

Abstract
Acute desensitization is a common property of Gi/o-coupled receptors. Recent data, however, suggest that, unlike μ-opioid receptors (MORs) located somatodendritically in neurons or expressed in heterologous systems, MORs in the presynaptic compartment of neurons are resistant to acute desensitization. It is not yet clear whether this differential desensitization is a shared property of many Gi/o-coupled receptors nor whether receptors located presynaptically and postsynaptically in a single cell type display differential desensitization. Here, whole-cell recordings were made from proopiomelanocortin (POMC) neurons in mouse brain slices. Agonists for μ-opioid, nociceptin, and GABAB receptors induced postsynaptic currents that desensitized within minutes, whereas inhibition of presynaptic transmitter release mediated by these receptors was maintained throughout agonist exposure. Expression of channelrhodopsin2 in POMC neurons allowed for light-evoked transmitter release from POMC neuron terminals, which was detected by recording postsynaptic currents in downstream neurons. Light-evoked currents were inhibited throughout the application of all agonists tested. Thus, the same receptors that desensitize when expressed in the postsynaptic compartment of POMC neurons resist desensitization when located in the presynaptic compartment. Pharmacologic knockdown of MORs revealed that depletion of receptor reserve does not account for presynaptic resistance to desensitization. In ∼25% of recordings with GABAB agonist application, presynaptic GABAB receptors desensitized, suggesting that resistance to desensitization is not due to an intrinsic property of the terminals themselves. Together, the results indicate that a variety of presynaptic receptors can continue to function after their postsynaptic counterparts desensitize and suggest that a compartment-specific modification may confer resistance to desensitization.
Introduction
Among inhibitory G-protein-coupled receptors (GPCRs), μ-opioid receptor (MORs) have been the subject of particularly intense study due to the role that these receptors play in analgesia and drug abuse. Consistent with the regulation of many GPCRs, continued activation of MORs can cause a reduction in cellular responses as agonist binding leads to desensitization of the receptor (Reiter and Lefkowitz, 2006). This desensitization of the receptor is thought to be an important early step in the development of tolerance to repeated or continued exposure to MOR agonists (Martini and Whistler, 2007; Christie, 2008). Therefore, much work has focused on determining the mechanisms underlying MOR receptor desensitization since limiting this desensitization could help preserve analgesic actions of MOR agonists during prolonged treatment.
To date, there is not a clear consensus on the mechanism of MOR desensitization. Like other GPCRs, agonist binding to MORs causes a conformational change in the receptor, activation of intracellular signaling cascades, and receptor phosphorylation with eventual β-arrestin binding and internalization, although internalization is not required for acute desensitization (Arttamangkul et al., 2006). In neurons, acute desensitization has often been studied by examining the ability of MOR agonists to activate postsynaptic receptors coupled to G-protein-coupled inwardly rectifying potassium (GIRK) channels. In many cell types, MOR agonists induce GIRK-mediated outward currents that decline within minutes of agonist exposure, representing desensitization of the receptor (Williams et al., 2001). MORs located on the presynaptic terminals of neurons can inhibit transmitter release through actions at voltage-gated potassium and calcium channels (Williams et al., 2001). Interestingly, recent studies in neurons maintained in intact brain slices indicate that presynaptic MORs can continue to inhibit transmitter release during an agonist exposure that causes desensitization of postsynaptic MORs (Blanchet and Lüscher, 2002; Fyfe et al., 2010; Pennock and Hentges, 2011). The resistance to desensitization of presynaptic MORs has not been thoroughly studied.
The present study examines the nature of the presynaptic resistance to desensitization in a defined population of neurons in the arcuate nucleus of the hypothalamus. The proopiomelanocortin (POMC) neurons in this region are regulated both presynaptically and postsynaptically by MORs as well as Gi/o-coupled κ-opioid, nociceptin, and GABAB receptors, allowing for a comparative study of several receptor types. Previous work indicates that the postsynaptic MORs on POMC neurons desensitize during acute exposure to the MOR agonist [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin (DAMGO), whereas the MORs on terminals presynaptic to these neurons do not (Pennock and Hentges, 2011). The present results show that similar to MORs, κ, nociceptin, and GABAB receptors all display distinct resistance to desensitization in the presynaptic but not postsynaptic compartment, suggesting that a common mechanism confers differential desensitization of many GPCRs. Current data also illustrate that presynaptic receptors can resist desensitization to various agonists and that resistance is independent of receptor reserve. The ability of presynaptic receptors to continue to signal once postsynaptic receptors have desensitized implies a functional switch from direct postsynaptic actions to presynaptic actions occurs during agonist exposure.
Materials and Methods
Animals.
Transgenic mice expressing discosoma red (DsRed) (Hentges et al., 2009) or Cre recombinase (Xu et al., 2005) driven by the POMC promoter were backcrossed onto the C57BL/6 background for >11 generations. Mice were housed at controlled temperatures (22–24°C) with a constant 12 h light/dark cycle. Standard rodent chow and tap water were provided ad libitum. Brain slices were prepared from both male and female mice (unless otherwise indicated) between 6 and 14 weeks of age. Standard PCR genotyping was performed to identify transgenic mice. All animal procedures were approved by the Colorado State University Institutional Animal Care and Use Committee and met the United States Public Health Services guidelines.
Brain slice preparations.
Mice were deeply anesthetized with isoflurane, and brains were rapidly removed and placed into ice-cold artificial CSF (aCSF) solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 glucose. All aCSF solutions were saturated with a 95% O2/5% CO2 mixture. Sagittal slices (240 μm) were prepared using a VT 1200S vibratome (Leica). Brain slices containing the arcuate nucleus were then transferred into warm (37°C) aCSF containing MK-801 (15 μm; Sigma-Aldrich). Slices were allowed to rest for ≥45 min before transfer to the recording chamber.
