptococcus, Actinomyces, Prevotella, Gemella) also are abundant in the oropharynx of these individuals93; indicating that either many constituents are swallowed from more proximal sites, or that close relatives of the oral microbiota colonize more distally. In contrast, among H. pylori-positive persons, H. pylori usually accounts for >90% of sequence reads from the gastric microbiota93, dramatically reducing the overall diversity of this microbiota. The ability of H. pylori to dominate indicates an evolved fitness for that specialized niche. H. pylori is a classical amphibiont; the presence (or absence) of an H. pylori dominated gastric microbiota is strongly associated with particular diseases with important age-related differences85. Its presence increases risk for developing peptic ulcer disease, gastric mucosa-associated lymphoid tissue (MALT) tumors, and gastric adenocarcinoma94 but also is associated with decreased reflux esophagitis95 and childhood-onset asthma96; demonstrating the complex biological interactions with our microbiota.
The colonic microbiota and colorectal cancer
The colonic microbiota has been suspected for a long time to be involved in the development of colorectal cancers97, possibly by synthesizing short-chain fatty acids (SCFAs) and other metabolites. SCFAs, in particular butyrate, may induce apoptosis, cell cycle arrest, and differentiation, via Wnt signaling98. Microbes may also be genotoxic to colonic epithelial cells, as demonstrated by the induction of aneuploidy and tetraploidy by Enterococcus faecalis99. The colonic microbiota also might promote colorectal cancer by eliciting host responses, for example, by stimulating exaggerated immune responses, potentially via Th17 cells99.
Further evidence of a link between colonic microbiota and colorectal cancer is suggested by the ability of antibiotic administration to alter the composition of the colonic microbiota and to affect the expression of host genes involved in cell cycle regulation, reducing epithelial proliferation100. Early studies evaluating specific microbes were limited to identifying culture-dependent species, such as Streptococcus bovis, but could not adequately assess anaerobic constituents. However, members of the anaerobic genus Fusobacterium have recently been associated with colorectal cancer; whole genome sequences of Fusobacterium species were compared between cases and matched controls using both quantitative PCR analysis and HTS101,102. Fusobacterium nucleatum is a mucosally adherent, pro-inflammatory microbe that was first identified in the mouth103. In colorectal cancer samples, F. nucleatum sequences were significantly enriched compared to samples obtained from control subjects, while both Bacteroidetes and Firmicutes were relatively depleted in those with Fusobacterium-rich malignancies102. The enrichment of Fusobacterium species (not limited to F. nucleatum) was confirmed when evaluating the mucosal microbiome of colorectal cancers compared to adjacent normal tissues from 99 subjects101. However, the causal direction of the association has not yet been ascertained.
The colon microbiota and inflammatory bowel disease
The microbiome is essential for the activation of host immune responses 104. For example, Th17 cell differentiation in the mouse lamina propria requires the presence of segmented filamentous bacteria (SFB)105, and polysaccharide A produced by Bacteroides fragilis mediates conversion of CD4+ T cells into regulatory T cells106. The inflammatory bowel diseases have long been considered to reflect interactions between microbes and the host. IBD susceptibility is associated with host polymorphisms in bacterial sensor genes such as nucleotide-binding oligomerization domain-containing protein 2 (NOD2) also known as caspase recruitment domain-containing protein 15 (CARD15)107,108 and toll-like receptor 4 (TLR4)109, and IBD patients sometimes improve following antibiotic treatment110. Early childhood antibiotic exposure has been associated with significantly increased risk for Crohn’s disease111, suggesting that gut microbiome perturbations may be critical for disease risk. Microbial diversity is significantly diminished in Crohn’s disease112, suggesting decreased gut microbiome resilience that could affect immune interactions. Gut microbiome population structures of patients with ulcerative colitis or Crohn’s disease19 depart from normality, but remain clustered by disease within their characteristic deviated patterns. Specific bacteria of the Enterobacteriaceae genus may act together with a disordered microbiome to increase the risk of ulcerative colitis113. Among twins discordant for ulcerative colitis, those affected had significantly reduced bacterial diversity, but increased proportions of Actinobacteria and Proteobacteria114. Patients with Crohn’s disease have over-representation of E. faecium and of several Proteobacteria compared to controls115. The microbial patterns observed for the conditions described above are preliminary and their specificity and causal direction have not been established.
