

Abstract
Despite advances in metabolic and postmetabolic labeling methods for quantitative proteomics, there remains a need for improved label-free approaches. This need is particularly pressing for workflows that incorporate affinity enrichment at the peptide level, where isobaric chemical labels such as isobaric tags for relative and absolute quantitation and tandem mass tags may prove problematic or where stable isotope labeling with amino acids in cell culture labeling cannot be readily applied. Skyline is a freely available, open source software tool for quantitative data processing and proteomic analysis. We expanded the capabilities of Skyline to process ion intensity chromatograms of peptide analytes from full scan mass spectral data (MS1) acquired during HPLC MS/MS proteomic experiments. Moreover, unlike existing programs, Skyline MS1 filtering can be used with mass spectrometers from four major vendors, which allows results to be compared directly across laboratories. The new quantitative and graphical tools now available in Skyline specifically support interrogation of multiple acquisitions for MS1 filtering, including visual inspection of peak picking and both automated and manual integration, key features often lacking in existing software. In addition, Skyline MS1 filtering displays retention time indicators from underlying MS/MS data contained within the spectral library to ensure proper peak selection. The modular structure of Skyline also provides well defined, customizable data reports and thus allows users to directly connect to existing statistical programs for post hoc data analysis. To demonstrate the utility of the MS1 filtering approach, we have carried out experiments on several MS platforms and have specifically examined the performance of this method to quantify two important post-translational modifications: acetylation and phosphorylation, in peptide-centric affinity workflows of increasing complexity using mouse and human models.
Mass spectrometry has rapidly evolved into a high throughput methodology for identifying differentially expressed proteins or post-translational modifications (for review, see Ref. 1). These data can be used to improve the understanding of regulatory pathways, to discover novel disease biomarkers, and to characterize molecular mechanisms of both normal and pathological processes. There are several methodologies available for carrying out differential proteomics using both stable isotope labeling or label-free workflows as reviewed in Ref. 2. For metabolic labeling, stable isotope labeling by amino acids in cell culture or SILAC1 is typically used in cell culture experiments (3), although this approach has been recently adapted for studies in animals (4, 5). As an alternative, isobaric labeling strategies that chemically tag peptides are carried out in a postmetabolic context after protein synthesis, isolation, and proteolytic digestion. iTRAQ (6) and more recently tandem mass tags (7, 8) are two widely used examples of this strategy capable of multiplexing up to six to eight separate samples, respectively. Other methods have also been described, such as 18O labeling during proteolysis, but have not been widely adopted because of technical challenges.
Despite these advances, the use of isobaric tags or SILAC for quantitation is not feasible for all experimental workflows. SILAC labeling, for example, may be cost prohibitive or even unfeasible in proteomic studies involving tissues from mammalian models (e.g. mice) or humans (e.g. plasma or tumor biopsies). Postmetabolic labeling strategies, such as iTRAQ, can in principle be used in mammalian systems, although it may be impractical in some cases. For example, in peptide-centric workflows that target post-translationally modified (PTM) peptides, antibody or other enrichment steps may be incompatible with the chemical tags. Our own effort to combine an anti-acetyllysine antibody enrichment step with iTRAQ was largely unsuccessful, with the enrichment efficiency of peptides containing this modification significantly lower than without chemical labeling.2
Label-free quantitative methods are better suited for proteomic experiments where SILAC labeling is not possible or where postmetabolic isobaric labeling approaches may result in substantial inefficiencies. In recent years several label-free approaches have been described including MS/MS spectral counting (9, 10) and MS ion intensity measurements (11). However, spectral counting approaches are typically used for relative protein quantitation over a small dynamic range and are not appropriate for affinity enrichment workflows that are by nature peptide-centric. In contrast, label-free quantitation approaches that extract ion abundances from MS1 scans are more compatible with quantifying PTM-modified peptides, because each peptide analyte is treated separately. Methods of this type include MSQuant (12), MaxQuant (13), Census (14), MASIC (15), SuperHirn (16), and Spectrum Mill (17), among others (for review see Ref. 11). However, existing MS1 extraction tools are often limited to a particular instrument platform or tailored to a specific laboratory's data pipeline. In addition, graphical outputs that allow ready evaluation of processed multireplicate data sets and run to run feature comparisons are often absent. Perhaps most importantly, the lack of a true platform-independent quantitation program that could be used across multiple laboratories is clearly lacking, thus limiting the ability for large scale proteomic experiments or direct comparison of data sets from different laboratories and instruments.
To address these limitations, we have adapted Skyline, a freely available open source software application (http://proteome.gs.washington.edu/software/skyline), to provide a label-free quantitation tool called Skyline MS1 filtering that efficiently extracts and processes ion intensity chromatograms from MS1 scans of peptide precursors across multiple experiments. We have extended the quantitative and graphical tools originally developed in Skyline for multiple reaction monitoring (MRM)-MS experiments (18) to support interrogation of multiple sample acquisitions for MS1 filtering. These tools in Skyline provide graphical displays for peak selection, replicate views for peak area and retention time, and after review Skyline allows for manual peak integration (when necessary), key features often lacking in existing software. In addition, Skyline MS1 filtering displays direct links of selected MS1 peak features to their underlying MS/MS data to ensure proper peak selection and integration, as well as facile raw data file import to a vendor independent format. Since its release, Skyline has proven to be a highly reliable and flexible program for targeted MRM-MS analysis (19–21). It is currently being used in the National Cancer Institute Clinical Proteomic Technology Assessment for Cancer program to assess reproducibility and accuracy of large scale proteomic experiments across multiple sites and laboratories2 (http://proteomics.cancer.gov/programs/cptacnetwork). To test the utility of Skyline MS1 filtering for label-free quantitative proteomics, we have carried out comparative experiments to examine the ability to process data from several instrument types, including QqTOF and LTQ FT-ICR mass spectrometers, and obtained appropriate linear and quantitative response curves for multiple peptides. Once these performance metrics were established, we then carried out a set of experiments that targeted two important PTMs, phosphorylation and N-ε-acetylation, that would provide a more rigorous evaluation of the performance and robustness of Skyline MS1 filtering, especially in peptide-centric workflows where quantitation methods are lacking. These experiments were carried out with samples of increasing complexity and workflow design, consisting of peptide affinity enrichment steps that targeted PTMs changes in breast cancer cell lines and mitochondria isolated from transgenic mouse models.
EXPERIMENTAL PROCEDURES
Materials
HPLC solvents including acetonitrile and water were obtained from Burdick and Jackson (Muskegon, MI). Reagents for protein chemistry including iodoacetamide, DTT, ammonium bicarbonate, formic acid, trifluoroacetic acid, acetic acid, dichloroacetic acid (DCA), dodecyl-maltoside, urea, as well as the protein standards bovine hemoglobin, BSA, rabbit phosphorylase B, and yeast enolase were purchased from Sigma-Aldrich. All protein standards were >95% purity. Tris(2-carboxyethyl)phosphine was purchased from Thermo (Rockford, IL), and HLB Oasis SPE cartridges were purchased from Waters (Milford, MA). Dialysis cassettes (MWCO 3 kDa) were obtained from Pierce, and proteomics grade trypsin was from Promega (Madison WI). Trypsin-predigested β-galactosidase (a quality control standard) was purchased from AB SCIEX (Foster City, CA).