Viral gene transfer.
To express channelrhodopsin-2 (ChR2) selectively in POMC neurons, a viral vector containing a floxed sequence for ChR2 with an mCherry tag [AAV2/9.EF1.dflox.hChR2(H134R)-mCherry. WPRE.hGH; obtained from the Penn Vector Core (University of Pennsylvania School of Medicine, Philadelphia, PA); 200 nl] was injected unilaterally into the arcuate nucleus of the hypothalamus of POMC-Cre transgenic mice (8 weeks of age). Brain slices were prepared 14–28 d after injection, which was a sufficient length of time to yield high levels of ChR2 expression.
Electrophysiology.
Brain slices placed in the recording chamber were continuously perfused with warm (37°C; ∼1.5 ml/min) aCSF saturated with 95% O2/5% CO2. Recording pipettes had tip resistances between 1.5 and 2.5 MΩ when filled with an internal solution containing the following (in mm): 57.5 K-methyl sulfate, 57.5 KCl, 20 NaCl, 1.5 MgCl2, 5 HEPES (K+ salt), 0.1 EGTA, 2 ATP, 0.5 GTP, 10 phosphocreatine, pH 7.3. POMC neurons were identified in the slice by the presence of DsRed or ChR2-mCherry. After obtaining a seal of >1 GΩ, negative pressure was applied to rupture the cell and enter whole-cell mode. Cells were held at −60 mV, and no series resistance compensation was applied. To electrically evoke transmitter release, a bipolar stimulating electrode was placed in the middorsal arcuate nucleus and pairs (100 ms delay between pulses) of 0.5 ms stimuli (10–800 μA as needed to reliably evoke IPSCs at ∼50% maximum amplitude) were applied every 20 s. Miniature IPSCs were collected at 10 kHz and digitally filtered at 1 kHz. Events were collected for 15 s/sweep with a 15 s delay between each sweep and detected using Axograph X software based on rise time kinetics. Events with a rise time of <100 μs were rejected. All mIPSC recordings were made in the presence of tetrodotoxin (300 nm) and 6,7-dinitroquinoxaline-2,3-dione (DNQX) (10 μm) and KCl and K-methylsulfate in the internal solution were replaced with CsCl and Cs-methanesulfonate.
Light-evoked transmitter release from POMC-ChR2-expressing neurons was accomplished by applying a brief (25 ms) blue light pulse to the slice every 20 s. Recordings were made in unidentified arcuate neurons within 100 μm of mCherry-expressing cells. The intensity of the light used to evoke transmitter release was between 5 and 11 mW/mm2, and was adjusted for each recording to achieve a consistent evoked PSC. All recordings were made using an Axoclamp 700B or 200B (Molecular Devices) amplifier. AxographX software (Axograph) was used for data collection. Recordings in which the series resistance exceeded 20 MΩ or changed significantly during the course of the experiment were not accepted for analysis. The AMPA receptor antagonist DNQX (10 μm) was constantly perfused onto the slice during the course of the experiment to ensure that all evoked PSCs recorded and analyzed were inhibitory (eIPSCs).
Chronic morphine treatment.
To treat mice chronically with morphine, POMC-DsRed transgenic mice (8- to 10-week-old males) were anesthetized and miniosmotic pumps (Alzet; DURECT Corporation) containing morphine were placed subcutaneously. Morphine-treated mice received 50 mg · kg−1 · d−1 of morphine for 5–7 d before being killed. Brain slices prepared from morphine-treated mice were prepared and collected in either morphine-free aCSF or aCSF containing morphine (1 μm). Slices collected into morphine-free aCSF were left for 2 h before being transferred to the recording chamber to allow morphine to wash from the slice. Slices collected into aCSF containing morphine were given at least 45 min to rest before being transferred to the recording chamber where they were constantly perfused with aCSF containing morphine. To precipitate withdrawal, the perfusion solution was changed from aCFS with morphine (1 μm) to aCSF containing naloxone (1 μm).
Drugs.
Stock solutions of DNQX (Sigma-Aldrich), (+)-MK-801 (Sigma-Aldrich), (+)-(5α,7α,8β)-N-methyl-N[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593) (Biomol International), and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845) (Tocris Bioscience) were prepared in DMSO as stock solutions (at 10,000× final concentration). [Met5]-enkephalin (Sigma-Aldrich), baclofen (Sigma-Aldrich), nociceptin (Sigma-Aldrich), the cyclized peptide d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) (Tocris Bioscience), nor-binaltorphimine (nor-BNI) (Sigma-Aldrich), morphine (Sigma-Aldrich), tetrodotoxin (TTX) (Tocris Bioscience), and β-chlornaltrexamine (β-CNA) (Sigma-Aldrich) were prepared as stock solutions in distilled water. All drugs were diluted in aCSF to achieve the working concentrations. β-CNA was divided into aliquots, and then stored at −80°C. For each experiment performed using β-CNA, an individual aliquot was thawed, diluted, and perfused onto the slice within 5 min to ensure no degradation of the compound before use. Dose–response curves were made for opioid and GABAB receptor agonists to determine the maximal and EC50 concentrations as used in the present studies.
Estimating presynaptic MOR receptor reserve.
