

Abstract
Epileptic seizures are a common feature associated with inherited mitochondrial diseases. This study investigated the role of mitochondrial oxidative stress in epilepsy resulting from mitochondrial dysfunction using cross-bred mutant mice lacking mitochondrial manganese superoxide dismutase (MnSOD or SOD2) and a lipophilic metalloporphyrin catalytic antioxidant. Video-EEG monitoring revealed that in the second to third week of postnatal life (P14–P21) B6D2F2 Sod2−/− mice exhibited frequent spontaneous motor seizures providing evidence that oxidative stress-induced mitochondrial dysfunction may contribute to epileptic seizures. To confirm the role of mitochondrial oxidative stress in epilepsy a newly developed lipophilic metalloporphyrin, AEOL 11207, with high potency for catalytic removal of endogenously generated reactive oxygen species was utilized. AEOL 11207-treated Sod2−/− mice showed a significant decrease in both the frequency and duration of spontaneous seizures but no effect on seizure severity. A significant increase in the average lifespan of AEOL 11207-treated Sod2−/− mice compared to vehicle-treated Sod2−/− mice was also observed. Indices of mitochondrial oxidative stress and damage (aconitase inactivation, 3-nitrotyrosine formation, and depletion of reduced coenzyme A) and ATP levels affecting neuronal excitability were significantly attenuated in the brains of AEOL 11207-treated Sod2−/− mice compared to vehicle-treated Sod2−/− mice. The occurrence of epileptic seizures in Sod2−/− mice and the ability of catalytic antioxidant therapy to attenuate seizure activity, mitochondrial dysfunction, and ATP levels suggest that ongoing mitochondrial oxidative stress can contribute to epilepsy associated with mitochondrial dysfunction and disease.
Keywords: Seizures, EEG, antioxidant, mitochondrial encephalopathy, glutathione, 3-nitrotyrosine, aconitase, complexes, ATP
INTRODUCTION
Epileptic seizures commonly occur in patients with inherited mitochondrial disease (Mecocci et al., 1993; Wallace et al., 1988) suggesting that mitochondrial dysfunction can contribute to seizures (Kunz, 2002; Patel, 2004). General and partial seizures with mitochondrial encephalopathy can be caused by mitochondrial dysfunction arising from mitochondrial mtDNA mutations (Shoffner et al., 1990; Wallace et al., 1988). Mitochondrial dysfunction is a consequence of many neurological insults such as neonatal or adult hypoxia, trauma and infections which are known risk factors for epilepsy development (Beal, 1998; Douglas et al., 2010; Jensen et al., 1991; Jensen et al., 1992; Mustafa et al., 2010). These data strongly suggest that mitochondrial dysfunction per se may be a common pathway contributing to epilepsy development. Mitochondria have important functions that include cellular ATP production, control of apoptotic/necrotic cell death, reactive oxygen species (ROS) formation and calcium homeostasis. Which of these critical mitochondrial functions contributes to increased seizure susceptibility associated with inherited or acquired epilepsies remains unknown. Results from this and other laboratories suggest that mitochondrial oxidative stress and resultant dysfunction are not only a consequence of seizure activity but also render the brain more susceptible to age-related epileptic seizures (Jarrett et al., 2008; Kudin et al., 2002; Liang et al., 2000; Liang and Patel, 2004; Waldbaum et al., 2010).
To understand the role of oxidative stress in epilepsy associated with mitochondrial disease it is useful to utilize an animal model in which spontaneous epileptic seizures arise due to increased steady-state mitochondrial ROS. Mutant mice lacking manganese superoxide dismutase (MnSOD or SOD2), a critical mitochondrial antioxidant, provide such a model. Mitochondrial disease has been characterized in SOD2 deficient mice generated in several background strains with phenotypes characteristic of increased steady-state mitochondrial ROS. Sod2−/− mice bred from a C57B6 background (B6 Sod2−/−) are neonatal lethal (Lebovitz et al., 1996), whereas CD-1 Sod2−/− mice develop and live approximately 8–10 days postnatal (Melov et al., 1999). Recently, Sod2−/− mutant mice from a mixed background (C57BL/6J X DBA/2J, B6D2) have been generated, which live approximately 3 weeks without pharmacological intervention (Huang et al., 2001). Behavioral tonic-clonic seizures have been anecdotally reported starting the second to third week of postnatal life in B6D2F1 Sod2−/− mice (Lynn et al., 2005). Therefore, increased life-span of the cross-bred B6D2 Sod2−/− mice provides a model in which the underlying role of oxidative stress in mitochondrial disease epilepsy can be investigated.
Metalloporphyrin catalytic antioxidants are small molecule mimics of superoxide dismutase (SOD) and/or catalase (CAT), and are also potent detoxifiers of lipid peroxides and peroxynitrite (ONOO−) (reviewed in (Day, 2004)). Because they are catalytic, and not merely bulk scavengers, these compounds are much more potent antioxidants than dietary additives such as vitamin E that act stoichiometrically. The manganese meso-porphyrin catalytic antioxidants combine the broad spectrum detoxification of reactive species like the stoichiometric antioxidants with the catalytic efficiency of the endogenous antioxidant enzymes. Additionally, these synthetic compounds can be chemically modified to increase their ability to cross the blood brain barrier (BBB), as well as their targeting to various subcellular compartments. Treatment of short-lived Sod2−/− mice in the CD-1 background with manganese tetrakis 5, 10, 15, 20-porphyrin (MnTBAP) ameliorated cardiomyopathy but not neurodegeneration (Melov et al., 1998) whereas EUK8 or EUK134 ameliorated spongiform encephalopathy and neurodegeneration (Melov et al., 2001). A major advancement in the field of catalytic antioxidants was the demonstration that AEOL 11207, a lipophilic metalloporphyrin, protected against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in vivo following oral administration (Liang et al., 2007). This compound belongs to a series of metalloporphyrins which were designed to have greater lipid solubility, oral bioavailability, and cross the BBB. The objective of this study was to determine the role of mitochondrial oxidative stress in the development of epileptic seizures using B6D2 Sod2−/− mice. Seizure activity was monitored by video-EEG methods in conjunction with indices of mitochondrial oxidative stress. To confirm the role of mitochondrial oxidative stress in epilepsy in Sod2−/− mice we asked whether treatment with a lipophilic metalloporphyrin antioxidant could attenuate seizure activity.