Amino Acid Analysis for Protein Standards (Utilized for LTQ FT-ICR Concentration Curve)
Amino acid analysis was performed by AAA Laboratory (Mercer Island, WA). Protein standards in 5% acetic acid (2 mg/ml, w/v) were subjected to hydrolysis in a mixture of 6 n HCl, 0.05% β-mercaptoethanol, 0.02% phenol for 24 h at 110 °C. Concentrations of amino acids except cysteine and tryptophan were then measured by UV-visible LC and using a 6-point standard curve.
Reagents for PTM Peptide Enrichment
Anti-acetylated lysine antibodies were purchased from ImmuneChem (ICP0380-100) and Cell Signaling (9441) for enrichment of acetylated peptides. Titanium dioxide chromatography for enrichment of phosphorylated peptides was performed using the Titanosphere Phos-TiO kit (200-μl columns, GL Sciences). Anti-pyruvate dehydrogenase 1α antibodies for Western blots were purchased from Abcam (whole protein), EMD (for pSer-293), or provided as a generous gift from Dr. J. Murray of Abcam (for Ser(P)-300 and Ser(P)-232). Acetylated and phosphorylated synthetic peptides containing stable isotope-labeled lysine or arginine residues (13C615N2-Lys and 13C615N4-Arg, respectively) were obtained from Thermo Fisher Scientific.
Data Accession
The data associated with this manuscript may be downloaded from the University of Washington, Seattle-hosted server at http://proteome.gs.washington.edu/supplementary_data/MS1_Filtering/. All of the details for peptide quantitation using MS1 filtering, including peptide peak areas for all MS replicates, are provided as Excel files in a subfolder titled “Quantitation Details” (and are also provided as supplemental Table S6, A–I).
Spectral Viewer: Skyline Spectral Library for Reviewing Data Quality of PTM-containing Peptides
An interactive Skyline spectral library file that contains all of the MS/MS spectra of PTM-containing peptides identified and that were subsequently used for MS1 filtering and quantitation is supplied as supplemental Document S1. Skyline is an open source program and can be downloaded at http://proteome.gs.washington.edu/software/skyline. All of the MS/MS spectral details underlying this data are displayed in supplemental Table S1 (A–F) with search engine parameters summarized in supplemental Methods S1 and Table S1G. PTM site assignment was initially suggested by search engines Protein Pilot and Mascot (for details see below) and confirmed by manual inspection using previously defined criteria (22). All of the PTM-containing peptides are provided for review as part of this Spectral Viewer Skyline file.
Skyline MS1 Filtering Tool Algorithm and Data Analysis
Skyline is an open source software project and can be freely installed (http://proteome.gs.washington.edu/software/skyline). Spectral libraries were generated in Skyline using the BiblioSpec algorithm (23) from database searches of the raw data files prior to MS1 filtering. Additional details and tutorials for creating spectral libraries and MS1 filtering can be viewed on the Skyline website (http://proteome.gs.washington.edu/software/skyline). For workflows focusing on PTM peptides, specific refinement features in Skyline were set to filter out nonacetylated or nonphosphorylated peptides from the Skyline peptide tree.
Briefly, a comprehensive spectral library was created from the raw data containing MS/MS spectra, including repetitive sampling of a single peptide within a run as well as sampling across multiple acquisitions (see Fig. 2). The use of a redundant library enabled MS/MS-directed MS1 peak picking/integration and peak identification following raw data file import and MS1 filtering (see Fig. 3C). For peak picking and ion intensity integration of peptide isotopes, Skyline dynamically selects a constant time interval to use for intensity values over the entire measured range. The time interval is chosen by first calculating the mode or modes of the intervals between the raw acquired points and then choosing the minimum mode value. After the interval is chosen, all of the intensity values are mapped by linear interpolation to consistently spaced times an interval width apart. Peak detection is performed on the interpolated points, and the peak areas are calculated using trapezoidal summation in seconds of intensity (s * i) units (24). The background area is subtracted from all peak areas, where background is defined as the rectangular area bounded by the integration boundaries, the x axis, and a line perpendicular to the minimum intensity at which the chromatogram crosses the integration boundaries. Typically, the precursor isotopic import filter was set to a count of three, (M, M + 1, and M + 2) at a resolution of 10,000–50,000 depending on mass spectrometric instrument platform (supplemental Fig. S1A). The use of multiple precursor isotopes per peptide enabled features that rank the theoretical and observed isotopic distribution using different novel metrics. These included the isotopic rank in the Skyline peptide tree and the “expected” isotopic distribution in the graphical interface (supplemental Fig. S2). In addition, an isotopic dot product score is generated from comparing the expected distribution to the observed distribution (scored from 0 to 1, where 1 is the highest) (supplemental Fig. S3). Skyline provides graphical tools to easily visually assess multiple precursors during MS1 filtering, to utilize that information to confirm confident peak picking, to identify potential interferences, and to visually assess extracted ion chromatogram quality (intensity and noise level) of the selected peak with its multiple precursor isoforms. Raw files were directly imported into Skyline in their native file format, which Skyline achieves using the ProteoWizard data access library (25).
Fig. 2.

Schematic of MS1 filtering proteomic data flow. New or significantly revised modules unique to Skyline MS1 filtering are indicated in text boxes (see text for explanation).
Fig. 3.

Skyline MS1 filtering graphical user interface. A, Skyline peptide tree for peptide YAPVAKacDLASR (Kac is acetyllysine, and R is 13C615N4-Arg) showing three extracted molecular ion isotope peaks M, M + 1, and M + 2. B, chromatograms and peak intensity traces for four acquisitions from two samples run in duplicates with heavy peptides spiked at 1 and 3 fmol (S1R1, S1R2, S2R1, and S2R2, respectively). The vertical lines with annotated retention times and identification (ID) mark underlying MS/MS sampling that initially directed MS1 peak picking. C, Skyline displays a library of MS/MS spectra for the selected peptide that provides underlying peptide identification information for a specific acquisition replicate. In this case, the underlying MS/MS spectra for two of the four replicates, S1R1 and S2R2 are shown, although all spectra can be displayed. D and E, established Skyline visual displays previously developed for targeted LC-MRM data processing, include peak area replicate views (D) and retention time replicates (E) (further details on Skyline graphical displays, settings, and parameters see supplemental Figs. S1–S3).
After data import, graphical displays of chromatographic traces (extracted ion chromatograms) were manually inspected for proper peak picking of MS1 filtered peptides. In some cases, the peak integration was adjusted manually in the chromatographic window. With the new MS1 filtering capabilities, other Skyline features were also updated and synchronized, such as the Skyline Custom Reports that can export additional relevant fields and parameters important for MS1 filtering analysis and quantitation (see tutorial section). All of the quantitations performed as part of this manuscript were done on the peptide level, because of the peptide centric approach that we took for PTM-containing peptides. All of the details for peptide quantitation using MS1 filtering, including peptide peak areas for all MS replicates, are provided as Excel files as supplemental Tables S6 (A–I). The Skyline website contains a detailed tutorial on MS1 filtering.