Dose–response curves representing the inhibition of eIPSC amplitude by MORs presynaptic to POMC neurons before and after treatment with an irreversible antagonist (β-CNA) were fitted using GraphPad Prism software. Values for the predicted concentration of ME necessary to reduce eIPSC amplitude by given values between 20 and 41% were obtained from these curves. Concentrations that were predicted to achieve an equivalent inhibition of eIPSC amplitude before ([ME]control) and after ([ME]β-CNA) β-CNA exposure were plotted against one another as a double reciprocal plot. The plot was fitted according to Furchgott's method (Furchgott, 1966) using the following equation:
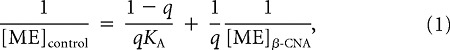 |
where q is equal to the fraction of MORs still functional after β-CNA treatment and KA is the dissociation constant of ME at MORs presynaptic to POMC neurons. Estimated values for q and KA were obtained using GraphPad Prism.
Statistics.
Comparisons between two groups were made using Student's t tests or paired t tests when a repeated measure was used. Datasets containing more than two groups were analyzed using repeated-measures ANOVA with Tukey's multiple-comparison post hoc tests or two-way ANOVA as indicated. Single-phase decay curves were fitted to postsynaptic desensitization data, and values for time constants and plateaus were obtained using GraphPad Prism software. Postsynaptic currents induced by a GPCR agonist were only included in analyses if the amplitude was >10 pA. All data are shown as the mean ± SEM, and differences between groups were considered significant if p < 0.05.
Results
Presynaptic, but not postsynaptic, MORs resist acute desensitization
To examine the desensitization of presynaptic and postsynaptic MORs during exposure to ME, whole-cell voltage-clamp recordings were made from fluorescently labeled POMC neurons. Presynaptic and postsynaptic effects of ME were measured simultaneously. The MOR selective opioid agonist ME (30 μm) induced a postsynaptic outward current (42 ± 14 pA; n = 6; Fig. 1A) that desensitized significantly within minutes of continued exposure (p < 0.001, one-way repeated-measures ANOVA; τ = 116 s; plateau, 46% of baseline; Fig. 1B). ME also caused a robust inhibition of the amplitude of eIPSCs (to 24 ± 3.5% of baseline; n = 14; p < 0.0001; Fig. 1C,D). Inhibition of eIPSC amplitude was maintained throughout the superfusion of ME (30 μm; p = 0.27, one-way repeated-measures ANOVA; minutes 2 through 10; Fig. 1C,D). Together, these results indicate that postsynaptic, but not presynaptic, MORs regulating POMC neurons acutely desensitize when exposed to a maximal concentration of ME similar to previous results with the agonist DAMGO (Pennock and Hentges, 2011).
Figure 1.

Postsynaptic, but not presynaptic, MORs desensitize acutely. A, Representative trace of an outward current induced by ME (30 μm), and the reduction in the amplitude of the current that occurs with prolonged ME exposure. Fast upward and downward deflections that occur at 20 s intervals represent stimulus artifacts that result from measuring presynaptic (eIPSC amplitude) and postsynaptic effects of MORs in a single recording. B, Compiled data from six recordings in which the amplitude of the outward current was measured at every minute for 4 min after the peak while under constant exposure to ME (30 μm). The curve fitted to the plot represents a single-phase exponential decay. The asterisks (*) represent points found to be significantly different from minute 0 using Tukey's multiple-comparison test. C, A representative plot of eIPSC amplitudes representing the effect of prolonged exposure to ME (30 μm) on eIPSC amplitude. Each point represents the average of three consecutive sweeps. Sample traces shown above the plot represent the average of three consecutive sweeps taken every third minute starting at the onset of agonist exposure. D, Compiled data representing the inhibition of eIPSC amplitude over the course of a 10 min exposure to ME (30 μm; n = 14). Error bars indicate SEM.
Presynaptic resistance to desensitization is a property of multiple Gi/o-coupled receptors
To determine whether differential presynaptic and postsynaptic desensitization may be a general property of Gi/o-coupled receptors, or rather a specific property of MORs, the ability of GABAB, nociceptin, and κ-opioid receptors (KORs) to undergo or resist desensitization was examined. Superfusion of a maximal concentration of nociceptin (500 nm) induced a postsynaptic outward current (38 ± 4.8 pA peak) that declined within minutes to a plateau of 40% of baseline (p < 0.0001, one-way repeated-measures ANOVA; τ = 210 s; n = 6; Fig. 2A). A maximal concentration of nociceptin (500 nm) also caused an inhibition of eIPSC amplitude (to 30 ± 7.7% of baseline; n = 7; Fig. 2B), but the inhibition of eIPSCs did not decline during the 10 min exposure to agonist (p = 0.47, one-way repeated-measures ANOVA; minutes 2 through 10; Fig. 2B,F). Similarly, the GABABR agonist baclofen (30 μm, maximal concentration) induced an outward current (36 ± 6.0 pA; n = 6; Fig. 2C) that declined during exposure (plateau, 53% of baseline; p < 0.0001, one-way repeated-measure ANOVA; τ = 84 s; Fig. 2C). Baclofen also caused an inhibition of eIPSC amplitude (to 36 ± 3.4% of baseline; n = 19; Fig. 2D) that was maintained throughout the exposure (p = 0.053, one-way repeated-measures ANOVA; minutes 2 through 10; Fig. 2D,F).
Figure 2.

Multiple Gi/o-coupled receptors presynaptic to POMC neurons resist acute desensitization. A, Compiled data representing the amplitude of the postsynaptic current induced by nociceptin at every minute, beginning at the peak, for 5 min during constant exposure to the agonist (n = 6). The inset shows a sample trace of a postsynaptic current induced by nociceptin. A similar plot and inset is shown in C for recordings made in the presence of baclofen (n = 6). The asterisks represent points found to be significantly different from minute 0 using Tukey's multiple-comparison test. B, A representative plot showing the effect of prolonged exposure to a maximal concentration of nociceptin (500 nm) on eIPSC amplitude. Each point represents the average of three consecutive sweeps. Similar plots are shown for recordings made in the presence of baclofen (D) and U69593 (E). F, Compiled data showing the inhibition of eIPSC amplitudes by U69593 (dashed line; n = 5), baclofen (gray line; n = 19), and nociceptin (black line; n = 7) during continued exposure to each agonist. Error bars indicate SEM.