MATERIALS AND METHODS
Animals
Animal studies were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23). All procedures were approved by the Institute Animal Care and Use Committee (IACUC) of the University of Colorado Denver (UCD), which is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. Heterozygous MnSOD (Sod2−/+) mutant mice on a C57BL/6J (B6) background were crossed with DBA/2J (D2) wild type mice to generate B6D2F1 Sod2−/+ mice, both male and female from B6D2F1 Sod2−/+ mice were used as breeding pairs to produce the F2 generation (B6D2F2) of Sod2−/− mice. The mutant mice were monitored on a daily basis to get an accurate birth and death data. Pups were not culled or handled before P5 to avoid maternal rejection. Pups were genotyped at P5 by PCR as previously described (Li et al., 1995).
Metalloporphyrin (AEOL 11207) administration
B6D2F2 Sod2−/− mice and their wild type littermates (Sod2+/+) used and designated as control mice were treated with AEOL 11207 (5 mg/kg) or vehicle by subcutaneous (s.c.) injection daily starting at 5 days of age until death or being sacrificed. AEOL 11207 was dissolved in dimethyl sulfoxide (DMSO) and diluted with sterilized phosphate buffered saline (PBS) to achieve the desired final concentration (1% DMSO). The animals were divided into four different groups: 1) B6D2F2 Sod2+/+ mice + vehicle; 2) B6D2F2 Sod2−/− mice + vehicle; 3) B6D2F2 Sod2+/+ mice + AEOL 11207; 4) B6D2F2 Sod2−/− mice + AEOL 11207. Both male and female Sod2+/+ and B6D2F2 Sod2−/− mice were used in this study. The ratios of the gender among the four groups were matched. The treated and untreated mice were sacrificed at P15–16 for pathology and biochemistry assays or until death for survival and seizure evaluation.
Behavioral seizure evaluation
Two groups of vehicle and AEOL11207-treated Sod2−/− mice that were positively confirmed by genotyping aged P16-P20 were digitally recorded (Q-See QD14B, Anaheim, CA) daily in Plexiglass cages for quantification of age-related changes in seizure parameters. During the weaning period (P16-P18) mice were recorded in the presence of their mothers, and individually thereafter. Video was digitally recorded (Panasonic DMR-ES15) and stored on DVD-R’s for observation and quantification of seizures.
Video-EEG recordings from Sod2−/− mice
Sod2−/− and Sod2+/+ mice that were positively confirmed by genotyping aged P18 receiving either vehicle or AEOL 11207 treatment were anesthetized via isoflurane continuous inhalation and placed in a mouse stereotoxic unit (MyNeuroLab, Leica Microsystems, Richmond, IL). Bilateral stainless steel electrodes were placed on the skull over the motor cortices and secured with dental cement to monitor brain electrical activity. A screw electrode behind lambda served as a ground and reference. Video-Electroencephalograph (EEG) activity was recorded using Stellate systems (Natus Medical Incorporated, San Carlos, CA) for a minimum of 24 hours and files analyzed and scored for seizure duration, severity, and frequency by an observer blind to genotype and treatment. Behavioral seizure severity was scored according to the following scale: 1=immobilization and staring, 2=head-nodding, shaking, 3=forelimb tonic/clonic activity, 4=continuous forelimb tonic/clonic activity with falling, running or jumping. In order to avoid overestimation and accurately define seizure events due to the occurrence of abnormal gait and posturing in Sod2−/− mice, only spontaneous motor seizures with scores > 3 were included for comparison between groups and all behavioral seizure activity correlated with EEG seizure events. Isolated electrographic seizures were differentiated from background noise and movement artifact by observing the corresponding video to verify motor seizures and by the appearance of rhythmic spike frequency activity that lasted a minimum of 5 seconds, had a clear beginning and resolution, and were separated from each other by a minimum of 30 s. Continuous seizure events were identified as rhythmic spike frequency activity that were separated from each other by less then 30 s and were confirmed with motor seizure behavior in the corresponding video.
Determination of metalloporphyrin levels
AEOL 11207 was measured by HPLC-UV following previously described methods (Liang et al., 2007).
Histochemical analyses
Mice were sacrificed at P15–16 and brain paraffin sections (10 µm) were cut coronally and stained with Hematoxylin and Eosin (H&E) following the company protocol (Sigma, St. Louis MO). Fluoro-Jade B (Histo-Chem Inc., Jefferson, AR) staining was performed following previously described methods (Hopkins et al., 2000; Liang et al., 2008). Images were captured using a Nikon Optiphot-2 80i microscope equipped with epifluorescense optics (Nikon Inc., Melville, NY). The Fluoro-Jade B positive signal of a given area was estimated with Image J (National Institutes of Health, Bethesda, MD), an open source image manipulation tool, in three sections 100 µm apart in the parietal cortex from both hemispheres of each animal. The average of the fluorescent relative density was expressed as percentage of the control.
Isolation of mitochondrial fractions
Mitochondria were isolated from the forebrain of mice according to previously described methods (Liang and Patel, 2006).
Aconitase and fumarase activity assay
Aconitase and fumarase activity were measured in mitochondrial fraction as previously described (Patel et al., 1996).
Measurement of metabolomics by HPLC
Glutathione (GSH), glutathione disulfide (GSSG), tyrosine, 3-nitrotyrosine (3-NT) and ascorbate assays were performed with an ESA (Chelmsford, MA) 5600 CoulArray HPLC equipped with eight electrochemical cells as previously described in the literature (Beal et al., 1990; Hensley et al., 1998; Liang et al., 2007). The potentials of the electrochemical cells were set at 0/150/300/450/570/690/800/850 mV. Analyte separation was conducted on a TOSOHAAS (Montgomeryville, PA) reverse-phase ODS 80-TM C-18 analytical column (4.6 mmx250 cm; 5 µm particle size). A two-component gradient elution system was used with component A of the mobile phase composed of 50 mM NaH2PO4 pH 3.2, and component B composed of 50mM NaH2PO4 and 40% methanol pH 3.2. Aliquot (50 µl) of the supernatant was injected to HPLC. The level of 3-NT was expressed as a ratio of 3-NT to tyrosine.
Measurement of Reduced coenzyme A (CoASH) and its GSH disulfide (CoASSG)
CoASH and CoASSG were measured by HPLC equipped with UV detection as previously described (Liang and Patel, 2006).