Skyline Step by Step Instructions for MS1 Filtering (Tutorial)
A new MS1 filtering “step by step” tutorial has been posted on the Skyline website. The tutorial guides users through the proper use of Skyline MS1 filtering settings, parameters and graphical tools (http://proteome.gs.washington.edu/software/Skyline/tutorials/ms1filtering.html). The tutorial provides a test data set that the user can download and then describes protocols for importing spectral libraries, choosing proper settings and filters, populating the Skyline peptide tree, importing mass spectrometric raw files for MS1 filtering and ion chromatogram extractions, and utilizing new graphical tools that aid in assessing and further processing data sets. New features added to Skyline Custom Reports (“PrecursorResult.IsotopeDotProduct,” “PrecursorResult.Identified,” “Transition.IsotopeDistIndex,” “Transition.IsotopeDistRank,” “Transition.IsotopeDistProportion,” etc.) are explained in another detailed web tutorial titled “Custom Reports & Results Grids” (http://proteome.gs.washington.edu/software/Skyline/tutorials.html). The periodic revisions and updates will occur as additional features are added to Skyline MS1 filtering.
Mass Spectrometric and Chromatographic Methods and Instrumentation
Three mass spectrometry platforms were used to acquire all data used for MS1 filtering experiments. The samples were analyzed by reverse phase HPLC-ESI-MS/MS using (i) an Eksigent Ultra Plus nano-LC two-dimensional HPLC system (Dublin, CA) connected to a quadrupole time-of-flight TripleTOF 5600 mass spectrometer (AB SCIEX), (ii) an Eksigent nano-LC two-dimensional HPLC system connected to a quadrupole time-of-flight QSTAR Elite mass spectrometer (AB SCIEX), and (iii) a LTQ FT-ICR mass spectrometer equipped with a 7-Tesla superconducting magnet (Thermo Fisher Scientific) connected to a Waters ultra high pressure nano-Aquity liquid chromatography system (Milford, MA). For all details describing mass spectrometric instrument parameters and settings and all chromatographic setups and gradient conditions, please refer to the supplemental Methods S1.
Bioinformatic Database Searches for TripleTOF 5600 and QSTAR Elite
In this study, mass spectral data sets were analyzed and searched with minor differences. For QSTAR Elite data sets, typically peak lists were generated using Analyst Mascot.dll v1.6b27 (AB SCIEX), and the data were searched using a Mascot (26) server version 2.3.02. In addition, mass spectrometric data from the TripleTOF 5600 and QSTAR Elite was also analyzed using the database search engine ProteinPilot (27) (AB SCIEX Beta 4.1.46, revision 460) with the Paragon algorithm (4.0.0.0, 459). All of the extensive details regarding search parameters, fixed and variable modifications, enzyme specificity, databases used, scoring, false discovery rate analysis, etc., are described in supplemental Methods S1 and are compiled in supplemental Table S1G.
Bioinformatic Database Searches for LTQ FT-ICR
MS1 and MS2 files were derived from the raw files using Make MS2 (28) and then searched using an in-house pipeline with the Sequest algorithm (version 2.7). Details for search parameters and decoy database searches can be found in supplemental Methods S1.
Sample Preparations for Standard Concentration Curves (TripleTOF 5600)
Six lysine-acetylated synthetic peptides containing 13C615N2-Lys and 13C615N4-Arg were used to generate standard concentration curves in a complex matrix (complex mitochondrial lysate, 0.3 μg on column) or simple matrix (Michrom bovine 6 protein mix, 25 fmol on column), respectively, spanning from 4.1 attomoles to 25 femtomoles over eight concentration points (0.0041, 0.0123, 0.037, 0.11, 0.33, 1, 3, and 25 fmol) for the following peptides: LVSSVSDLPKacR (hydroxymethylglutaryl-CoA synthase 2 protein), MVQKacSLAR (hydroxymethylglutaryl-CoA synthase 2 protein), AFVDSCLQLHETKacR (long chain specific acyl-CoA dehydrogenase protein), YAPVAKacDLASR (SDHA protein), LFVDKacIR (ATP synthase-coupling factor 6 protein), and AFGGQSLKacFGK (SDHA protein). Three concentration curves, each with injections from lowest to highest spike concentrations, were acquired for each curve in complex and simple matrix, respectively. Additional details are listed in supplemental Table S2.
Sample Preparations for Standard Concentration Curves (LTQ-FT-ICR)
An equal molar protein standard mixture consisting of bovine hemoglobin, BSA, rabbit phosphorylase B, and yeast enolase was prepared, reduced, alkylated, and digested with trypsin as described in detail in supplemental Methods S1.
Methods for Phosphorylation and Acetylation Affinity Enrichment Workflows
To demonstrate the efficiency and functionality with different features of Skyline MS1 filtering a variety of biological projects was investigated, specifically focusing on peptide centric workflows and analyzing post-translationally modified peptides ranging from “simple” to “complex” biological experiments. All of the experimental details for these biological test case experiments are described in supplemental Methods S1, including phosphopeptide enrichment (TiO2 chromatography), sample preparation for pyruvate dehydrogenase E1 α kinase inhibitor study, preparation of conditioned media for breast cancer biomarker workflow, affinity purification of lysine-acetylated peptides, and sample preparation for mouse skeletal muscle mitochondrial protein lysate.
Graphical Methods (Plots and Linear Regressions)
Calibration curve plots and linear regressions were generated with a combination of Python 2.7 (www.python.com) and GNU R 2.12.2 (http://www.r-project.org/). Interoperability was provided with rpy2 2.1.9-1 (http://rpy.sourceforge.net/rpy2.html). The source code is provided. After integrating peaks in Skyline, custom Skyline reports were generated containing fields such as SampleName, PeptideSequence, PrecursorCharge, ProductMz, FragmentIon, and Area. The data were parsed with a custom python program and then passed to R for plot generation and statistics. In some cases, the data set contained outliers or otherwise nonlinear data points. These anomalies could be explained by interference, saturation, or some other phenomenon related to the assay. To work with the data, a robust weighted linear regression was fitted in R using the lrm function from the MASS package (29). The robust regression iteratively discards outliers in an effort to calculate a more robust and proper fit. The regression was weighted proportionally to the inverse of the variance.
Determining LOD and LOQ
The LOD was calculated on the peak area scale. It was calculated from the variance of the blank sample (sample b, with no spiked peptides) and the variance of the lowest spiked in concentration (sample S, 4 amol spiked peptide) (30). Assuming a type I error rate α = 0.05 for deciding that the peptide is present when it is not and a type II error rate β = 0.05 for not detecting the peptide when it is present, LOD was calculated as LOD = meanb + t1 − β * (S.D.b + S.D.S)/2, where the t1 − β term is equal to the (1 − β) percentile of the standard t distribution on n degrees of freedom, where n is equal to the number of replicates. Y values were then transformed and calculated into concentrations using the linear regressions described above. This method relies on LOD being in a region of the regression where the response is still linear. Once the LOD was determined, the LOQ was calculated using the customary relation: LOQ = 3 * LOD (31).