Activation of the KOR inhibits neurotransmitter release onto POMC neurons but has no apparent postsynaptic effects (Pennock and Hentges, 2011). Superfusion of a maximal concentration of the KOR agonist U69593 (500 nm) resulted in inhibition of the amplitude of eIPSCs (to 66 ± 9.3% of baseline; n = 5; p < 0.0001; Fig. 2E,F). Similar to the MOR, GABABR, and nociceptin receptors, the KOR-mediated inhibition of eIPSC amplitude showed no desensitization during >10 min exposure to U69593 (p = 0.071, one-way repeated-measures ANOVA; minutes 3 through 13; n = 5; Fig. 2F). Nor-BNI (100 nm) was added upon the termination of U69593 superfusion to enhance wash/reversal (Fig. 2E). Together, these data show that resistance to acute desensitization may be a common characteristic of Gi/o-coupled receptors located on presynaptic terminals.
Differential desensitization of presynaptic and postsynaptic Gi/o-coupled receptors in POMC neurons
It is plausible that the cell type on which a receptor is located, not the subcellular compartment in which it is found, determines whether or not that receptor will be resistant to desensitization. To determine whether receptors located in presynaptic and postsynaptic compartments of a single cell type show differential acute desensitization, experiments were performed in which desensitization of presynaptic receptors within POMC neurons was studied using an optogenetic approach. A viral vector containing a floxed ChR2 sequence was injected into the arcuate nucleus of mice expressing Cre recombinase under the control of the POMC promoter. In slices prepared from these mice, brief pulses of blue light depolarized the POMC neurons resulting in the release of neurotransmitter onto unidentified neighboring neurons from which recordings were made. Superfusion of ME (30 μm) resulted in a robust inhibition of light-evoked IPSC amplitude (to 36 ± 10% of baseline; n = 6; p < 0.0001; Fig. 3A), which was reversed by the MOR antagonist CTAP (500 nm). Inhibition of the light-evoked IPSCs was maintained during the exposure to ME (p = 0.14, one-way repeated-measures ANOVA; minutes 3 through 10; n = 6; Fig. 3B), indicating that, unlike postsynaptic MORs in POMC neurons, MORs directly regulating transmitter release from POMC neurons do not undergo acute desensitization.
Figure 3.

Gi/o-coupled receptors on the axon terminals of POMC neurons resist desensitization. A, A plot of light-evoked IPSCs recorded from a neuron within the arcuate nucleus innervated by a POMC neuron(s) expressing ChR2. Each point represents the average of three consecutive sweeps. Sample traces of light-evoked currents from minutes 0, 5, and 10 of ME superfusion are shown in the inset. Each trace is the average of three consecutive sweeps. Similar plots for recordings made in the presence of baclofen and U69593 are shown in C and E. Compiled data from recordings representing the average inhibition of light-evoked IPSC amplitude by ME (n = 6), baclofen (n = 6), and U69593 (n = 4) are shown in B, D, and F, respectively. Error bars indicate SEM.
Baclofen (30 μm) also inhibited presynaptic release from POMC terminals as indicated by the reduction in light-evoked IPSC amplitude (to 27 ± 7.3% of baseline; n = 6; p < 0.0001; Fig. 3C). This inhibition was maintained during continued agonist exposure (p = 0.051, one-way repeated-measures ANOVA; minutes 2 through 10; n = 6; Fig. 3D). Thus, both μ-opioid and GABAB receptors undergo desensitization in the postsynaptic (Figs. 1, 2), but not presynaptic compartments (Fig. 3A–D).
Although KOR activation does not induce a GIRK-mediated current in POMC neurons (Pennock and Hentges, 2011), the KOR agonist U69593 (500 nm) significantly inhibited the amplitude of light-evoked IPSCs (to 45 ± 14% of baseline; n = 4; p < 0.0001; Fig. 3E). The inhibition of transmitter release was maintained during 14 min of continuous U69593 perfusion (p = 0.94, one-way repeated-measures ANOVA; minutes 4 through 14; n = 4; Fig. 3F). The inhibition of light-evoked IPSC amplitude was completely reversed by the KOR-selective antagonist nor-NBI (100 nm; Fig. 3E). Together, the data from the light-evoked release studies demonstrate that the ability of some receptors to resist desensitization is likely dependent on the location of the receptor within the neuron.
Decreasing receptor number does not induce presynaptic desensitization
To determine whether receptor reserve may account for the lack of desensitization observed for MORs, the irreversible MOR antagonist β-CNA was used to reduce receptor reserve. β-CNA (50 nm) was superfused over the slice for 2 min following the washout of a brief application of ME (30 μm; Fig. 4A,B). The pulse of ME before β-CNA application was used to determine the maximal inhibition possible for ME before receptor reserve was reduced. After the superfusion of β-CNA and a washout period (>10 min), ME (30 μm) was again applied. β-CNA reduced the inhibition of eIPSC amplitude caused by ME to 60 ± 11% of its original value (83 ± 3.4% inhibition of eIPSC amplitude before β-CNA vs 49 ± 8.0% inhibition of eIPSC amplitude after β-CNA; n = 3; p = 0.039, paired t test; Fig. 4A,B). Although β-CNA treatment reduced the inhibition of eIPSC amplitude by ME, there was still no decrease in ME-mediated inhibition (desensitization) over the course of the drug application (p = 0.065; n = 3; one-way repeated-measures ANOVA; minutes 2 through 11; Fig. 4C).
Figure 4.