Measurement of AMP, ADP and ATP by HPLC
The forebrains were dissected out, quickly frozen with liquid nitrogen, weighed and sonicated in 10 % w/v (e.g. 20 mg/200 µl) 0.42 M perchloric acid. (The homogenates can be store at −80 °C). The homogenates were centrifuged at 13000 g 4°C for 15 min. One hundred µl supernatant was removed to a new tube and neutralized with 10 µl 4 N KOH. The neutralized supernatant was mixed well and left on −20°C for at least 10 min to ensure removal of perchlorate (as KClO4). After centrifugation at 8500 g, 4°C for 10 min, equal volumes (100 µl) of supernatants and (100 µl) 50 mM KH2PO4 were mixed, an aliquot of 50 µl of the mixture was injected into the HPLC system. The levels of AMP, ADP and ATP were quantified by HPLC-UV set at 258 nm following previous described methods (Botker et al., 1994; Sellevold et al., 1986). Analyte was separated by 5 µM, 4.6×250 cm C-18 reversed-phase column. Mobile phase is composed of 50 mM KH2PO4, 10% methanol, 3 mM tetrabutyl ammonium sulphate (TBAS), pH 6.0 and flow rate set at 0.8 ml/min.
Complex Activity Assay
Complex I (NADH: ubiquinone oxidoreducase, EC 1.6.99.3) activity was measured in mitochondrial fractions as previously described (Birch-Machin and Turnbull, 2001; Tieu et al., 2003). The oxidation of NADH was monitored spectrophotometrically at 340 nm for 2 min at 30°C. Activity was further monitored for 2 min following the addition of rotenone (2µg/ml). The difference between the rate of oxidation, before and after the addition of rotenone, was used to calculate Complex I activity. Complex II (Succinate-Ubiquinone Oxidoreductase,) activity was measured following previously described methods (Birch-Machin and Turnbull, 2001) by reduction of 2,6-dichlorophenolindophenol (DCPIP) and monitoring the absorbance at 600 nm (extinction coefficient 19.1 mM−1cm−1).
Measurement of mitochondrial respiration rates from cortical synaptosomes
Cortical tissue was dissected from Sod2−/− mice and homogenized using a Dounce tissue grinder (Wheaton, Millville, NJ) in 5–10% w/v (50–100mg tissue/ml) and homogenizing buffer (320 mM sucrose, 1 mM EDTA, 0.25 mM DTT, pH 7.4). The homogenate was centrifuged at 3600 g for 10 min at 4°C. The supernatant was centrifuged at 32500 g for 10 min at 4°C on a 3%/10%/23% Percoll density gradient. The synaptosomes were isolated at the band between the 10% and 23% solutions and spun at 15000 g for 15 min to remove Percoll. Synaptosomes were re-suspended in an ionic buffer (20 mM HEPES, 10 mM glucose, 1.2 mM Na2HPO4, 1 mM MgCl2, 5 mM NaHCO3, 5 mM KCl, 140 mM NaCl, pH 7.4) and diluted to a .2 mg/ml solution. 50 µl was aliquoted into each well of a PEI-coated XF24 plate and spun down at 2300 g for 1 hour at 4°C. The ionic buffer was removed and replaced with an incubation buffer (3.5 mM KCl, 120 mM NaCl, 1.3 mM CaCl2, .4 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgSO4, 15 mM glucose, 4 mg/ml BSA, 10 mM pyruvate, pH 7.4). The plate was then run on a XF24 Analyzer and synaptosomes were subjected to consecutive injections of oligomycin (ATP synthase inhibitor), FCCP (Carbonyl cyanide 4-(trifluoromethoxyl) phenyl-hydrazone, uncoupler), and antimycin A (inhibitor of complex III) to obtain oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) indicating mitochondrial respiration and glycolytic rates respectively.
Statistical analyses
Survival analysis was performed using the Kaplan-Meier method. For all biochemical analyses, two-way ANOVA was used with GraphPad Prism 5 software. P values less than 0.05 were considered significant.
RESULTS
Seizure activity in Sod2−/− mice
Forty-six Sod2−/− mice were initially digitally recorded for a minimum of 8 hr/day beginning on P16 to establish seizure parameters and any age-related changes in behavioral seizure activity. A significant increase in seizure duration and numbers from P17 to P20 in vehicle or AEOL11207 treated Sod2−/− mice was observed (Figure 1), suggesting that elevated mitochondrial ROS over time may contribute to epilepsy progression. In order to obtain a higher degree of quantification and accuracy than possible with exclusive behavioral monitoring, a group of Sod2+/+ and Sod2−/− mice were then implanted with bilateral dural electrodes for 24 h a day video/EEG monitoring to quantify seizure parameters (Figure 2). Only seizures scoring > 3 on the severity scale were included for analysis. EEG implantation prior to P18 and long-term recordings from Sod2−/− mice were not feasible due to their weaning age and sensitivity to electrode implantations, respectively. Sod2+/+ mice implanted with dural EEG electrodes showed no evidence of seizure activity (Figure 2 A). Two types of seizures were evident in vehicle treated Sod2−/− mice, isolated seizures (Figure 2C) that were separated from each other by a minimum of 30 s and continuous seizures (Figure 2B) that appeared one after the other and were separated from each other by less then 30 s. Continuous seizure events that lasted longer than 60 s comprised approximately 3.6% of the total seizure events in vehicle treated Sod2−/− mice (Table 3, Supplemental Materials). All implanted vehicle-treated Sod2−/− mice exhibited spontaneous seizure activity with an average inter-seizure interval and duration of 2.93 ±0.51 min and 18.73 ± 1.56 s, respectively (Figure 2 E,F).
Figure 1. Behavioral seizure parameters in Sod2−/− mice.

The average duration (A) and number (B)of spontaneous behavioral seizures monitored by digital recordings from vehicle or AEOL11207 treated Sod2−/− mice from 17 to 20 days old. Bars represent mean or mean + S.E.M, *p < 0.05, compared to the same treatment of 17 and 18 day old mice and # p < 0.05 compared to age-matched vehicle-treated mice. Data were analyzed by one way ANOVA (Figure 1A) n=46 mice (23 each vehicle- or AEOL 11207-treated mice).