The gut microbiota and diseases of the liver
The gut microbiota may be involved in hepatologic conditions, including non-alcoholic fatty liver disease (NAFLD)116, alcoholic steatosis, and hepatocellular carcinoma. The liver is the first solid organ exposed to the metabolic products generated by the gut microbiome, including acetaldehyde, ammonia, and phenols. Compared to germ-free mice, the presence of a microbiome in conventional mice leads to suppression of intestinal epithelium angiopoietin-related protein 4, which inhibits lipoprotein lipase, increasing downstream triglyceride accumulation in the hepatic parenchyma and adipocytes117. Chronic ethanol exposure disturbs the gut microbiome118,119, but roles for the microbiome in steatosis are unresolved. Particular murine colonic commensals (e.g. H. hepaticus) promote the development of hepatocellular carcinoma120. Patients with cirrhosis have a substantially altered microbiome, including community-wide changes at multiple taxonomic levels, with enrichment of Proteobacteria and Fusobacteria (phyla), and Enterobacteriaceae, Veillonellaceae, and Streptococacceae (family) 121. Although many observations suggest links between microbiome composition and liver disease, definitive associations in humans are lacking.
The gut microbiota and pbesity
Genetically obese (ob/ob) mice have decreased Bacteroidetes/Firmicutes ratios compared with lean (ob/+ and +/+ wild-type) siblings31. Transplantation of gut microbiota from the obese (ob/ob) to germ-free mice conferred an obese phenotype, demonstrating transmissibility of metabolic phenotypes17; the transferred microbiomes had increased capacity for energy harvest. In humans, relative Bacteroidetes proportions increase with weight loss122. In mono- and dizygotic twins, obesity was associated with decreased Bacteroidetes and diminished bacterial diversity, with enrichment of genes related to lipid and carbohydrate metabolism. Despite substantial taxonomic variation, functional metagenomic differences were minor37. Changing exposures/selection pressures on the early-life microbiome may contribute to development of obesity. Antibiotic use in human infancy, before age 6 months, was significantly associated with obesity development123. In contrast, perinatal administration of a Lactobacillus rhamnosus GG-based probiotic decreased excessive weight gain during childhood124. These early studies provide support for the concept that microbiota alterations could lead to childhood-onset obesity, which could be modifiable. Alterations in the gut microbiome also occur with interventions used to treat obesity. Roux-en-Y surgery significantly increases levels of Proteobacteria, while altering specific metabolic markers, such as the production of urinary amines and cresols125.
The gut microbiota and rheumatoid arthritis
Disregulation of host responses secondary to dysbiosis within the gut lumen could affect distant anatomical sites through activation of host immune responses. This may be a mechanism in rheumatoid arthritis, another chronic idiopathic inflammatory condition. In mice, the presence of SFBs in the gut microbiome causes local expansion of Th17 cells126 that then migrate to peripheral immune compartments and activate B cells into antibody-producing plasma cells. Antibody production leads to immune-mediated destruction of the joints that mirrors rheumatoid arthritis127.
Cause or effect?
Microbiome analysis in humans has been largely based on observation, with associations of disease phenotypes with particular microbiota constituents. But which is causal: does factor A cause factor B, does factor B cause factor A, or does factor C cause both A and B? Hill developed criteria to address the questions: “In what circumstances can we pass from this observed association to a verdict of causation? Upon what basis should we proceed to do so?”128 The criteria include the strength of association, its consistency, specificity, temporality, and biological plausibility, and whether biological gradients are present, experimental support exists, and support can be extrapolated from known causal relationships. Although these criteria were advanced largely to unravel epidemiological relationships, they are applicable to genetics as well and to metagenomics, in particular. Sometimes successful treatment trials with amelioration or cure of a particular condition provide the critical evidence for a causal relationship. The changed natural history of peptic ulcer disease following elimination of H. pylori129 demonstrated its pathogenic role.
For understanding causation and pathogenesis, model organisms provide an important approach. Animal models approximate some human diseases (e.g. asthma, atherosclerosis), but many diseases are not well reproduced (e.g. psoriasis). For diseases that can be studied in model organisms, microbiota roles can be explored within the constraints of particular model systems (Table 2). Standard models of inbred mice are limited by their uncontrolled microbiome diversity. Certain disease states are well-studied in these models, such as the effects of SFBs on Th17 development or the susceptibility to type I diabetes in non-obese diabetic (NOD) mice. The use of gnotobiotic mice eliminates the above-mentioned microbiome variability, but the animals are expensive and require specialized facilities and expertise limiting their widespread use. The recent availability of gnotobiotic animals from commercial sources permits conventionalization of animals with experimental or control microbiota without needing xenobiotic facilities; such approaches allow for the direct observation of microbiota effects on the host. The extension of this concept to humanized model organisms130 allows better approximation of the effects of the human microbiome on disease processes in tractable animal models.
Table 2.