RESULTS
Skyline MS1 Filtering Method
Full scan MS1 filtering in Skyline was developed to extract quantitative information from data-dependent acquisitions of multiple HPLC-MS/MS experiments (Fig. 1A). Native MS instrument output files from multiple vendors can be imported directly into Skyline, which Skyline achieves using the ProteoWizard data access library (25). The MS1 filtering method is fully implemented in Skyline version 1.2 to extract retention times and intensity chromatograms for selected peptide precursor ions. For high resolution MS1 scans, m/z peaks of the precursor isotope distribution (M, M + 1, M + 2, etc.) are filtered into separate chromatograms for greater selectivity and chromatogram peak identity confirmation (Fig. 1B). MS1 filtering settings are based on MS instrument resolution. During chromatogram extraction Skyline sums intensities within a window of 2× full width at half-maximum to either side of the theoretical, predicted mass to charge ratio (Fig. 1C). Raw data from several major MS instrument vendors have been tested, including AB SCIEX, Thermo, Waters, and Agilent. Specific Skyline MS1 filtering settings and raw file import settings are provided in supplemental Fig. S1, A and B. To ensure consistent peak integration areas that account for dynamic mass spectrometric acquisition switching between MS1 and MS/MS scans throughout data acquisition, Skyline dynamically picks a constant time interval to use for intensity values over the entire range measured. The time interval is chosen by first calculating the mode or modes of the intervals between the raw acquired points and then choosing the minimum mode value. After the interval is chosen, all of the intensity values are mapped by linear interpolation to consistently spaced times an interval width apart. Peak detection is performed on the interpolated points, and consistent peak areas are calculated using trapezoidal summation in seconds of intensity (s * i) units (24).
Fig. 1.
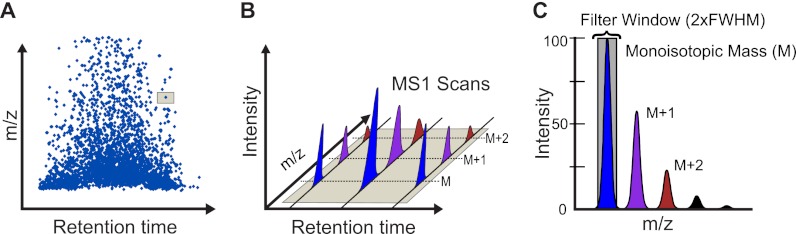
Schematic of MS1 filtering. A, heat map of MS1 signal across the chromatographic gradient for the entire m/z range as acquired on a high resolution mass spectrometer. B, “zoom-in” display of an isotopic envelope for the molecular ion of a typical peptide with peaks at M, M + 1, and M + 2 selected showing changes in MS1 intensity over time. C, high resolution data allow specific filtering of molecular ions and separation of individual peaks within the isotope distribution. Skyline sums intensities within a single resolution to either side of the predicted mass to charge ratio allowing for a filter window of twice the theoretical resolution, predicted full width at half-maximum (2×FWHM). The resolution setting can be selected by the user depending on MS instrument type and preferred isotope m/z acquisition range.
A schematic of the Skyline MS1 filtering workflow is shown in Fig. 2 that emphasizes the new or markedly revised Skyline modules and tools. Mass spectrometric raw data is obtained from multiple HPLC MS/MS experiments to generate both MS1 and MS/MS scans. The MS/MS raw data are initially processed by one of several possible search algorithms, and an associated Skyline spectral library is created. This spectral library is then subjected to analysis by the Skyline BiblioSpec module. This module allows one to populate the Skyline peptide tree, a list of isotope precursor masses and charge states, and set up the Skyline document for MS1 filtering. Raw files are then directly imported into Skyline using Skyline-ProteoWizard and the novel Skyline “Full Scan Filter” module where peptide MS1 ion chromatograms (extracted ion chromatograms) are generated. Automatic peak integration within Skyline is guided by MS/MS-directed peak picking. A set of new MS1 filtering visual tools, such as identification annotations, expected isotope distribution, isotopic rank, and isotopic dot product score (as described below), are then used to aid the assessment of data quality and to make adjustments of peak integration if necessary.
The graphical interface in Skyline for inspecting MS1 filtered data is shown in Fig. 3 with the Skyline peptide tree shown in A. Ion chromatograms of one or more isotopic peaks for a peptide of interest are displayed with annotations that indicate the retention times at which an MS/MS spectrum was acquired and matched to the peptide (Fig. 3, B and C). Peptide search results containing all MS/MS identifications, and their corresponding retention times are obtained from each MS acquisition and imported into Skyline to create a comprehensive spectral library. Spectral libraries can be generated from a variety of peptide search engines, and several data formats are supported (18). Raw MS acquisition files are then imported, and chromatograms are extracted that correlate MS1 features to the correct peptide sequence based on sampled MS/MS spectra available from the library.
For support of proper peak selection, the extracted ion chromatogram contains an identification indicator at the retention time of a sampled MS/MS spectrum for a specific peptide (Fig. 3B). The spectral library also retains each identified MS/MS spectrum acquired across sample replicates and can be viewed upon selecting the identification annotation within the MS1 ion chromatogram window (Fig. 3B) or dropdown menu within the spectrum window (Fig. 3C). In addition, multiple isotopic precursors are extracted from the data to more fully encompass the isotopic envelope (i.e. M, M + 1, M + 2, etc.), providing additional confirmation of proper integration of the selected peak and identification of potential interferences (supplemental Fig. S2). Skyline then ranks the expected relative abundance of the preselected isotope peaks (isotopic rank) and generates an isotopic similarity score (isotopic dot product score) between the experimental and calculated isotope distributions for each peptide (supplemental Figs. S2 and S3). For most peptides, including less abundant isotopes (isotopic rank 2, 3, and lower), the overall integrated ion intensity measurements improves the accuracy of quantitation, especially for larger peptides where higher mass isotopes make significant contributions to the precursor envelope (supplemental Fig. S4). Even though using multiple isotopes for quantitation has been discussed previously by Zhang (32), the multiple precursor features implemented into Skyline MS1 filtering provide beneficial information for the user during data analysis and offer great flexibility for interrogating peak integration. The Skyline multiple precursor ion support provides the user with tools to visually assess multiple precursors and utilizes that information to confirm confident peak picking, to identify potential interferences, and to visually assess extracted ion chromatogram quality (intensity, noise level) of the selected peak with its multiple precursor isoforms. Lastly, individual retention times and isotopic peak area contributions are displayed for each of the four acquisitions obtained from two samples in duplicates with heavy isotope peptides added in at 1 or 3 fmol (Fig. 3, D and E). Contributions from each isotope per replicate are displayed to assess variability and consistency.