Figure 2. Spontaneous electrographic seizure parameters in Sod2−/− mice.

Dural EEG recording from a P18 (A) Sod2+/+ mouse showing no seizure activity as a result of electrode implantation, (B) vehicle-treated Sod2−/− mouse showing a period of continuous seizure activity, (C) vehicle-treated Sod2−/− mouse showing a period of isolated seizure activity, (D) AEOL 11207-treated Sod2−/− mouse showing the attenuation of seizure activity. (E) Averaged inter-seizure interval and (F) seizure duration of electrographically recorded spontaneous seizures in vehicle or AEOL 11207-treated Sod2−/− mice. n=5 per treatment group. Bars represent mean + S.E.M, **p<0.01; Mann-Whitney non-parametric test.
Histopathological analysis
Serial coronal sections from the brains of Sod2−/− mice at P15–16 were examined by H&E and Fluoro-Jade B staining in order to determine the pathological damage related to the observed neurological symptoms. Neuronal damage was not observed in any brain region of Sod2+/+ animals by H&E staining (Figure 3 panel 1, A). Sod2−/− mice developed “vacuolar degeneration” characteristic of different strains of this model (Melov et al., 1998, 2001) in regions of the cerebral cortex, predominantly in the parietal cortex (Figure 3 panel 1, B), and to lesser extents in the frontal and piriform cortex, brainstem, thalamus, and in the pyramidal layer of hippocampus. Vacuoles ranged from 4 to 40 µm and impinged on neighboring structures such as neurons and blood vessels. These neuropathological results are consistent with those identified in patients with mitochondrial encephalopathy (spongiform encephalopathy) (Hauser, 1997) and with previous experimental observations (Lynn et al., 2005; Melov et al., 2001).
Figure 3. Neuropathological damage in Sod2−/− mice.
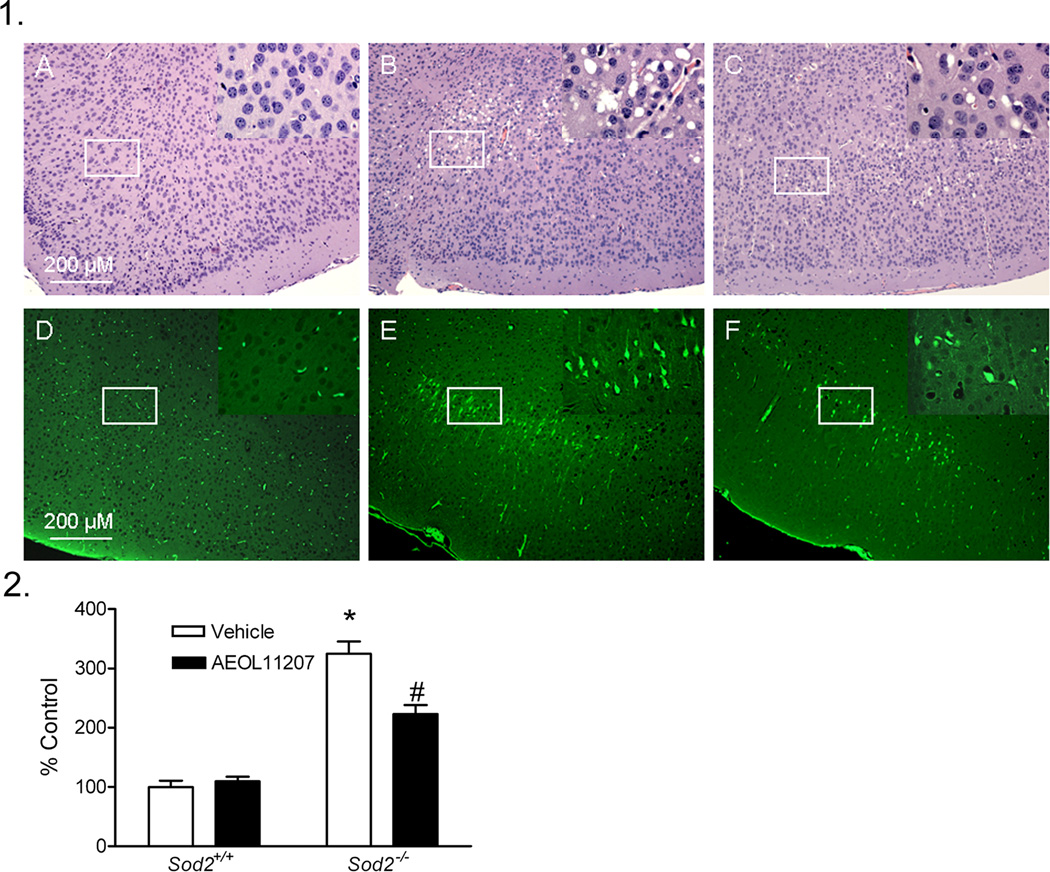
Panel 1: Representative H&E and Fluoro-Jade B staining images in the parietal cortex of P15–16 Sod2+/+ or Sod2−/− mice receiving either vehicle or AEOL 11207 treatment. H&E staining (A, B, C) and Fluoro-Jade B staining (D, E, F). Control (A, D), vehicle- (B, E) and AEOL 11207-treated (C, F). The insets on the upper right corner of each picture are the enlarged image from the white rectangle.
Panel 2: Quantitative analysis of Fluoro-Jade B fluorescence in the parietal cortex of P15–16 Sod2+/+ or Sod2−/− mice receiving either vehicle or AEOL 11207 treatment. The Fluoro-Jade B positive signal in a given area of parietal cortex from both hemispheres of each animal was estimated with Image J. Bars represent mean + S.E.M, *p<0.01 vs. Sod2+/+ mice with same treatment; #p<0.05 vs. vehicle-treated Sod2−/− mice; two way ANOVA, n=6 mice per group.
Fluoro-Jade B staining, a sensitive marker assessing degeneration of neuronal cell bodies and processes (Schmued and Hopkins, 2000), was performed on control and Sod2−/− mice. Fluoro-Jade B staining was not observed in brain regions of Sod2+/+ mice (Figure 3 panel 1, D). However, prominent staining (degeneration) was observed in the cell bodies and terminals in regions of the cerebral cortex from vehicle-treated Sod2−/− mice, most notably in the parietal cortex (Figure 3 panel 1, E). The relative fluorescence density quantified by Image J increased ~225% in the parietal cortex of Sod2−/− mice compared to the Sod2+/+ group (Figure 3 panel 2).