The use of mouse models in microbiome studies.
| Model | Advantages | Disadvantages |
|---|---|---|
| Inbred mice138 | Relatively inexpensive Often well-characterized Genetically homogeneous Allow the study of pathogenetic mechanisms |
Poorly controlled microbial variability Limited translation potential to humans |
| Gnotobiotic mice139 | Well-controlled microbial variability Allow for better understanding of specific microbe interactions Genetically homogeneous Allow mechanistic studies |
Expensive Difficult to maintain Limited translation potential to humans Physiologically less well- understood than conventional animals |
| Humanized mice130 | More relevant to human disease states Genetically homogeneous |
Expensive Difficult to maintain Physiologically less well- understood than non-chimeric animals |
| Conventionalized gnotobiotic mice140 |
Well-controlled microbial variability Allow for better understanding of specific microbe interactions Genetically homogeneous Allow mechanistic studies Do not require specialized xenobiotic facilities |
Physiologically less well- understood than conventional animals “Artificial” colonization using known microbiota may not be representative of real-world situation |
Perspectives
Inherent complexities in the composition of the microbiome may preclude investigations of microbe-associated diseases using classical approaches such as Koch’s postulates. Instead of single organisms associated with disease, community characteristics (composition and metagenomic functionality) may be more relevant. The principles of host interaction with pathogens and commensals contain many parallel features, which can help tutor the new field, but the nature of the selection for commensalism is more complex and highly dynamic. The scale of the interface suggests that microbiome–host interactions have important bearings on disease susceptibility, and the microbial effects on the balance of host metabolism and immunity131 provides an excellent model for the broader phenomenon of disease susceptibility. Modifying disease risk by altering metabolic, immunological, or developmental pathways are obvious strategies.
Given the ongoing extinction of our ancient commensal organisms, the future of a healthy human microbiome may include restoration of our ancestral microbial ecology. There are two types of restoration. The first involves restoring ancient organisms (or pathways) in healthy hosts lacking them, as prophylaxis against future imbalances. The second type of restoration could be therapeutic, when the etiologic extinctions and imbalances are recognized. This scientific frontier will require understanding the biology of re-introductions, as well as developing microbial breeding programs. In addition to the technical problems associated with restoring particular organisms to specific hard-to-reach niches, such as the distal ileum, there also will be substantial biological problems related to understanding how reintroductions affect the population structure of the extant organisms, and host interactions.
To better understand the implications of microbiota and metagenome variation in human health and disease, the field needs improved informatics tools, including new approaches for understanding the complexity of the metadata132. The multidimensionality of the human and microbial phenotypes and the dynamic, non-linear interactions challenge deterministic solutions. For example, in analyses of 16S-defined operational taxonomic units (OTU) populations in mice receiving traditional or Western-type diets, Reshef et al., examined top-scoring non-linear abundance relationships133. These often involved “noncoexistence”, sometimes related to known factors (e.g. diet, host gender), but were often unexplained. Although incomplete, such work leads to new approaches to understand the underlying complexity.
We also will need new tools to implement the health implications presented by metagenomic analyses. Although the known principles of evolution and genetics apply, study of the microbiome provides new applications, and will lead to new understandings of complex traits. Important questions to pursue are listed in Box 2. As such, this is a frontier for human preventive medicine, and for medical management of chronic diseases.
Box 1: 10 areas of microbiome inquiry that should be pursued.
Understanding microbiome characteristics in relation to families: what is inherited and what is not? *
Understanding secular trends in microbiome composition: what has been lost or gained? #
For diseases that have changed markedly in incidence in recent decades are changes in the microbiome playing a role? Notable examples include childhood-onset asthma, food allergies, type 1 diabetes, obesity, inflammatory bowel disease, autism. *#
Do particular signatures of the metagenome predict risk for specific human cancers and other diseases associated with aging? Can these signatures be pursued to better understand oncogenesis (work on Helicobacter pylori provides a clear example of this)? *
How do antibiotics perturb the microbiome—in the short-term and long term? Does the route of administration matter [the route is not discussed in the text so you might want to provide more context here]? *
How does the microbiome affect the pharmacology of medications? Can we “micro-type” people to improve pharmacokinetics and/or reduce toxicity? Can we manipulate the microbiome to improve pharmacokinetic stability? *#
Can we harness knowledge of the microbiome to improve diagnostics for disease status and susceptibility? *
Can we harness the close mechanistic interactions between the microbiome and host to provide hints for the creation of new drugs? #
Specifically, can we harness the microbiome to create new narrow-spectrum antibiotics? #
Can we use knowledge of the microbiota to develop true probiotics (and prebiotics)? *#
* Areas currently under investigation
# Proposed areas for investigation
Online Summary.
The human microbiome and its relationship to disease is a new and rapidly evolving field of study.
Co-evolution of hosts and their microbiomes has led to cooperative interactions in metabolism and homeostasis.
Concepts from community ecology such as resilience, community disturbances, and extinction are useful in understanding the microbiome.
New computational and statistical tools are being actively developed to analyze the large sequence datasets generated by the increasingly powerful technologies.
The taxonomic composition and functional characteristics of the microbiome may allow individuals to be categorized into different microbial patterns, called “enterotypes”, in the gastrointestinal tract. Although low-level taxonomy varies substantially among individuals, higher level taxonomy and functional characteristics appear largely preserved.
Many factors affect the composition of the microbiome over the course of a human lifetime. These include inheritance, mode of infant delivery, diet, and age-related changes in adults.