Decreasing the number of functional receptors presynaptic to POMC neurons does not induce acute desensitization. A, Sample traces indicating the inhibition of eIPSC amplitude in response to ME (30 μm) before (top traces) and after receptor knockdown with β-CNA (bottom traces). The sample traces in A represent the average of three consecutive sweeps taken under the conditions indicated. B, A plot of eIPSC amplitudes showing the effect of β-CNA on the inhibition of eIPSC amplitude by ME. Each point represents the average of three consecutive sweeps. C, Compiled data representing the effect of prolonged exposure to ME (30 μm) on eIPSC amplitude after treatment with β-CNA (solid line) (n = 3). D, Dose–response curves for ME-induced inhibition of eIPSC amplitude before (squares) and after (circles) treatment with β-CNA (50 nm; 2 min). The sample size for each point of the curve is shown in parentheses. E, A reciprocal plot of agonist concentrations that produced an equivalent inhibition of eIPSC amplitude before (vertical axis) and after (horizontal axis) β-CNA treatment was obtained using Furchgott's method (Eq. 1). Estimates of the fraction of receptors remaining after β-CNA treatment and the dissociation constant (in micromolar concentration) of ME at MORs presynaptic to POMC neurons are represented by q and KA, respectively. The dashed lines represent the 95% confidence limits of the regression line. Error bars indicate SEM.
To estimate the size of the MOR receptor reserve presynaptic to POMC neurons, dose–response curves were constructed before and after β-CNA treatment and analyzed using Furchgott's method (Furchgott, 1966) (Fig. 4D,E). ME (30 μm) caused a much larger inhibition of eIPSC amplitude in control conditions compared with the inhibition after a 2 min exposure to β-CNA (79 ± 2.1% inhibition of eIPSC amplitude before β-CNA vs 42 ± 2.4% inhibition of eIPSC amplitude after β-CNA; p < 0.0001; n = 36, 12; Fig. 4D). Additionally, there was an approximately threefold shift in the EC50 for ME after β-CNA (444 nm before β-CNA vs 1.336 μm after β-CNA). To estimate the fraction of the total number of presynaptic receptors inhibited by β-CNA, a double reciprocal plot of the agonist concentration needed to achieve a given inhibition of eIPSC amplitude before and after β-CNA treatment was constructed according to Furchgott's method (Eq. 1). This analysis revealed that the 44% decrease in the maximal inhibition of eIPSC amplitude by ME corresponded to a 59–68% reduction in the total number of presynaptic MORs (95% confidence interval for q [0.3189, 0.4137]; Fig. 4E) and provided an estimated KA value near that of the EC50 (95% confidence interval [0.693 μm, 1.315 μm]; Fig. 4E) under control conditions. These data suggest that presynaptic resistance to desensitization is not due to receptor reserve since removing enough receptors to potently blunt the maximal response did not cause presynaptic receptors to display desensitization. Furthermore, the finding that removing ∼50% of the surface receptors reduces the functional response by ∼50% indicates that terminals regulating POMC neurons do not have a significant receptor reserve even under baseline conditions.
Morphine does not cause desensitization of MORs in the presynaptic compartment
MORs activated by morphine may desensitize through a mechanism distinct from those activated by ME or DAMGO in some systems (Johnson et al., 2006; Kelly et al., 2008). Thus, it is possible that the environment of the presynaptic terminal may confer resistance to desensitization to a ME-activated MOR but not a morphine-activated receptor. To determine whether resistance to desensitization by presynaptic MORs may be agonist specific, eIPSC amplitude was measured during a 15 min superfusion of morphine (20 μm). Morphine caused a reduction in the amplitude of eIPSCs (to 52 ± 12% of baseline; n = 4; p < 0.0001) that was maintained throughout exposure to morphine (p = 0.91; n = 4; one-way repeated-measures ANOVA; minutes 3 through 13; Fig. 5A,B), indicating that presynaptic receptors do not undergo acute desensitization whether bound by full or partial agonists that likely confer different conformational states of the receptor.
Figure 5.

Presynaptic MORs resist desensitization when activated with morphine. A, Plot of eIPSC amplitudes during acute morphine exposure (20 μm). CTAP (500 nm) was added upon the cessation of morphine superfusion to speed wash time. Each point represents three consecutive sweeps. B, Compiled data representing the inhibition of eIPSC amplitude over 11 min of morphine (20 μm) superfusion (n = 4). Error bars indicate SEM.
Chronic morphine treatment (CMT) can reduce the efficacy of MOR agonists at presynaptic terminals (North and Vitek, 1980; Fyfe et al., 2010) and enhance the extent and rate of acute desensitization of postsynaptic MORs (Dang and Williams, 2005; Ingram et al., 2008). To determine whether CMT can modify presynaptic MOR signaling to enhance acute desensitization of these receptors in terminals presynaptic to POMC neurons, mice were treated with morphine (50 mg · kg−1 · d−1) or saline for 5–7 d using subcutaneous miniosmotic pumps before slice preparation. When slices were collected into morphine (1 μm) and maintained in morphine, application of naloxone (1 μm) caused a greater increase in the eIPSC amplitude (1.8 ± 0.13-fold increase in control; 4.4 ± 1.0-fold increase in CMT; n = 4–6; p = 0.01; Fig. 6A,C) and decrease in the paired-pulse ratio in cells from CMT mice compared with saline-treated mice (0.68 ± 0.04 in control; 0.51 ± 0.05 in CMT; n = 4–6; p = 0.01; Fig. 6A,B), indicating that morphine was effectively reaching the synapses and inhibiting transmitter release. Application of ME (30 μm) to slices from CMT mice that were collected and maintained in morphine-free aCSF resulted in a robust decrease in eIPSC amplitude (to 32 ± 3.2% of baseline; n = 20; p < 0.0001; Fig. 6D,E) that was maintained over the course of an 11 min exposure (p = 0.32; n = 5; one-way repeated-measures ANOVA; minutes 2 through 11) similar to that observed for tissue from untreated mice. Thus, presynaptic MORs resist desensitization after both acute and chronic exposure to morphine.