Mitochondrial oxidative stress
To confirm mitochondrial oxidative stress in the brains of Sod2−/− mice, the levels of CoASH, CoASSG, aconitase, and 3-NT were examined in the forebrain at P15–16. CoASH and CoASSG are primarily compartmentalized within mitochondria where they exchange thiols with GSH and GSSG. The measurement of CoASH and CoASSG in intact tissue provides a reliable assessment of redox status in the mitochondria and overcomes artifactual changes in GSH and GSSG associated with subcellular fractionation (Liang and Patel, 2006; O'Donovan et al., 2002; Waldbaum et al., 2010). The level of CoASH in vehicle-treated Sod2−/− mice was depleted ~50% and CoASSG was increased ~210% resulting in a CoASH/CoASSG ratio that was decreased to 18% of control in the forebrain (Figure 4 A, B). It has been suggested that the mitochondrial GSH pool plays a more important role in maintaining cell viability following toxic insults compared to the cytoplasmic pool (Meredith and Reed, 1982) and we observed that the level of GSH was not changed in forebrain cytosol fractions of Sod2−/− mice (data not shown). To determine if mitochondrial oxidative stress in Sod2−/− mice played a role in seizure severity, regression analysis of CoASH/CoASSG and behavioral seizure activity in Sod2−/− mice was conducted. In fact, decline of CoASH/CoASSG measured in a subset of mice analyzed in Figure 1 showed a significant correlation with seizure numbers (r2=0.6922, p=0.0008, linear regression analysis, n=12 Sod2−/− mice) suggesting that mitochondrial oxidative stress and epilepsy may be causally related.
Figure 4.

Markers of oxidative stress in Sod2−/− mice were attenuated by AEOL 11207 treatment. (A) CoASH, (B) CoASSG, (C) aconitase activity, and (D) 3-NT levels in mitochondrial fractions of P15–16 Sod2+/+ or Sod2−/− mice receiving either vehicle or AEOL 11207 treatment. Bars represent mean + S.E.M, *p<0.01 vs. Sod2+/+ mice with same treatment; #p<0.05 vs. vehicle-treated Sod2−/− mice; two-way ANOVA, n=6–12 mice per group.
Aconitase has been reported to be highly sensitive to O2.− - and ONOO− -mediated inactivation (Gardner et al., 1997; Gardner and Fridovich, 1992; Patel et al., 1996). The activity of aconitase in mitochondria from vehicle-treated Sod2−/− mice was significantly decreased 65% compared to Sod2+/+ mice (Figure 4C), and consistent with previously reported results (Melov et al., 1999). By contrast, the activity of aconitase in the cytosol and fumarase in the mitochondria showed no significant alteration in the forebrain of vehicle-treated Sod2−/− mice. 3-NT is an indicator of free nitro tyrosine residues in proteins following the reaction with nitrating oxidants. The concentration of 3-NT was significantly increased 15 fold in forebrain mitochondrial fractions of vehicle-treated Sod2−/− mice compared with Sod2+/+ mice (Figure 4D). Ascorbate, an endogenous antioxidant, showed no alterations in the forebrain of vehicle-treated Sod2−/− mice. In summary, decreases in aconitase, CoASH, and increased CoASSG and 3-NT (Figure 4) together confirm mitochondrial oxidative stress in the brains of Sod2−/− mice.
Mitochondrial respiration, complex I and II activities and ATP levels
To evaluate the vulnerability of mitochondrial ETC enzymes to mitochondrial oxidative stress, the activity of complex I and II was assessed. Complex I activity was reduced 25% and complex II activity was dramatically reduced 87% in forebrain mitochondrial fractions from Sod2−/− mice compared to wild-type Sod2+/+ mice (Figure 5A and 5B). These results indicate significant differences in the sensitivity of key ETC enzymes to endogenous oxidative stress and confirm previous studies showing greater complex II deficiency than complex I in mitochondria isolated from skeletal muscle and heart of Sod2−/− mice (Hinerfeld et al., 2004; Melov et al., 1999). A primary function of the mitochondria is to synthesize ATP and the measurement of ATP production is a commonly utilized marker for mitochondrial function, particularly in the brain where glycolysis provides much less ATP than in other organs. A 70% reduction in ATP levels was observed in forebrain homogenates of vehicle-treated Sod2−/− mice compared with Sod2+/+ mice (Figure 5C). We further determined whether mitochondria isolated from Sod2−/− mice show respiratory deficits in cortical synaptosomes. No differences were observed in baseline respiration rates between wild-type (Sod2+/+) and Sod2−/− mice. However, an 86% decrease in maximal respiratory rate was observed after treatment with 4 µM FCCP (n=3, p=.0123), indicative of a deficit in mitochondrial respiration in Sod2−/− mice as compared to wild-type Sod2+/+ mice.
Figure 5.

AEOL 11207 partially rescued altered bioenergetics and mitochondrial ETC complexes in Sod2−/− mice. (A) complex I activity, (B) complex II activity and (C) ATP levels from Sod2+/+ plus vehicle or AEOL 11207-treated Sod2−/− mice. Bars represent mean + S.E.M, *p<0.01 vs. Sod2+/+ mice with same treatment; #p<0.05 vs. vehicle-treated Sod2−/− mice; two way ANOVA, n=5–8 mice per group.
AEOL 11207 levels in Sod2−/− mice brains
To confirm the role of mitochondrial ROS in epilepsy occurring in SOD2 mutant mice, we first determined whether AEOL 11207 penetrated the mouse BBB and mitochondria. The concentrations of AEOL 11207 in mouse forebrain homogenates and mitochondria were measured at P15–16 following treatment (5 mg/kg, s.c. daily from P5) to determine its bioavailability. The concentration of AEOL 11207 was observed to be 371.0 ± 30.4 nM in the mouse forebrain and 2.99 ± 0.18 pmol/mg protein in the mitochondria. No significant difference in AEOL 11207 concentration was observed between Sod2+/+ and Sod2−/− mice. No SOD2 protein band detectable by Western blot analyses was present from Sod2−/− mice regardless of treatment.