The relationships between the microbiome and several human diseases are being intensively studied for conditions that include colorectal cancer, inflammatory bowel disease, and immunologically-mediated skin diseases.
Causal relationships for many of the associations between the microbiome and disease states have yet to be proven.
Understanding the links between the microbiome and human disease may provide prophylactic or therapeutic tools to improve human health.
Acknowledgements
This work was supported by R01GM63270, R01DK090989, UH2 AR057506, 5 P30 CA016087, and 1UL1RR029893 from the National Institutes of Health, the Diane Belfer Program for Human Microecology, the Michael Saperstein Medical Scholars Fund, and the Levin Fellowship in Gastroenterology.
Glossary
- Microbiota
The microbial organisms that constitute the microbiome. The composition of the microbiota within a community can vary substantially between environmental sites and host niches in health and disease
- 16S rRNA
A component of the 30S small subunit of prokaryotic ribosomes. Sequencing of the 16S rRNA has been used to identify prokaryotic taxonomy in complete environmental samples such as the microbiome
- Metagenome
The genetic information of a complex population, typically from microbes in an environmental or host niche sample, that is constituted by the genomes of many individual organisms. The metagenome provides information about the functional genetic potential of the aggregate population
- Microbiome
The totality of microbes, their genetic information, and the milieu in which they interact. Microbiomes typically consist of environmental or biological niches containing complex communities of microbes
- Extinction
The loss, usually of a species, within an ecosystem
- Enterotype
A recently proposed classification unit of animals based on the bacteriological composition of their gut microbiome. There are reported to be at least three distinct enterotypes independent of ethnic background and diet
- Nash equilibrium
A concept from game theory in which players know the strategies of the others, and in which any change from their strategy puts them in a less favourable position.
- Resilience
A term in ecology indicating the capacity of a system to absorb disturbance and reorganize itself while undergoing change, so as to retain essentially the same function, structure, and identity.
- Extirpation
The loss of species in a locality (e.g. an individual host)
- Allelopathy
A phenomenon in which a microbe uses chemical means to aid in its competition with a group of microbe(s). Allelopathy may involve manipulation of third parties (e.g. host) to favor competition
- Mating preference
The selection or choice of sexual partners that is often based on traits of a potential mate. Genetic differences between selected and non-selected hosts are a source of selectable variation
- Antecubital fossa
The triangular area on the anterior (or flexor) aspect of the elbow joint
- Popliteal fossae
The shallow depression found on the flexor aspect of the knee joint
- Pilosebaceous units
The anatomic structure around each hair shaft that consists of the hair shaft and follicle, the sebaceous gland, and the erector pili muscle
- Amphibiont
An organism (e.g. microbe) that may have a pathogenic or symbiotic relationship with another organism (e.g. its host), depending on context. A more accurate term than commensal
- Lamina propria
A thin layer of loose connective tissue that lies underneath the epithelium and with it constitutes the mucosa that lines various lumens within the body; it is a dense locus of immunological and inflammatory cells
- Steatosis
The pathological accumulation and retention of lipids within liver parenchymal cells. Substantial steatosis can compromise the function of the cell and is associated with disease processes, including alcoholism, diabetes, and hyperlipidemia
- Commensal
An organism (e.g. microbe) that is involved in a form of symbiosis in which one organism derives a benefit while the other is unaffected
- Probiotics
Living microorganisms that are thought to confer a benefit to the host.
- Roux-en-Y surgery
A type of gastric bypass surgery primarily used for the treatment of morbid obesity. In Roux-en-Y surgeries, a portion of the small bowel is bypassed to decrease the absorption of nutrients
- Dysbiosis
A condition in which the normal microbiome population structure is disturbed, often through external pressures such as disease states or medications
- Gnotobiotic
Describes an animal that is colonized solely with known strains of bacteria or other microorganisms. The term also describes germ-free animals, as the status of their microbial communities is known
- Operational taxonomic unit (OTU)
The smallest phylogenetic unit described by variations in 16S rRNA sequencing. Dissimilarity of <1% in 16S rRNA sequences has commonly been used to define an OTU but <3% and <5% have also been used
- Noncoexistence
An exclusivity scenario in which the abundance of one species leads to another species being less abundant than would be expected by chance
- Prebiotics
Food ingredients that confer specific changes in the gut microbiome and lead to beneficial effects in the host
Biographies
Ilseung Cho received his MD and MS from New York University and completed his post-graduate training in Internal Medicine and Gastroenterology at New York University (NYU), USA. He is an Assistant Professor of Medicine at the NYU Langone Medical Center, USA. His current research focuses on the role of the gut microbiome in drug metabolism and availability.
Martin J. Blaser received his MD from New York University (NYU), USA. He received post-graduate training at the University of Colorado at Boulder (CO, USA), Centers for Disease Control in Atlanta (Georgia, USA), Rockefeller University (NY, USA), and at the Institut Pasteur in Paris (France). He is a Professor of Medicine and Microbiology at the NYU Langone Medical Center, USA. His current research focuses on antibiotic-induced changes in the human and mouse microbiome and their effects on disease development.