Figure 6.

Chronic morphine treatment does not enhance desensitization of presynaptic MORs. A, Sample traces from recordings made in control and CMT (50 mg · kg−1 · d−1) mice that were prepared and maintained in 1 μm morphine until application of 1 μm naloxone (NLX) to induce precipitated withdrawal. IPSCs were evoked using paired stimuli 100 ms apart. B, NLX precipitated withdrawal decreased the paired-pulse ratio (P2/P1) to a greater extent in CMT mice than in controls. C, NLX also caused a larger increase in the amplitude of the IPSC evoked by the first of the paired stimuli (P1) in recordings made in slices prepared from CMT animals than in those prepared from controls. D, Plot of eIPSCs from a recording in which a slice prepared from an animal that had received CMT was exposed to ME (30 μm) for a prolonged period. Each point represents the average of three consecutive sweeps. E, Compiled data representing the effect of prolonged ME exposure on recordings made from slices prepared from mice that had received CMT (n = 5). Error bars indicate SEM.
A fraction of presynaptic GABABRs acutely desensitize
It is plausible that a property of the presynaptic environment could somehow prevent receptor desensitization. However, in 7 of 26 recordings made from POMC neurons in which baclofen (30 μm) was applied while recording eIPSC amplitude, the inhibition of eIPSC amplitude declined during baclofen application (p < 0.0001, one-way repeated-measures ANOVA; minutes 2 through 10; n = 7; Fig. 7A,B). Inhibition peaked by the second minute of agonist exposure, on average, and desensitized by ∼50% by minute 10 (25 ± 6.9% of baseline at minute 2 vs 60 ± 7.4% of baseline at minute 10). In some recordings in which GABAB receptor desensitization was observed, it was possible to subsequently perfuse a maximal concentration of ME for >10 min. In these recordings, only the GABAB-mediated inhibition of eIPSC amplitude desensitized, whereas ME-mediated inhibition was maintained (data not shown). Additionally, GABAB-mediated inhibition of miniature IPSC (mIPSC) frequency was examined to determine whether ∼25% of all inputs onto POMC neurons contain GABAB receptors that are able to undergo desensitization, or whether only ∼25% of POMC neurons receive inputs with GABAB receptors that are able to undergo desensitization. Similar to what was observed when using eIPSCs to measure GABAB-mediated inhibition of neurotransmitter release, desensitization of GABAB-mediated inhibition of mIPSC frequency was only observed in 25% of recordings made (3 of 12 recordings; Fig. 7C,D). This suggests that 25% of POMC neurons receive input from terminals on which GABAB receptors are able to undergo desensitization. If resistance or susceptibility of GPCRs to desensitization is due to a property of the terminals on which they are found, desensitization should have been observed in a similar fraction of recordings using agonists for the other receptors examined. Together, it appears that resistance to desensitization is likely conferred by a property of the receptor itself and not an intrinsic property of the terminals on which the receptors are found.
Figure 7.

A fraction of GABABRs presynaptic to POMC neurons do desensitize. A, A plot of eIPSC amplitudes from a recording in which the baclofen-induced inhibition of eIPSC amplitude declined robustly during application. Each point represents the average of three consecutive sweeps. The eIPSC traces shown above the plot are the average of three consecutive sweeps. Sample traces were taken at minutes 0 (baseline) and 4 (maximal inhibition) of baclofen superfusion and then every third minute thereafter. B, Compiled data showing the inhibition of eIPSC amplitude by baclofen at each minute during continuous exposure (n = 7). C, A plot of mIPSC frequency from a recording in which baclofen-induced inhibition of mIPSC frequency declined during baclofen application. Representative traces taken at various time points during a recording are shown above the plot. D, Compiled data showing the inhibition of mIPSC frequency by baclofen at each minute during continuous exposure (n = 3). The asterisks denote points that were found to be significantly different from the maximal inhibition caused by baclofen (minute 2) using Tukey's multiple-comparison test. Error bars indicate SEM.
Discussion
The present data demonstrate that multiple Gi/o-coupled receptors located presynaptic to POMC neurons as well as on POMC terminals resist acute desensitization in response to agonist exposure, while the same receptors located on the somatodendritic region of POMC neurons robustly desensitize under identical conditions. MORs display resistance to desensitization in the presence of various agonists and after receptor number is drastically reduced. The current data together with previous reports of differential presynaptic and postsynaptic desensitization indicate that resistance to desensitization is a common and robust property of presynaptic Gi/o-coupled receptors. Comparative studies between presynaptic and postsynaptic receptor regulation could provide important insight into mechanisms underlying desensitization and could help explain complex physiologic responses to prolonged agonist exposure.