Effects of AEOL 11207 on seizures, histopathology, and mitochondrial oxidative stress in Sod2−/− mice
AEOL11207 treated Sod2−/− mice showed a decrease in the average number and duration of spontaneous behavioral seizures based on daily observation from 17 to 20 days old compare to vehicle-treated Sod2−/− mice (Figure 1). Only 60% of AEOL 11207-treated Sod2−/− mice exhibited spontaneous seizure activity compared to 100% of vehicle-treated Sod2−/− mice. Seizures from AEOL11207-treated Sod2−/− mice (Figure 2D) occurred less frequently and had a decreased duration as compared to vehicle-treated Sod2−/− mice (Figure 2E,F). Furthermore, the continuous seizure event those last longer than 60 seconds was not observed in AEOL 11207 treated Sod2−/− mice (Table 3, Supplemental Materials). The average duration and inter-seizure interval for individual mice are shown in Tables 1–3 (Supplemental Material). The AEOL 11207 group showed no detectable adverse events up to 20 days of treatment and no mortality resulting from treatment. No differences were observed in seizure severity between vehicle- and AEOL 11207-treated Sod2−/− mice.
Histopathology revealed damage including increased vacuole size and number observed in vehicle-treated Sod2−/− mice that was diminished by AEOL 11207 treatment (Figure 3 panel 1, C). Fluoro-Jade B staining was used to quantify the neuroprotective effects of AEOL 11207. The histopathological neuronal degeneration seen in vehicle-treated Sod2−/− mice was significantly attenuated in the AEOL 11207 group (Figure 3 panel 1, 3F). The relative fluorescence density quantified by Image J in vehicle-treated Sod2−/− mice was significantly attenuated 50% by AEOL 11207 administration (Figure 3 panel 2). AEOL 11207 treatment resulted in significant attenuation of SOD2 deficiency-induced oxidative stress markers aconitase, CoASH, and increased CoASSG and 3-NT (Figure 4). The observed decreases in hippocampal ATP levels and complex II activity in the forebrain of vehicle-treated Sod2−/− mice were also significantly attenuated by AEOL 11207 treatment, although complex I activity was not (Figure 5). Linear Similar to previously reported results (Huang et al., 2001), B6D2F2 Sod2−/− mice had an average lifespan of 14.42± 0.44 days (n=73). Sod2−/− mice treated daily with AEOL 11207 (5 mg/kg) had a significant increase in their average lifespan compared to vehicle-treated Sod2−/− mice to 20.383 ± 0.43 days (n=21). A prominent feature of the determined survival curve was the dramatic increase in the percentage of Sod2−/− mice living beyond 2 weeks of age with AEOL 11207 treatments. Only ~48% of vehicle-treated Sod2−/− mice survived beyond 15 days, whereas 100% of those treated with AEOL 11207 survived beyond that time point (Figure 6). Daily AEOL 11207 treatment at a lower dose (2.5 mg/kg) had no significant increase in average lifespan (data not shown).
Figure 6.

Survival curve of Sod2−/− mice treated with AEOL 11207 or vehicle. Lifespan from 72 vehicle and 21 AEOL 11207 treated Sod2−/− mice was analyzed by a Kaplan-Meier survival curve. *p<0.01 vehicle vs AEOL 11207.
DISCUSSION
The present study used SOD2 deficient mice and a novel lipophilic metalloporphyrin catalytic antioxidant, AEOL 11207, to establish the role of mitochondrial oxidative stress as a contributing mechanism in epilepsy resulting from mitochondrial dysfunction. Specifically, the results demonstrated that: (1) Sod2−/− mice from a mixed genetic background (B6D2F2) lived approximately 3 weeks, exhibited frequent spontaneous seizures by the second to third week postnatal, and possessed significant brain pathology and neuronal death; (2) AEOL 11207 treatment attenuated seizure frequency and duration in Sod2−/− mice, extended their lifespan, and protected against neuronal death; and (3) Sod2−/− mice brains expressed decreased levels of CoASH, CoASH/CoASSG, aconitase, ATP, complex I and II activity, and increased levels of 3-NT which were all partially rescued by AEOL 11207 with the exception of complex I. Together, these results suggest that mitochondrial oxidative stress contributes to epileptic seizures in Sod2−/− mice.
Mechanisms of epilepsy in Sod2−/− mice
Mitochondrial oxidative stress and dysfunction are contributing factors in many neurological disorders. Although mitochondrial oxidative stress is known to occur as a consequence of seizures (Bruce and Baudry, 1995; Chuang et al., 2004; Gluck et al., 2000; Liang et al., 2000), its role as a contributor to epilepsy is still emerging (Jarrett et al., 2008; Liang and Patel, 2004; Liang and Patel, 2006; Waldbaum et al., 2010). We have previously demonstrated a role of mitochondrial oxidative stress in lowering seizure threshold from studies in Sod2−/+ mice that are partially deficient in SOD2 and develop age-dependent spontaneous seizures (Liang and Patel, 2004). The present study suggests that mitochondrial oxidative stress is a contributing mechanism to increased seizure susceptibility and epilepsy in Sod2−/− mice demonstrated by increased oxidative stress markers, depletion of cellular antioxidants, and alterations to bioenergetics potentially affecting targets that control neuronal excitability. The use of AEOL 11207, a lipophilic metalloporphyrin with high potency for catalytic removal of ROS, attenuated these markers and decreased seizure frequency and duration in Sod2−/− mice suggesting a role for mitochondrial oxidative stress as a contributing mechanism to the increased seizure susceptibility. It is estimated that the majority of mitochondrial O2.− is consumed by SOD2 based on the estimation of steady-state [O2.−] in the picomolar range (Imlay and Fridovich, 1991) and SOD’s extremely high reaction rate of (1.8 X 109 M−1s−1) coupled with its micromolar subcellular concentrations. It has been demonstrated that ONOO−, a reaction product of NO and O2.−, is likely a primary source and major contributor to tyrosine nitration in physiological and pathological events in vivo (Sawa et al., 2000) indicated by increased 3-NT levels. The concentration of 3-NT was significantly increased in vehicle-treated Sod2−/− mice suggesting that OONO− production is markedly amplified in the absence of SOD2, likely due to increased steady-state [O2.−]. Therefore, instantaneous formation of more potent oxidants, such as H2O2, ONOO− and OH.− from short-lived O2.− may play an important role in the mitochondrial oxidative stress, dysfunction, and epilepsy observed in Sod2−/− mice.