Footnotes
Websites:
Human Microbiome Project: http://commonfund.nih.gov/hmp
Metagenomics of the Human Intestinal Tract (MetaHIT): http://www.metahit.eu
Metagenomics Rapid Annotation using Subsystem Technology (MG-RAST): http://metagenomics.anl.gov
greengenes: http://greengenes.lbl.gov
Quantitative Insights Into Microbial Ecology (QIIME): http://www.qiime.org
QIIME database: http://www.microbio.me/qiime
Ribosomal Database Project (RDP): http://rdp.cme.msu.edu
Nature Reviews Genetics Series on Applications of Next-Generation Sequencing: http://www.nature.com/nrg/series/nextgeneration/index.html
References
- 1.Baumann P, Moran NA. Non-cultivable microorganisms from symbiotic associations of insects and other hosts. Antonie van Leeuwenhoek. 1997;72:39–48. doi: 10.1023/a:1000239108771. [DOI] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich SD. MetaHIT : The European Union Project on Metagenomics of the Human Intestinal Tract. In: Nelson KE, editor. Metagenomics of the Human Body. Springer; 2011. pp. 307–316. [Google Scholar]
- 4. Ravel J, et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America. 2011;108 Suppl 1:4680–4687. doi: 10.1073/pnas.1002611107. Describes vaginal microbiome differences and similarities in women of reproductive age who vary by ethnicity, and explores factors related to bacterial vaginosis
- 5. Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. Proposes enterotype classifications that are defined by the intrinsic characteristics of the gut microbiome, and that seem to be independent of ethnic or dietary factors
- 6.Morris SC, Peel JS. The earliest annelids: Lower Cambrian polychaetes from the Sirius Passet Lagerstatte, Peary Land, North Greenland. Acta Palaeontologica Polonica. 2008;53:135–146. [Google Scholar]
- 7. Ley R, Lozupone CA, Hamady M, Knight R, Gordon J. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. A review that contrasts the microbial communities in the vertebrate gut with each other and with free-living microbial communities
- 8.Ochman H, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS biology. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran NA, Munson MA, Baumann P, Ishikawa H. A Molecular Clock in Endosymbiotic Bacteria Is Calibrated Using the Insect Hosts. Proceedings of the Royal Society of London Series B-Biological Sciences. 1993;253:167–171. [Google Scholar]
- 10.Benson AK, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wikoff WR, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. A comparison of germ-free and conventional animals to show that the microbiome has substantial effects on host blood metabolites, including the metabolism of amino acids and organic acids
- 12.Petchey OL, Eklof A, Borrvall C, Ebenman B. Trophically unique species are vulnerable to cascading extinction. The American naturalist. 2008;171:568–579. doi: 10.1086/587068. [DOI] [PubMed] [Google Scholar]
- 13. Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. Proposes that co-evolved bacteria in human hosts establish homeostases that conform to the principles of Nash equilibria. Understanding such equilibria may provide insight into shifts in microbial communities in health and disease
- 14.Maynard Smith J, et al. Models in ecology. Cambridge Eng.: University Press; 1974. pp. xii–145. [Google Scholar]
- 15.Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO reports. 2006;7:956–960. doi: 10.1038/sj.embor.7400812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tringe SG, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 17. Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. A seminal paper describing the ability of the gut microbiome to extract energy from dietary sources
- 18.Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 19. Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. The authors report on the identification of a library of microbial genes foundwithin the human gut microbiome using massive parallel high-throughput metagenomic sequencing
- 20.Nelson KE, et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuczynski J, et al. Experimental and analytical tools for studying the human microbiome. Nature reviews. Genetics. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Academy of Sciences of the United States of America. 2011 Dec 19; doi: 10.1073/pnas.1116053109. A new method of comparing metagenomic data that involves analysing metabolic networks and their associated genes to describe changes that occur with disease states (such as obesity or inflammatory bowel disease).