To demonstrate these features, MS1 filtering was used to examine experimental replicates of trypsin-digested β-galactosidase (a quality control standard) over 1 month of instrument operation for peak area, peak width, and retention time variations. The resulting tryptic peptides were found to show high reproducibility in both chromatography (retention time) and mass spectrometry (peak area) performance characteristics (supplemental Fig. S5A). In support of this analysis, one can readily create a custom results report that documents selected key parameters and metrics, such as peak area and retention time, for each acquisition in csv format using Skyline export (see supplemental Fig. 5, B and C, and Table S6, A–I).
Standard Concentration and Response Curves
To assess the reproducibility and robustness of Skyline MS1 filtering, standard response curves were generated in three replicates (Fig. 4, A and B, and supplemental Fig. S6). For the TripleTOF 5600, six heavy labeled acetyllysine peptides containing 13C615N2-lysine or 13C615N4-arginine (supplemental Table S2) were spiked into a mitochondrial protein lysate (complex matrix) and a six-protein standard digestion (simple matrix) for eight final concentrations ranging from 4 amol to 25 fmol. These curves demonstrate an excellent linear response with good replicate reproducibility except at the lowest concentrations. For each peptide monitored weighted linear regressions ranged from 0.93 to 1.03 and from 1.00 to 1.13 in complex and simple matrix, respectively (supplemental Fig. S6G). Coefficient of variation (CV) values, calculated at each concentration point across the three replicates, are under 20% CV except at the lowest concentrations (≤37 amol) in the complex matrix (Fig. 3C). LOQs, calculated for each acetyllysine-containing peptide when spiked into the complex mitochondrial matrix, ranged from 52 to 185 amol when only the first precursor isotope (M) was integrated (supplemental Table S3). These same samples were also analyzed on an older QSTAR Elite (QqTOF) yielding similar linear response curves, although the detection limits were not as low (data not shown). To evaluate the reproducibly of Skyline MS1 filtering across a larger number of peptides, we examined a subset of 366 confidently identified peptides from the complex background matrix (a mitochondrial mouse liver extract) in these same samples over 27 replicate injections. Of the 366 peptides, 295 had peak area CVs less than 20%, and only 12 out of the 366 peptides had CVs in excess of 30% (Fig. 4D). Although there was a moderate improvement in peak area CV with increased peak abundance, good reproducibility was achieved even at low peak area (supplemental Fig. S7).
Fig. 4.

Standard concentration curves for stable isotope-labeled and acetyllysine containing peptides. A and B, YAPVAKacDLASR (A, succinate dehydrogenase complex, subunit A) and LVSSVSDLPKacR (B, 3-hydroxy-3-methylglutaryl-CoA synthase 2) spanning a concentration range from 4 attomoles to 25 femtomoles. Peptides were diluted into either a simple matrix (red triangles, 25 fmol “six protein mix”) or a complex matrix (blue circles, mitochondrial lysate from mouse liver, 0.3 μg on column), and acquired on a TripleTOF 5600 mass spectrometer in triplicates. MS1 filtered peak area curves for precursor ions M with m/z 621.83952+ (A, heavy YAPVAKacDLASR) and m/z 626.86042+ (B, heavy LVSSVSDLPKacR). Weighted regression lines and calculated LOQ are indicated (also see supplemental Table S3), and weighted linear regression slopes were determined as 1.03 and 1.03 for YAP and 0.99 and 1.00 for LVS peptides in the different matrices. C, peak area CVs across three replicates for each concentration point (0.037–25 fmol) in complex matrix are shown for six targeted acetyllysine peptides with 20 and 30% CV cutoffs indicated (also see supplemental Figs. S6, C and F). D, high throughput analysis showing peak area CVs for 366 peptides across 27 independently acquired runs in a complex mitochondrial lysate background matrix. Peak area CVs are displayed against MS1 filtered precursor m/z.
To demonstrate that Skyline MS1 filtering is also compatible with data obtained from other instrument types and vendors, a separate standard concentration curve was generated and acquired in triplicate on a high resolution LTQ FT-ICR mass spectrometer (Thermo). Thirteen serial dilutions of four digested standard proteins were generated and spiked into a trypsin-digested Caenorhabditis elegans lysate. As before, the resulting response curves showed good linear responses (the slopes for all weighted linear regressions are shown in supplemental Fig. S8 and supplemental Appendix A). In addition to AB SCIEX and Thermo instruments, Skyline MS1 filtering is capable of importing and analyzing raw data from Waters and Agilent platforms (data not shown).
Quantitation of Phosphorylation Sites in Mitochondrial Pyruvate Dehydrogenase
To test the ability of MS1 filtering to quantify biologically relevant PTMs in a more challenging matrix and workflow, we performed a kinase inhibitor study on mouse liver tissue and assessed changes in the phosphorylation status of pyruvate dehydrogenase E1α subunit (PDHE1α). Mouse liver mitochondria were isolated by gradient purification and treated with the pyruvate analog DCA that leads to specific inhibition of the pyruvate dehydrogenase kinases and loss of phosphorylation on three regulatory sites (Ser(P)-232, Ser(P)-293 and Ser(P)-300) of PDHE1α (33). Following the DCA time course, equal amounts of mitochondria were digested, phosphopeptides were selectively enriched by TiO2 chromatography, and equal amounts of two heavy labeled (13C615N4-Arg) tryptic phosphopeptides encompassing either Ser(P)-293 or Ser(P)-300 were added. Phosphopeptides were then analyzed in triplicate at each time point by LC-MS/MS, allowing us to confirm by MS/MS several endogenous phosphopeptides corresponding to the three known phosphorylation sites on PDHE1α (supplemental Fig. S9). MS1 filtering of the results showed an immediate decrease during the first 10 min of the diphosphopeptide containing both Ser(P)-293 and Ser(P)-300, which subsequently drops below detection by 30 min (Fig. 5A). Interestingly, we were able to monitor an initial increase in the abundance of the co-eluting monophosphopeptides corresponding to either Ser(P)-293 or Ser(P)-300 (Fig. 5B) that we attributed to the rapid dephosphorylation of the corresponding diphosphopeptide. However, as expected, the monophosphopeptide abundance was significantly decreased by 30 min of the DCA treatment. In addition to the above quantitation that was exclusively based on MS1 filtered precursor peak area, we were able to normalize the endogenous phosphorylated peptides to their corresponding heavy peptide analogs and obtained nearly identical results (supplemental Fig. S10). The phosphopeptide containing Ser(P)-232 initially remained unchanged and then decreased after 30 min (Fig. 5C). To demonstrate these results were PDHE1α-specific, an additional 24 phosphopeptides that were expected to be unaffected by inhibitor treatment were monitored during the DCA time course by MS1 filtering with 88% of phosphopeptides showing CVs below 30% (supplemental Fig. S11). Western blots demonstrate no change in total PDHE1α protein levels and phosphosite-specific antibodies results were consistent with those obtained by MS1 filtering (Fig. 5D).
Fig. 5.