Differential presynaptic and postsynaptic desensitization of multiple Gi/o-coupled receptors
The ability of presynaptic, but not postsynaptic MORs to resist desensitization on POMC neurons is similar to the differential regulation of MORs previously reported in the periaqueductal gray (PAG) (Fyfe et al., 2010) and the locus ceruleus (Blanchet and Lüscher, 2002). Thus, sustained presynaptic MOR signaling is not unique to terminals within the arcuate nucleus. Differential desensitization of presynaptic and postsynaptic receptors also does not appear to be a phenomenon specific to the MOR since similar presynaptic resistance and postsynaptic susceptibility to desensitization was also found to occur for nociceptin and GABAB receptors regulating POMC neurons. κ-Opioid receptors, which were only found on presynaptic terminals, were also resistant to acute desensitization. Although KOR-induced postsynaptic currents were not observed in the present study, they likely possess the property of differential desensitization between presynaptic and postsynaptic receptors since KORs expressed in heterologous systems do display acute desensitization (Henry et al., 1995). GABABRs and adenosine A1 receptors (A1Rs) located in the presynaptic but not postsynaptic compartment of cultured hippocampal neurons also resist acute agonist-induced desensitization (Wetherington and Lambert, 2002a,b), and a similar phenomenon has been described for GABABRs regulating dopaminergic neurons in the VTA (Cruz et al., 2004). Thus, the ability to maintain signaling may be a general property of a variety of presynaptic receptors located throughout the brain.
Presynaptic resistance to desensitization is independent of receptor reserve
The apparent lack of acute desensitization observed for presynaptic MORs could simply reflect that there is a high receptor reserve presynaptically such that there is always a sufficient pool of non-desensitized receptors to mediate maximal inhibition of transmitter release. This does not appear to be the case, however, since reducing MOR receptor reserve with β-CNA did not unmask any acute presynaptic MOR desensitization, although inhibition of transmitter release was significantly reduced. These results are consistent with a previous study examining presynaptic MORs in the PAG that also resist desensitization after knockdown with β-CNA (Fyfe et al., 2010). The analysis of dose–response curves constructed under control conditions and after β-CNA exposure revealed that not only does resistance to desensitization by presynaptic MORs occur independent of receptor reserve, but that minimal MOR reserve is present on terminals presynaptic to POMC neurons to begin with.
CMT also can reduce receptor reserve (Christie et al., 1987) and enhance both the extent (Ingram et al., 2008) and rate (Dang and Williams, 2005) of MOR desensitization following a subsequent acute application of agonist. However, in the present study, CMT did not induce desensitization of presynaptic MORs nor reduce the presynaptic inhibition induced by μ receptor agonists as has been observed in other studies (North and Vitek, 1980; Fyfe et al., 2010). The presynaptic inhibition in the present study may be linked to the observation that terminals presynaptic to POMC neurons do not have a substantial receptor reserve even under basal conditions. Interestingly, GABABRs and A1Rs located on the presynaptic terminals of hippocampal neurons also resist desensitization in a manner that is independent of receptor reserve (Wetherington and Lambert, 2002a,b). These similarities across multiple cell types and multiple systems imply that not only is resistance to desensitization a common property of presynaptic Gi/o-coupled receptors but that this resistance may occur through similar mechanisms. Furthermore, the fact that even prolonged exposure to agonist is not sufficient to reduce the presynaptic inhibition to a subsequent application of μ receptor agonist suggests that MORs in the presynaptic compartment resist desensitization by a specific mechanism rather than simply undergoing a postsynaptic-like desensitization in a slower manner.
Contribution of presynaptic environment in the resistance to acute receptor desensitization
MORs and GABABRs undergo acute desensitization in the postsynaptic but not presynaptic compartment of POMC neurons as determined in the ChR2 experiments. This is consistent with previous studies showing that when hippocampal neurons form synapses onto themselves in culture, GABAB and adenosine A1 receptors in the somatodendritic compartment undergo acute receptor-specific (not effector-dependent) desensitization, but those receptors located in the presynaptic terminal do not desensitize (Wetherington and Lambert, 2002a,b). Together, the data suggest that the cellular compartment in which a receptor resides may confer the tendency to undergo desensitization. However, the finding that presynaptic GABABRs displayed acute desensitization in 10 of the 38 experiments (including both mIPSC and eIPSC experiments) in the present study indicates that compartmentalization alone may not be sufficient to confer resistance to desensitization. Furthermore, GABABRs in CA3 neurons of the neonatal rat hippocampus located on the postsynaptic membrane or in presynaptic glutamatergic terminals do not desensitize with acute agonist exposure, although GABABRs receptors located on GABAergic terminals desensitize readily (Tosetti et al., 2004). Resistance to desensitization also occurs in postsynaptic GABABRs located on GABAergic, but not dopaminergic, neurons of the VTA (Cruz et al., 2004). These discrepancies between the patterns of presynaptic and postsynaptic desensitization of GABABRs and the other receptors examined suggests that, although there is a strong correlation between compartmentalization and desensitization, compartmentalization alone is not sufficient to explain differential desensitization between presynaptic and postsynaptic Gi/o-coupled receptors.
Such discrepancies between the GABABR and the other receptors examined may be the result of variable expression of the GABAbR1a/b splice variants. Although these splice variants are preferentially targeted to either terminal regions (R1a) or somatodendritic regions (R1b) of neurons, studies using R1a and R1b knock-outs suggest that either variant can be expressed at GABAergic terminals and postsynaptic membranes (Vigot et al., 2006). Variable expression patterns of the GABABR1 splice variants may explain the discrepancies between the patterns of presynaptic and postsynaptic desensitization of the GABABR and other receptors that have been examined. Determining whether and why a certain GABABR heterodimer composition is resistant to desensitization may provide insight into the mechanism by which other receptors resist acute desensitization.