- 23.Eckburg PB, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogaert D, et al. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS ONE. 2011;6:e17035. doi: 10.1371/journal.pone.0017035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. Describes temporal and topographical variations in the human microbiome at a range of anatomic sites
- 26.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuczynski J, et al. Direct sequencing of the human microbiome readily reveals community differences. Genome Biology. 2010;11:210. doi: 10.1186/gb-2010-11-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. Describes the substantial alterations that occur in the gut microbiome after exposure to antibiotics. It highlights varied taxonomic changes betweenindividuals
- 29.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huse SM, et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS genetics. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley RE, et al. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linz B, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douglass JM, Li Y, Tinanoff N. Association of mutans streptococci between caregivers and their children. Pediatric dentistry. 2008;30:375–387. [PubMed] [Google Scholar]
- 34.Li Y, Ismail AI, Ge Y, Tellez M, Sohn W. Similarity of bacterial populations in saliva from African-American mother-child dyads. Journal of Clinical Microbiology. 2007;45:3082–3085. doi: 10.1128/JCM.00771-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2117–2122. doi: 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McNulty NP, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Science translational medicine. 2011;3:106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS biology. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. Describes the taxonomic developments that occur in the infant microbiome and their relationships with environmental exposures
- 40. Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nature Reviews Microbiology. 2009;7:887–894. doi: 10.1038/nrmicro2245. Proposes that changes in modern life have led to the extinction of certain microbes, and that their disappearance may have deleterious effects on human health
- 41.Sjolund M, Wreiber K, Andersson DI, Blaser MJ, Engstrand L. Long-term persistence of resistant Enterococcus species after antibiotics to eradicate Helicobacter pylori. Annals of internal medicine. 2003;139:483–487. doi: 10.7326/0003-4819-139-6-200309160-00011. [DOI] [PubMed] [Google Scholar]
- 42.Blaser MJ. Antibiotic overuse: Stop the killing of beneficial bacteria. Nature. 2011;476:393–394. doi: 10.1038/476393a. [DOI] [PubMed] [Google Scholar]
- 43.Evans AS. Causation and disease: the Henle-Koch postulates revisited. The Yale journal of biology and medicine. 1976;49:175–195. [PMC free article] [PubMed] [Google Scholar]
- 44.Muegge BD, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huston MA, et al. Biological diversity : the coexistence of species on changing landscapes. Cambridge; New York, NY, USA: Cambridge University Press; 1994. pp. xix–681. [Google Scholar]
- 46.Kennedy TA, et al. Biodiversity as a barrier to ecological invasion. Nature. 2002;417:636–638. doi: 10.1038/nature00776. [DOI] [PubMed] [Google Scholar]
- 47.Strogatz SH. Exploring complex networks. Nature. 2001;410:268–276. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- 48.Paine RT. Food Web Complexity and Species Diversity. The American Naturalist. 1966;100:65–75. [Google Scholar]
- 49.Sole RV, Montoya JM. Complexity and fragility in ecological networks. Proceedings. Biological sciences / The Royal Society. 2001;268:2039–2045. doi: 10.1098/rspb.2001.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borrvall C, Ebenman B. Early onset of secondary extinctions in ecological communities following the loss of top predators. Ecology letters. 2006;9:435–442. doi: 10.1111/j.1461-0248.2006.00893.x. [DOI] [PubMed] [Google Scholar]
- 51.Bik EM, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maldonado-Contreras A, et al. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. The ISME journal. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Caufield PW, Dasanayake AP, Wiener HW, Vermund SH. Mode of delivery and other maternal factors influence the acquisition of Streptococcus mutans in infants. Journal of dental research. 2005;84:806–811. doi: 10.1177/154405910508400905. [DOI] [PubMed] [Google Scholar]
- 54. Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. Shows that infants have largely undifferentiated microbiota across multiple anatomic sites immediately after birth, and that delivery mode determines the types of bacteria that are the earliest colonizers of the infant microbiome
- 55. Savage DC, Dubos R, Schaedler RW. The gastrointestinal epithelium and its autochthonous bacterial flora. The Journal of experimental medicine. 1968;127:67–76. doi: 10.1084/jem.127.1.67. One of the pioneering studies of the early-life features of bacterial colonization of the gastrointestinal tract
- 56.Gronlund MM, Lehtonen OP, Eerola E, Kero P. Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. Journal of pediatric gastroenterology and nutrition. 1999;28:19–25. doi: 10.1097/00005176-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Grant BR, Grat PR. Cultural inheritance of song and its role in the evolution of Darwin's finches. Evolution. 1996;50:2471–2487. doi: 10.1111/j.1558-5646.1996.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 58.Hunt J, Simmons LW. Maternal and paternal effects on offspring phenotype in the dung beetle Onthophagus taurus. Evolution; international journal of organic evolution. 2000;54:936–941. doi: 10.1111/j.0014-3820.2000.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 59.Raymond J, et al. Genetic and transmission analysis of Helicobacter pylori strains within a family. Emerging infectious diseases. 2004;10:1816–1821. doi: 10.3201/eid1010.040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. Reports the discovery of a large network of gene exchange that occurs in microbial communities that allows rapid genetic information transfer to occur in the microbiome. The authors speculate that such networks may have roles in specific human diseases