Time course of phosphorylation changes at Ser-293, Ser-300, and Ser-232 in PDHE1α following kinase inhibition with DCA. A, relative quantitation over three injection replicates for diphosphopeptide, YHGHpS293MSDPGVpS300YR, with precursor ion M extracted at m/z 876.8155. B, relative quantitation for the corresponding phosphopeptides, YHGHpS293MSDPGVSYR and/or YHGHSMSDPGVpS300YR, with precursor ion M extracted at m/z 836.8323. C, phosphopeptide YGMGTpS232VER with precursor ion M extracted at m/z 540.2150. D, independent Western blot analysis of PDHE1α phosphorylation with phosphosite-specific and total protein antibodies in response to DCA treatment.
Quantitation of Phosphopeptides from Conditioned Media of Breast Cancer Cell Lines
To increase the number of peptide targets to be interrogated, we employed MS1 filtering to quantify relative differences in protein phosphorylation between secretomes of cell lines derived from several subtypes of breast tumors. Cancer cells with metastatic potential secrete proteins into the extracellular microenvironment that modify cell adhesion, intercellular communication, motility, and invasiveness (34). Some of these proteins have a potential to reach the bloodstream and become biomarkers suitable for early noninvasive diagnosis. To identify changes in these secreted proteins, we analyzed phosphoproteins obtained from the conditioned media (CM) of 10 breast cancer cell lines (five luminal and five basal, including four basal B, the latter corresponding to a triple negative tumor type) that reflect the molecular characteristics of primary breast tumor subtypes (35). Proteins secreted by these cancer cells into the CM were concentrated, digested with trypsin, and enriched for phosphopeptides using TiO2 chromatography after sample fractionation by hydrophilic interaction liquid chromatography. The enriched phosphopeptide fractions were then analyzed by LC-MS/MS in two biological and technical replicates (Fig. 6A). Next, MS1 filtering was used to assess differences in the abundance of phosphopeptides derived from proteins secreted by the two tumor subtypes. Vimentin, a known mesenchymal marker specific for basal-type tumors and correlated with adverse pathologic parameters (36), yielded the phosphopeptide ETNLDpS430LPLVDTHSKR440, the abundance of which was found to be significantly increased in three different basal cell lines but undetected in the luminal cell lines (Fig. 6, B and C). Phosphorylation of Ser-430 was previously implicated in the regulation of intermediate filament assembly (37), and because vimentin has been suggested to be a specific marker of a mesenchymal phenotype acquisition by tumor cells (38), measuring relevant phosphopeptides from vimentin may potentially increase sensitivity of this marker. In addition, MS1 filtering results demonstrate increased phosphorylation at Ser-7 in luminal cell line CM for the epithelial cell marker cytokeratin 18 (39, 40) (supplemental Fig. S12, A and E). However, phosphopeptides from tumor protein D54 (41) and calnexin, two other cancer-related proteins, were present at ∼3-fold higher concentrations in basal-type cell lines (supplemental Fig. S12, F and G, and Table S4).
Fig. 6.

Relative quantitation of phosphopeptides from conditioned media from subtype specific breast cancer cell lines. A, workflow for phosphopeptide enrichment from CM, including hydrophilic interaction liquid chromatography separation steps prior to TiO2 phosphopeptide enrichment and subsequent LC-MS/MS analysis. B, Skyline peak area replicate view of phosphopeptide, ETNLDpSLPLVDTHSKR, derived from vimentin and extracted by precursor ion M at m/z 635.642. The peak was monitored for CM from five luminal, one basal A, and four basal B cancer cell lines (two biological and two injection replicates each) with measured peak area means at 126, 33, and 8538, respectively. The asterisks at individual replicates indicate sampling of an MS/MS and identification of the phosphopeptide. C, plot of MS1 filtered peak areas clustered by breast cancer subtype, with blue for luminal, brown for basal A, and green for basal B CM samples (two biological and two acquisition replicates per individual cell line maintain the same symbol).
Identification and Quantitation of Acetylation Sites in Skeletal Muscle Mitochondrial Lysates from SIRT3 Knockout Animals
Mice deficient for the protein deacetylase SIRT3 show hyperacetylation of a wide variety of mitochondrial proteins (42). Recently, this mitochondrial sirtuin has been implicated in regulating the enzymatic activity of a number of mitochondrial proteins including isocitrate dehydrogenase (IDHP), succinate dehydrogenase subunit A (DHSA), and stress 70 protein (Grp75) (43–45), as well as being implicated in various metabolic diseases including diabetes. To evaluate the utility of MS1 filtering to identify changes in PTMs in a complex tissue background, we monitored lysine acetylation changes at multiple sites within a set of proteins that were obtained from isolated mitochondria. Skeletal muscle mitochondria from four wild type (WT) and four SIRT3−/− (KO) mice were enriched by differential centrifugation and gradient purification. We developed a robust workflow for immunoenrichment of acetylated peptides from mitochondria using a combination of two anti-acetyllysine antibodies (Fig. 7A). Equal amounts of acetyllysine peptides were analyzed in duplicate by HPLC-MS/MS. From this data set, we were able to identify 2 acetylation sites in Grp75, 3 in DHSA, and 16 in IDHP (supplemental Table S5). We subsequently generated a spectral library for tryptic acetylated peptides in Skyline and analyzed relative peptide abundance of each identified acetylated peptide by MS1 filtering across all samples. Using this approach, the relative ratio (KO:WT) of each acetyllysine peptide from skeletal muscle was measured across all three proteins (Fig. 7B). Regulation of IDHP by SIRT3 has been previously shown in mice to play a role in oxidative stress-mediated hearing loss during caloric restriction (45) without identifying specific sites of regulation. Compared with WT, we measured a significant increase by more than 2-fold in acetylation at Lys-45, Lys-272, and Lys-299 of IDHP in skeletal muscle SIRT3 KO mice (Fig. 7B). In addition, DHSA and Grp75 each had one site with a significant increase in acetylation at Lys-179 and Lys-288, respectively (Fig. 7B). Notably, for each protein some acetylation sites were observed without dramatic changes between WT and KO, suggesting that not all sites are potential substrates of SIRT3. Overall, these results demonstrate that MS1 filtering is able to quantify changes in lysine acetylation following a complex series of tissue fractionation and enrichment steps. In this case, we showed that the loss of SIRT3 leads to increased acetylation of several mitochondrial proteins and indicate potential sites of enzymatic regulation in vivo.
Fig. 7.

Monitoring the acetylation profile of mitochondrial proteins in SIRT3 KO mice. A, schematic of workflow for acetylpeptide enrichment from skeletal muscle mitochondria from WT and SIRT3 KO animals, including anti-acetyllysine enrichment of Lys-acetylated peptides, LC-MS/MS of two injection replicates, and generation of a spectral library in Skyline for MS1 filtering and relative quantitation. B, ratio (KO:WT) of peak area intensities for individual tryptic acetyllysine peptides at different sites across three proteins from skeletal muscle samples for four WT and four SIRT3 KO mice. # indicates significance: p < 0.05 by two-tailed Student's t test; n = 4 for WT and SIRT3−/− (KO) with two injection replicates per sample.