Mitochondrial oxidative stress and subsequent dysfunction likely leads to epilepsy development in Sod2−/− mice through mechanisms which initiate cellular dysfunction via key bioenergetic and metabolic processes demonstrated in this study that could impact neuronal excitability. The mitochondrial redox status in Sod2−/− mice as demonstrated by altered CoASH/CoASSG suggests a persistent oxidative environment which may damage sensitive targets involved in the control of neuronal excitability and seizure susceptibility leading to epilepsy. Although CoASH and its redox couple, CoASSG, have been used to monitor mitochondrial redox status from whole tissue samples in a variety of models (Liang and Patel, 2006; Waldbaum et al., 2010), this is the first study to our knowledge that validates the use of the CoASH/CoASSG redox couple for assessing mitochondrial redox status. We have previously demonstrated that the expression of astroglial glutamate transporters, EAAT2 (GLT-1), GLAST, and EAAC-1 decreased in epileptic Sod2−/+ and Sod2+/+ mice at increasing ages (Liang and Patel, 2004). Increased O2.− production in the mitochondrial matrix of Sod2−/− mice and consequent generation of H2O2 which is freely permeable across cellular membranes may result in oxidative damage of plasma membrane-bound targets such as astroglial GLT-1 or Na+, K+-ATPase (EC 3.6.3.9), a neuronal membrane ion channel responsible for maintaining membrane potential which have been demonstrated to be sensitive to oxidative damage (Boldyrev and Kurella, 1996; Dobrota et al., 1999; Trotti et al., 1998). In fact, the expression of GLT-1 and activity of Na+, K+-ATPase in Sod2−/− mice were examined and demonstrated to significantly decreased by 50% and 42% respectively, in the hippocampus of vehicle-treated Sod2−/− mice compared to Sod2+/+ mice (data not shown) suggesting that these alterations may contribute to an increase in neurotoxic extracellular glutamate levels and increased neuronal excitability producing increased seizure susceptibility and epilepsy in Sod2−/− mice.
Impaired mitochondrial electron transport chain (ETC) function as a result of elevated ROS may lead to Ca2+-dependent depolarization of the mitochondrial membrane potential resulting in incomplete oxygen consumption, reduced ATP production, overproduction of ROS and oxidative damage (Giulivi et al., 1995; Kovacs et al., 2002; Nicholls and Ward, 2000; Patel, 2002). As altered function of the ETC produces excess ROS, which are direct inhibitors of ETC and tricarboxylic acid cycle (TCA) enzymes, a vicious cycle can result leading to oxidative cell damage (Giulivi et al., 1995; Patel, 2004). Increased mitochondrial oxidative stress and subsequent oxidative damage to the mitochondrial ETC in Sod2−/− mice presumably impairs electron flux for ATP synthesis affecting energy dependent cellular processes such as the Na+, K+-ATPase and altered neuronal excitability. Further, oxidative damage to aconitase affecting an important component of the TCA cycle, which provides reduced NADH and FADH to the mitochondrial ETC to synthesis ATP, could explain its depletion in the forebrains of Sod2−/− mice. It is likely that the AEOL 11207 treatment protocol used in this study was unable to fully protect against oxidative damage, altered bioenergetics and total life-span in Sod2−/− mice pups due to the chronic alterations that began and continued in utero prior to the first treatment as well as other ongoing mechanisms. Nonetheless, treatment with AEOL 11207 beginning the first week postnatal significantly attenuated altered metabolic processes, seizure activity and mean life-span in Sod2−/− mice suggesting their roles as contributing mechanisms to the development of neurological disease in these mice.
A threshold was presumably reached at approximately 3 weeks postnatal whereby the increase in mitochondrial oxidative stress became incompatible with life, as no vehicle-treated animals survived beyond that age. It is possible that the distribution of seizure events from Sod2−/− mice was affected by seizure clustering that is known to occur in other epilepsy models (Goffin et al., 2007; Grabenstatter et al., 2005; Williams et al., 2009), however mitochondrial oxidative stress-induced mechanisms underlying the seizure activity in these mice may provide more evenly distributed ictal events then chemoconvulsant-induced models. AEOL 11207 did not affect seizure severity in this model; although this may be due to the exclusion of less severe seizure events from analysis.
Targeting mitochondrial dysfunction with catalytic antioxidant therapy
An alternative approach to standard therapies aimed at decreasing neuronal excitability in epileptic patients is the targeting of epileptogenesis with drugs intended to affect the underlying processes that lead to the development of epilepsy. Pharmacological removal of ROS using compounds such as catalytic antioxidants may provide therapeutic strategies for epilepsy and other pathological manifestations of mitochondrial diseases. The demonstration that catalytic antioxidant therapy attenuates seizure parameters in Sod2−/− mice suggests potential implications for inherited mitochondrial disorders resulting in epilepsy such as mitochondrial encephalopathy with ragged red fibers (MERFF), mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) and other metabolic disorders associated with seizures. The AEOL class of metalloporphyrin compounds contains a manganese center that is capable of detoxifying a wide range of ROS including O2.−, H2O2, ONOO−, and lipid peroxide radicals (Day, 2004). AEOL 11207 has potent H2O2 scavenging activity and relatively modest SOD activity (Castello et al., 2007; Liang et al., 2007) and is a potent inhibitor of mitochondrial ROS production (Trova et al., 2003). It should be noted however, AEOL11207 may have off-target effects that were not assessed here and that ROS play an important physiological role in cell signaling and their removal with antioxidants may have deleterious consequences on cellular functions during chronic administration that remain to be elucidated. These findings suggest that mitochondrial oxidative stress and dysfunction play a mechanistic role in epilepsy occurring in Sod2−/− mice and treatment with a lipophilic metalloporphyrin catalytic antioxidant decreases seizure parameters by inhibiting mitochondrial oxidative damage.
Highlights.
The role of mitochondrial oxidative stress in epilepsy was investigated. > Mice deficient in mitochondrial antioxidant (Sod2−/−) showed spontaneous electrographic and motor seizures. > Seizures, oxidative stress and bioenergetic parameters in Sod2−/− mice were attenuated by a catalytic antioxidant. > Mitochondrial oxidative stress is a contributing mechanism to epilepsy in Sod2−/− mice.