- 61.Wirth T, et al. Distinguishing human ethnic groups by means of sequences from Helicobacter pylori: lessons from Ladakh. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4746–4751. doi: 10.1073/pnas.0306629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharon G, et al. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20051–20056. doi: 10.1073/pnas.1009906107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leyden JJ, McGinley KJ, Holzle E, Labows JN, Kligman AM. The microbiology of the human axilla and its relationship to axillary odor. The Journal of investigative dermatology. 1981;77:413–416. doi: 10.1111/1523-1747.ep12494624. [DOI] [PubMed] [Google Scholar]
- 64.Dobzhansky T. Further Data on the Variation of the Y Chromosome in Drosophila Pseudoobscura. Genetics. 1937;22:340–346. doi: 10.1093/genetics/22.3.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mayr E. Systematics and the origin of species from the viewpoint of a zoologist. New York: Columbia University Press; 1942. pp. xiv–334. incl. illus. (incl. maps) tables, diagrs. [Google Scholar]
- 66.Brailsford SR, et al. The microflora of the erupting first permanent molar. Caries Research. 2005;39:78–84. doi: 10.1159/000081661. [DOI] [PubMed] [Google Scholar]
- 67.Cephas KD, et al. Comparative analysis of salivary bacterial microbiome diversity in edentulous infants and their mothers or primary care givers using pyrosequencing. PLoS ONE. 2011;6:e23503. doi: 10.1371/journal.pone.0023503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaedler RW. The relationshp between the host and its intestinal microflora. The Proceedings of the Nutrition Society. 1973;32:41–47. doi: 10.1079/pns19730013. [DOI] [PubMed] [Google Scholar]
- 69.Jukes TH. Antibiotics in Feeds. Science. 1979;204:8. doi: 10.1126/science.204.4388.8. [DOI] [PubMed] [Google Scholar]
- 70.Robinson CJ, Young VB. Antibiotic administration alters the community structure of the gastrointestinal micobiota. Gut microbes. 2010;1:279–284. doi: 10.4161/gmic.1.4.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wlodarska M, et al. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infection and immunity. 2011;79:1536–1545. doi: 10.1128/IAI.01104-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gemmell NJ, Slate J. Heterozygote advantage for fecundity. PLoS ONE. 2006;1:e125. doi: 10.1371/journal.pone.0000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cauci S, et al. Prevalence of bacterial vaginosis and vaginal flora changes in peri- and postmenopausal women. Journal of Clinical Microbiology. 2002;40:2147–2152. doi: 10.1128/JCM.40.6.2147-2152.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Osborne NG, Wright RC, Grubin L. Genital bacteriology: a comparative study of premenopausal women with postmenopausal women. American journal of obstetrics and gynecology. 1979;135:195–198. doi: 10.1016/0002-9378(79)90342-9. [DOI] [PubMed] [Google Scholar]
- 75.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature reviews. Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 76.Giannakis M, Chen SL, Karam SM, Engstrand L, Gordon JI. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4358–4363. doi: 10.1073/pnas.0800668105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li XX, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS ONE. 2009;4:e7985. doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mariat D, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC microbiology. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nordling CO. A new theory on cancer-inducing mechanism. British journal of cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vanhoutvin SA, et al. Butyrate-induced transcriptional changes in human colonic mucosa. PLoS ONE. 2009;4:e6759. doi: 10.1371/journal.pone.0006759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hamilton WD. The moulding of senescence by natural selection. Journal of theoretical biology. 1966;12:12–45. doi: 10.1016/0022-5193(66)90184-6. A pioneering paper that describes how several key factors — fertility, mortality, and age —affect population dynamics
- 82.Perry S, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE. 2010;5:e8804. doi: 10.1371/journal.pone.0008804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Higgins PD, et al. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: Mucosal crosstalk between stomach and distal intestine. Inflammatory Bowel Diseases. 2011;17:1398–1408. doi: 10.1002/ibd.21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arnold IC, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. The Journal of clinical investigation. 2011;121:3088–3093. doi: 10.1172/JCI45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. The Journal of clinical investigation. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blaser MJ, Webb G. American Society for Microbiology Conference of Beneficial Microbes. Lake Tahoe, NV: 2005. Host demise as a beneficial function of indigenous microbiota in multicellular hosts. [Google Scholar]
- 87.Patel RV, Lebwohl M. In the clinic. Psoriasis. Annals of internal medicine. 2011;155 doi: 10.7326/0003-4819-155-3-201108020-01002. ITC2-1,àíICT2-15; quiz ITC2-16. [DOI] [PubMed] [Google Scholar]
- 88.Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE. 2008;3:e2719. doi: 10.1371/journal.pone.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Grice EA, Segre JA. The skin microbiome. Nature reviews. Microbiology. 2011;9:244–253. doi: 10.1038/nrmicro2537. A comprehensive review of the skin microbiome and its connection to several associated diseases
- 90.McDowell A, et al. A novel multilocus sequence typing scheme for the opportunistic pathogen Propionibacterium acnes and characterization of type I cell surface-associated antigens. Microbiology. 2011;157:1990–2003. doi: 10.1099/mic.0.049676-0. [DOI] [PubMed] [Google Scholar]
- 91.Price LB, et al. Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: impact of diabetes and antibiotics on chronic wound microbiota. PLoS ONE. 2009;4:e6462. doi: 10.1371/journal.pone.0006462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grice EA, et al. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14799–14804. doi: 10.1073/pnas.1004204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Andersson AF, et al. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McColl KE. Clinical practice. Helicobacter pylori infection. The New England journal of medicine. 2010;362:1597–1604. doi: 10.1056/NEJMcp1001110. [DOI] [PubMed] [Google Scholar]