DISCUSSION
The label-free peptide quantitation experiments described in these studies have taken advantage of both existing and expanded sets of targeted proteomics tools in Skyline that were integrated into a new workflow, MS1 filtering. Skyline MS1 filtering was shown to be capable of supporting large scale extraction of ion chromatograms from MS1 full scan spectra into the established Skyline user interface and quantifying multiple peptides in a variety of proteomic workflows (supplemental Fig. S13). The graphical interface tools allow easy inspection of data with direct links to underlying MS/MS identifications, allowing one to process data accurately and make corrections if necessary. Moreover, experiments are highly scalable; in this study, we have used MS1 filtering to quantify three phosphosites in a targeted analysis of PDHE1α, to several hundred mitochondrial peptides for assessment of reproducibility. Currently, we are using MS1 filtering to examine thousands of peptides as part of a large scale study to assess changes in PTM networks.3 Lastly, this label-free method is robust and supports direct import of raw data files in a platform-independent manner, features that are increasingly needed in the proteomics community given the large number of multicenter clinical studies underway (30). It is also noteworthy that MS1 filtering is built within the Skyline open source environment (18, 20), which has recorded over 7,000 downloads since its release and over 350 registered users. Skyline MS1 filtering was designed such that additional software modules can be added and utilized as needed. As examples, programs developed as part of the National Cancer Institute Clinical Proteomic Technology Assessment for Cancer consortium, such as “Retention Time Viewer” (http://www.gibsonproteomics.org/resources/rt-viewer) and other tools can readily work with data from the editable data report format of Skyline. We fully expect that Skyline MS1 filtering will also undergo further changes with the incorporation of additional tool features, such as chromatographic alignment algorithms to enhance peak picking, once users become familiar with its overall structure and utility.
The quantitation features of MS1 filtering were validated in several experimental designs. Response curves were acquired of stable isotope-labeled acetyllysine peptides spiked into a complex mitochondrial lysate matrix using a TripleTOF 5600 mass spectrometer (AB SCIEX), as well as protein digestion concentration curves spiked into C. elegans lysate using an LTQ FT-ICR mass spectrometer (Thermo). In both cases, linear responses were obtained over several orders of magnitude (i.e. TripleTOF 5600 regression slopes in complex matrix = 0.93–1.03) with LODs and LOQs in the low attomole range and CVs typically below 20%. To examine the robustness of this label-free approach in a more challenging setting where the biological significance would be critically examined, we tested this method in several affinity-based workflows. In the simplest case, phosphorylation site changes in PDHE1α were quantified after DCA inhibitor treatment and phosphopeptide-specific enrichment in a time course experiment and independently validated by Western analysis. In a more rigorous experimental design as part of an ongoing cancer biomarker study, we described phosphosite quantitation across several proteins, e.g. vimentin and keratin type I cytoskeletal 18, in the conditioned media of breast cancer cell lines. Tryptic phosphopeptides were affinity enriched and compared across 10 cell lines, yielding several statistically significant differences. Lastly, the acetylome of mitochondrial protein hydrolysates from SIRT3 KO and WT mice using antibody affinity enrichment of acetyllysine peptides was investigated. Previously, we had attempted to carry out an isobaric labeling approach but found that it compromised the efficiency of immunoprecipitation using two different commercial antibodies. Here, we successfully analyzed these complex mixtures using MS1 filtering by targeting the analysis of a set of acetylation sites in several key mitochondrial enzymes, of which IDHP and DHSA were known to be regulated by SIRT3, whereas Grp75 has not been previously reported to be regulated by SIRT3. Moreover, we not only identified the acetylation sites but also demonstrated for the first time how these sites change quantitatively with respect to loss of SIRT3 activity. Taken together, these experiments show Skyline MS1 filtering is capable of quantifying changes in multiple peptide analytes with increasing levels of sample and workflow complexity. Moreover, we have demonstrated a high degree of linearity and reproducibility in our sampling of MS1 ion intensity data, even under conditions of multiple sample comparisons.
One especially important use of MS1 filtering would be providing the initial large scale and quantitative profiling of multiple peptides for developing MRM-MS assay. Skyline itself was originally developed for this latter purpose, and therefore peptide targets and associated MS/MS spectral libraries acquired in these profiling experiments can be seamlessly transferred for developing MRM-MS verification-type studies. In fact, for both the mouse acetylome and cancer biomarker conditioned medium studies described here, MS1 filtering data provided the rationale for MRM-MS candidate selection to interrogate tissue-specific changes in SIRT3 substrates and identify novel biomarkers for breast cancer plasma analysis, respectively.
Acknowledgments
We thank Drs. Christie Hunter (AB SCIEX), Sean Seymour (AB SCIEX), D. R. Mani (Broad Institute of MIT and Harvard), James Murray (Abcam), and John Cottrell (Matrix Science) for valuable help and advice and Dr. Christopher Benz (Buck Institute) for breast cancer cell lines.
Footnotes
* This work was supported by National Institutes of Health Grants PL1 AG032118 (to B. W. G.), R24 DK085610 (to E. V.), T32AG000266 (to M. J. R.), R01 RR032708 (to M. J. M.), and P41 RR011823 (to M. J. M.) and by National Cancer Institute Grant U24 CA126477 (to B. W. G., subcontract to UCSF), which is part of the National Cancer Institute Clinical Proteomic Technologies for Cancer initiative. This work was also supported in part by NCRR Shared Instrumentation Grant S10 RR024615 (to B. W. G.) and by funds from AB SCIEX for evaluation of the TripleTOF 5600 at the Buck Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
 This article contains supplemental material.
This article contains supplemental material.
2 M. J. Rardin, and B. W. Gibson, unpublished results.
3 M. J. Rardin and B. W. Gibson, unpublished data.
1 The abbreviations used are:
- SILAC
- stable isotope labeling with amino acids in cell culture
- iTRAQ
- isobaric tag for relative and absolute quantitation
- MS/MS
- tandem mass spectrometry
- MS1
- full scan mass spectrum
- PTM
- post-translationally modified
- MRM
- multiple reaction monitoring
- DCA
- dichloroacetic acid
- LOD
- limit of detection
- LOQ
- limit of quantitation
- CV
- Coefficient of variation
- CM
- conditioned media
- SIRT3
- sirtuin 3
- IDHP
- isocitrate dehydrogenase
- DHSA
- succinate dehydrogenase subunit A
- WT
- wild type
- KO
- knock-out.
Author Contributions
B. S., M. J. R., A. M. Z., and B. W. G. designed studies and wrote the manuscript; M. J. R., A. M. Z., B. S., and M. S. B. performed experiments; B. S. and M. J. R. acquired mass spectrometric data on the QSTAR Elite and TripleTOF 5600; B. S., M. J. R., and A. M. Z. analyzed and interpreted the data; B. X. M. and M. J. M. designed and developed the Skyline software and edited the manuscript; B. S. and M. J. R. assisted in feature development for Skyline MS1 filtering; B. E. F. provided Skyline spectral library implementations; C. C. W. assisted in Skyline development; M. P. C. provided statistical analysis and wrote software for custom graphical plots; D. J. S. assisted with data analysis; M. S. B. prepared the LTQ FT-ICR concentration curves and acquired the FT-ICR mass spectrometric data; E. J and C. R. K. provided skeletal muscle samples; and C. R. K. and E. V. assisted in the design of the acetylome study.