Implications of differential presynaptic and postsynaptic receptor desensitization
The physiologic consequences of differential presynaptic and postsynaptic desensitization for specific receptors remain to be determined, but the data suggest that acute and chronic agonist exposure can differently affect cellular activity as postsynaptic responses may desensitize while presynaptic actions are maintained. In POMC neurons, for example, the initial action of MOR agonists is direct postsynaptic inhibition, whereas the prolonged effect of MOR activation would likely be disinhibition since the majority of inputs to POMC neurons are GABAergic (Pinto et al., 2004; Hentges et al., 2009). Similar differential desensitization of GABABRs located on GABAergic terminals and postsynaptic GABABRs in the neonatal hippocampus has been suggested to underlie the induction of epileptiform discharges in this region (Vardya et al., 2010). The lack of presynaptic MOR desensitization may explain the observation that synaptic transmission increases upon withdrawal of MOR agonist in multiple brain regions (Bonci and Williams, 1997; Hack et al., 2003; Bie and Pan, 2005), consistent with a continued action at presynaptic MORs until removal of drug. This increase in synaptic transmission after the cessation of chronic opioid exposure has been implicated in withdrawal symptoms such as hyperalgesia (Bie and Pan, 2005; Bie et al., 2005; Heinl et al., 2011). Thus, the differential presynaptic and postsynaptic desensitization of certain GPCRs may have significant functional consequences.
Conclusions
Whereas the postsynaptic actions of MORs and other Gi/o-coupled receptors decline within minutes of agonist exposure, these same receptors on the presynaptic terminals of POMC neurons and presynaptic to POMC neurons continue to inhibit transmitter release. Thus, during continued agonist exposure, postsynaptic responses will likely decline while presynaptic actions will be maintained. The mechanisms limiting presynaptic receptor desensitization remain to be determined, but the data here suggest that compartment-specific modification of the receptor may underlie the resistance.
Footnotes
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK0798749 (S.T.H.). We thank Connie King for technical assistance in managing the mouse colony and stereotaxic injections.
The authors declare no competing financial interests.
References
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT. Separation of μ-opioid receptor desensitization and internalization: endogenous receptors in primary neuronal cultures. J Neurosci. 2006;26:4118–4125. doi: 10.1523/JNEUROSCI.0303-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Pan ZZ. Increased glutamate synaptic transmission in the nucleus raphe magnus neurons from morphine-tolerant rats. Mol Pain. 2005;1:7. doi: 10.1186/1744-8069-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Peng Y, Zhang Y, Pan ZZ. cAMP-mediated mechanisms for pain sensitization during opioid withdrawal. J Neurosci. 2005;25:3824–3832. doi: 10.1523/JNEUROSCI.5010-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet C, Lüscher C. Desensitization of μ-opioid receptor-evoked potassium currents: initiation at the receptor, expression at the effector. Proc Natl Acad Sci U S A. 2002;99:4674–4679. doi: 10.1073/pnas.072075399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C. Bi-directional effects of GABAB receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- Dang VC, Williams JT. Morphine-induced μ-opioid receptor desensitization. Mol Pharmacol. 2005;68:1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. Metabolic factors that influence contractility of vascular smooth muscle. Bull N Y Acad Med. 1966;42:996–1006. [PMC free article] [PubMed] [Google Scholar]
- Fyfe LW, Cleary DR, Macey TA, Morgan MM, Ingram SL. Tolerance to the antinociceptive effect of morphine in the absence of short-term presynaptic desensitization in rat periaqueductal gray neurons. J Pharmacol Exp Ther. 2010;335:674–680. doi: 10.1124/jpet.110.172643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45:575–584. doi: 10.1016/s0028-3908(03)00205-3. [DOI] [PubMed] [Google Scholar]
- Heinl C, Drdla-Schutting R, Xanthos DN, Sandkühler J. Distinct mechanisms underlying pronociceptive effects of opioids. J Neurosci. 2011;31:16748–16756. doi: 10.1523/JNEUROSCI.3491-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C. Kappa-opioid receptors couple to inwardly rectifying potassium channels when coexpressed by Xenopus oocytes. Mol Pharmacol. 1995;47:551–557. [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29:13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Macey TA, Fossum EN, Morgan MM. Tolerance to repeated morphine administration is associated with increased potency of opioid agonists. Neuropsychopharmacology. 2008;33:2494–2504. doi: 10.1038/sj.npp.1301634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of μ-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153(Suppl 1):S379–S388. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of μ opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- North RA, Vitek LV. The effect of chronic morphine treatment of excitatory junction potentials in the mouse vas deferens. Br J Pharmacol. 1980;68:399–405. doi: 10.1111/j.1476-5381.1980.tb14553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci. 2011;31:281–288. doi: 10.1523/JNEUROSCI.4654-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Tosetti P, Bakels R, Colin-Le Brun I, Ferrand N, Gaiarsa JL, Caillard O. Acute desensitization of presynaptic GABAB-mediated inhibition and induction of epileptiform discharges in the neonatal rat hippocampus. Eur J Neurosci. 2004;19:3227–3234. doi: 10.1111/j.0953-816X.2004.03413.x. [DOI] [PubMed] [Google Scholar]
- Vardya I, Drasbek KR, Gibson KM, Jensen K. Plasticity of postsynaptic, but not presynaptic, GABAB receptors in SSADH deficient mice. Exp Neurol. 2010;225:114–122. doi: 10.1016/j.expneurol.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigot R, Barbieri S, Bräuner-Osborne H, Turecek R, Shigemoto R, Zhang YP, Luján R, Jacobson LH, Biermann B, Fritschy JM, Vacher CM, Müller M, Sansig G, Guetg N, Cryan JF, Kaupmann K, Gassmann M, Oertner TG, Bettler B. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron. 2006;50:589–601. doi: 10.1016/j.neuron.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington JP, Lambert NA. Differential desensitization of responses mediated by presynaptic and postsynaptic A1 adenosine receptors. J Neurosci. 2002a;22:1248–1255. doi: 10.1523/JNEUROSCI.22-04-01248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington JP, Lambert NA. GABAB receptor activation desensitizes postsynaptic GABAB and A1 adenosine responses in rat hippocampal neurones. J Physiol. 2002b;544:459–467. doi: 10.1113/jphysiol.2002.023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]