- 95.el-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer and reflux disease. Gut. 1998;43:327–333. doi: 10.1136/gut.43.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Archives of internal medicine. 2007;167:821–827. doi: 10.1001/archinte.167.8.821. [DOI] [PubMed] [Google Scholar]
- 97.Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host & Microbe. 2011;10:324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. International journal of cancer. Journal international du cancer. 2004;110:523–531. doi: 10.1002/ijc.20152. [DOI] [PubMed] [Google Scholar]
- 99.Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature medicine. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reikvam DH, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE. 2011;6:e17996. doi: 10.1371/journal.pone.0017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Castellarin M, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Research. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kostic AD, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Research. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Krisanaprakornkit S, et al. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infection and immunity. 2000;68:2907–2915. doi: 10.1128/iai.68.5.2907-2915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host & Microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ivanov II, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host & Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. An important study that describes the immunological interplay between segmented filamentous bacteria and Th-17 cells in the distal small bowel
- 106.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ogura Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 108.Hugot JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 109.Franchimont D, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ewaschuk JB, Tejpar QZ, Soo I, Madsen K, Fedorak RN. The role of antibiotic and probiotic therapies in current and future management of inflammatory bowel disease. Current gastroenterology reports. 2006;8:486–498. doi: 10.1007/s11894-006-0039-z. [DOI] [PubMed] [Google Scholar]
- 111.Hviid A, Svanstrom H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- 112.Manichanh C, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host & Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lepage P, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141:227–236. doi: 10.1053/j.gastro.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 115.Mondot S, et al. Highlighting new phylogenetic specificities of Crohn's disease microbiota. Inflammatory Bowel Diseases. 2011;17:185–192. doi: 10.1002/ibd.21436. [DOI] [PubMed] [Google Scholar]
- 116.Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nature reviews. Gastroenterology & hepatology. 2010;7:691–701. doi: 10.1038/nrgastro.2010.172. [DOI] [PubMed] [Google Scholar]
- 117.Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mutlu E, et al. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcoholism, clinical and experimental research. 2009;33:1836–1846. doi: 10.1111/j.1530-0277.2009.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yan AW, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fox JG, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59:88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Y, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- 122.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 123.Ajslev TA, Andersen CS, Gamborg M, Sorensen TI, Jess T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. International journal of obesity. 2011;35:522–529. doi: 10.1038/ijo.2011.27. [DOI] [PubMed] [Google Scholar]
- 124.Luoto R, Kalliomaki M, Laitinen K, Isolauri E. The impact of perinatal probiotic intervention on the development of overweight and obesity: follow-up study from birth to 10 years. International journal of obesity. 2010;34:1531–1537. doi: 10.1038/ijo.2010.50. [DOI] [PubMed] [Google Scholar]
- 125.Li JV, et al. Metabolic surgery profoundly influences gut microbial-host metabolic cross-talk. Gut. 2011;60:1214–1223. doi: 10.1136/gut.2010.234708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nature reviews. Rheumatology. 2011;7:569–578. doi: 10.1038/nrrheum.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hill AB. The Environment and Disease: Association or Causation? Proceedings of the Royal Society of Medicine. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hentschel E, et al. Effect of ranitidine and amoxicillin plus metronidazole on the eradication of Helicobacter pylori and the recurrence of duodenal ulcer. The New England journal of medicine. 1993;328:308–312. doi: 10.1056/NEJM199302043280503. [DOI] [PubMed] [Google Scholar]
- 130.Devoy A, Bunton-Stasyshyn RK, Tybulewicz VL, Smith AJ, Fisher EM. Genomically humanized mice: technologies and promises. Nature reviews. Genetics. 2012;13:14–20. doi: 10.1038/nrg3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shulzhenko N, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nature medicine. 2011;17:1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 133.Reshef DN, et al. Detecting novel associations in large data sets. Science. 2011;334:1518–1524. doi: 10.1126/science.1205438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer prevention research. 2008;1:329–338. doi: 10.1158/1940-6207.CAPR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57:561–567. doi: 10.1136/gut.2007.133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tana C, et al. Altered profiles of intestinal microbiota and organic acids may be the origin of symptoms in irritable bowel syndrome. Neurogastroenterology and motility : the official journal of the European Gastrointestinal Motility Society. 2010;22:512–519. e114–e115. doi: 10.1111/j.1365-2982.2009.01427.x. [DOI] [PubMed] [Google Scholar]
- 137.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Larsson E, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2011 Nov;23 doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Backhed F, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Turnbaugh PJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Science translational medicine. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]