REFERENCES
- 1. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 2. Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
- 3. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 4. Krüger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fässler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 5. Zanivan S., Krueger M., Mann M. (2012) In vivo quantitative proteomics: The SILAC mouse. Methods Mol. Biol. 757, 435–450 [DOI] [PubMed] [Google Scholar]
- 6. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 7. Dayon L., Turck N., Kienle S., Schulz-Knappe P., Hochstrasser D. F., Scherl A., Sanchez J. C. (2010) Isobaric tagging-based selection and quantitation of cerebrospinal fluid tryptic peptides with reporter calibration curves. Anal. Chem. 82, 848–858 [DOI] [PubMed] [Google Scholar]
- 8. Pichler P., Köcher T., Holzmann J., Möhring T., Ammerer G., Mechtler K. (2011) Improved precision of iTRAQ and TMT quantification by an axial extraction field in an Orbitrap HCD cell. Anal. Chem. 83, 1469–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 10. Zybailov B., Coleman M. K., Florens L., Washburn M. P. (2005) Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 77, 6218–6224 [DOI] [PubMed] [Google Scholar]
- 11. Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., Haynes P. A. (2011) Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 [DOI] [PubMed] [Google Scholar]
- 12. Mortensen P., Gouw J. W., Olsen J. V., Ong S. E., Rigbolt K. T., Bunkenborg J., Cox J., Foster L. J., Heck A. J., Blagoev B., Andersen J. S., Mann M. (2010) MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J. Proteome Res. 9, 393–403 [DOI] [PubMed] [Google Scholar]
- 13. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 14. Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Monroe M. E., Shaw J. L., Daly D. S., Adkins J. N., Smith R. D. (2008) MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M. Y., Vitek O., Aebersold R., Müller M. (2007) SuperHirn: A novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 17. Addona T. A., Shi X., Keshishian H., Mani D. R., Burgess M., Gillette M. A., Clauser K. R., Shen D., Lewis G. D., Farrell L. A., Fifer M. A., Sabatine M. S., Gerszten R. E., Carr S. A. (2011) A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 29, 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maclean B., Tomazela D. M., Abbatiello S. E., Zhang S., Whiteaker J. R., Paulovich A. G., Carr S. A., Maccoss M. J. (2010) Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal. Chem. 82, 10116–10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stergachis A. B., MacLean B., Lee K., Stamatoyannopoulos J. A., MacCoss M. J. (2011) Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods 8, 1041–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H., Liu Q., Zimmerman L. J., Ham A. J., Slebos R. J., Rahman J., Kikuchi T., Massion P. P., Carbone D. P., Billheimer D., Liebler D. C. (2011) Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol. Cell. Proteomics 10, 10.1074/mcp.M110.006593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danielson S. R., Held J. M., Schilling B., Oo M., Gibson B. W., Andersen J. K. (2009) Preferentially increased nitration of α-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson's disease. Anal. Chem. 81, 7823–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frewen B., MacCoss M. J. (2007) Using BiblioSpec for creating and searching tandem MS peptide libraries. Curr. Protoc. Bioinformatics, 13.7.1–13.7.12 [DOI] [PubMed] [Google Scholar]
- 24. Wang G., Wu W. W., Zeng W., Chou C. L., Shen R. F. (2006) Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: Reproducibility, linearity, and application with complex proteomes. J. Proteome Res. 5, 1214–1223 [DOI] [PubMed] [Google Scholar]
- 25. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pappin D. J., Hojrup P., Bleasby A. J. (1993) Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3, 327–332 [DOI] [PubMed] [Google Scholar]
- 27. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 28. McDonald W. H., Tabb D. L., Sadygov R. G., MacCoss M. J., Venable J., Graumann J., Johnson J. R., Cociorva D., Yates J. R., 3rd (2004) MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom 18, 2162–2168 [DOI] [PubMed] [Google Scholar]
- 29. Venables W. N., Ripley B. D. (2002) Modern Applied Statistics with S, 4th Ed., Springer, New York [Google Scholar]
- 30. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marazzi A. (1993) Algorithms, Routines and S Functions for Robust Statistics, CRC Press, Boca Raton, FL [Google Scholar]
- 32. Zhang Z. (2009) Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal. Chem. 81, 8354–8364 [DOI] [PubMed] [Google Scholar]
- 33. Rardin M. J., Wiley S. E., Naviaux R. K., Murphy A. N., Dixon J. E. (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 389, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caccia D., Zanetti Domingues L., Miccichè F., De Bortoli M., Carniti C., Mondellini P., Bongarzone I. (2011) Secretome compartment is a valuable source of biomarkers for cancer-relevant pathways. J. Proteome Res. 10, 4196–4207 [DOI] [PubMed] [Google Scholar]
- 35. Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J. P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., Kuo W. L., Stilwell J. L., Pinkel D., Albertson D. G., Waldman F. M., McCormick F., Dickson R. B., Johnson M. D., Lippman M., Ethier S., Gazdar A., Gray J. W. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen M. H., Yip G. W., Tse G. M., Moriya T., Lui P. C., Zin M. L., Bay B. H., Tan P. H. (2008) Expression of basal keratins and vimentin in breast cancers of young women correlates with adverse pathologic parameters. Mod. Pathol. 21, 1183–1191 [DOI] [PubMed] [Google Scholar]
- 37. Eriksson J. E., He T., Trejo-Skalli A. V., Härmälä-Braskén A. S., Hellman J., Chou Y. H., Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 38. Sarrió D., Rodriguez-Pinilla S. M., Hardisson D., Cano A., Moreno-Bueno G., Palacios J. (2008) Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 68, 989–997 [DOI] [PubMed] [Google Scholar]
- 39. Weigelt B., Peterse J. L., van 't Veer L. J. (2005) Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 5, 591–602 [DOI] [PubMed] [Google Scholar]
- 40. Woelfle U., Sauter G., Santjer S., Brakenhoff R., Pantel K. (2004) Down-regulated expression of cytokeratin 18 promotes progression of human breast cancer. Clin. Cancer Res. 10, 2670–2674 [DOI] [PubMed] [Google Scholar]
- 41. Boutros R., Fanayan S., Shehata M., Byrne J. A. (2004) The tumor protein D52 family: Many pieces, many puzzles. Biochem. Biophys. Res. Commun. 325, 1115–1121 [DOI] [PubMed] [Google Scholar]
- 42. Lombard D. B., Alt F. W., Cheng H. L., Bunkenborg J., Streeper R. S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M. D., Bronson R. T., Haigis M., Guarente L. P., Farese R. V., Jr., Weissman S., Verdin E., Schwer B. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 27, 8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cimen H., Han M. J., Yang Y., Tong Q., Koc H., Koc E. C. (2010) Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schwer B., Eckersdorff M., Li Y., Silva J. C., Fermin D., Kurtev M. V., Giallourakis C., Comb M. J., Alt F. W., Lombard D. B. (2009) Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., Prolla T. A. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]