Supplementary Material
Acknowledgements
This work is supported by grants from the NIH RO1NS039587 and R21NS53548 (MP), RO1AG24400 (TTH), CURE Innovator Award (MP), EFA (SW) and CCTSI (SW). The authors wish to thank Dr. Yogendra Raol and the UCD In Vivo Neurophysiology Core and Dr. Andrew White for help analyzing and interpreting EEG data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure:
B.J.D. holds equity and serves as a consultant for Aeolus Pharmaceuticals which is developing metalloporphyrins as therapeutics. M.P. has been a recipient of research grants from Aeolus Pharmaceuticals. M.P. and L.L. have a patent pending for therapeutic use of metalloporphyrins in epilepsy.
REFERENCES
- Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochimi Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- Beal MF, et al. Kynurenine pathway measurements in Huntington's disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Boldyrev A, Kurella E. Mechanism of oxidative damage of dog kidney Na/K-ATPase. Biochem Biophys Res Commun. 1996;222:483–487. doi: 10.1006/bbrc.1996.0770. [DOI] [PubMed] [Google Scholar]
- Botker HE, et al. Analytical evaluation of high energy phosphate determination by high performance liquid chromatography in myocardial tissue. J Mol Cell Cardiol. 1994;26:41–48. doi: 10.1006/jmcc.1994.1006. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Baudry M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med. 1995;18:993–1002. doi: 10.1016/0891-5849(94)00218-9. [DOI] [PubMed] [Google Scholar]
- Castello PR, et al. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YC, et al. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia. 2004;45:1202–1209. doi: 10.1111/j.0013-9580.2004.18204.x. [DOI] [PubMed] [Google Scholar]
- Day BJ. Catalytic antioxidants: a radical approach to new therapeutics. Drug Discov Today. 2004;9:557–566. doi: 10.1016/S1359-6446(04)03139-3. [DOI] [PubMed] [Google Scholar]
- Dobrota D, et al. Na/K-ATPase under oxidative stress: molecular mechanisms of injury. Cell Mol Neurobiol. 1999;19:141–149. doi: 10.1023/A:1006928927480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RM, et al. Neuronal death during combined intermittent hypoxia/hypercapnia is due to mitochondrial dysfunction. Am J Physiol Cell Physiol. 2010;298:C1594–C1602. doi: 10.1152/ajpcell.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PR, et al. Nitric oxide sensitivity of the aconitases. J Biol Chem. 1997;272:25071–25076. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- Giulivi C, et al. Hydroxyl radical generation during mitochondrial electron transfer and formation of 8-hyrdoxydeoxyguanosine in mitochondrial DNA. Arch Biochem Biophys. 1995;316:909–916. doi: 10.1006/abbi.1995.1122. [DOI] [PubMed] [Google Scholar]
- Gluck MR, et al. CNS oxidative stress associated with the kainic acid rodent model of experimental epilepsy. Epilepsy Res. 2000;39:63–71. doi: 10.1016/s0920-1211(99)00111-4. [DOI] [PubMed] [Google Scholar]
- Goffin K, et al. Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat. Exp Neurol. 2007;205:501–505. doi: 10.1016/j.expneurol.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Grabenstatter HL, et al. Use of chronic epilepsy models in antiepileptic drug discovery: the effect of topiramate on spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia. 2005;46:8–14. doi: 10.1111/j.0013-9580.2005.13404.x. [DOI] [PubMed] [Google Scholar]
- Hauser WA. Epidemiology of seizures and epilepsy in the elderly. In: RR Rowan AJ, editor. Seizures and epilepsy in the elderly. Boston: Butterworth-Heinemann; 1997. pp. 7–18. [Google Scholar]
- Hensley K, et al. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinerfeld D, et al. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- Hopkins KJ, et al. Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res. 2000;864:69–80. doi: 10.1016/s0006-8993(00)02137-5. [DOI] [PubMed] [Google Scholar]
- Huang T, et al. Genetic modificaiton of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free Radic Biol Med. 2001;31:1101–1110. doi: 10.1016/s0891-5849(01)00694-3. [DOI] [PubMed] [Google Scholar]
- Imlay JA, Fridovich I. Assay of metabolic superoxide production in Escherichia coli. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- Jarrett SG, et al. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol Dis. 2008;30:130–138. doi: 10.1016/j.nbd.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FE, et al. Epileptogenic effect of hypoxia in the immature rodent brain. Ann Neurol. 1991;29:629–637. doi: 10.1002/ana.410290610. [DOI] [PubMed] [Google Scholar]
- Jensen FE, et al. Age-dependent changes in long-term seizure susceptibility and behavior after hypoxia in rats. Epilepsia. 1992;33:971–980. doi: 10.1111/j.1528-1157.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Kovacs R, et al. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J Neurophysiol. 2002;88:2909–2918. doi: 10.1152/jn.00149.2002. [DOI] [PubMed] [Google Scholar]
- Kudin AP, et al. Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. Eur J Neurosci. 2002;15:1105–1114. doi: 10.1046/j.1460-9568.2002.01947.x. [DOI] [PubMed] [Google Scholar]
- Kunz WS. The role of mitochondria in epileptogenesis. Curr Opin Neurol. 2002;15:179–184. doi: 10.1097/00019052-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liang LP, et al. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–570. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- Liang LP, et al. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. J Neurosci. 2007;27:4326–4333. doi: 10.1523/JNEUROSCI.0019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang LP, et al. Chelation of mitochondrial iron prevents seizure-induced mitochondrial dysfunction and neuronal injury. J Neurosci. 2008;28:11550–11556. doi: 10.1523/JNEUROSCI.3016-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free Radic Biol Med. 2004;36:542–554. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med. 2006;40:316–322. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Lynn S, et al. Selective neuronal vulnerability and inadequate stress response in superoxide dismutase mutant mice. Free Radic Biol Med. 2005;38:817–828. doi: 10.1016/j.freeradbiomed.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Mecocci P, et al. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- Melov S, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, et al. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Meredith MJ, Reed DJ. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J Biol Chem. 1982;257:3747–3753. [PubMed] [Google Scholar]
- Mustafa AG, et al. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med. 2004;37:1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Patel M, et al. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- Patel MN. Oxidative stress, mitochondrial dysfunction, and epilepsy. Free Radic Res. 2002;36:1139–1146. doi: 10.1080/1071576021000016391. [DOI] [PubMed] [Google Scholar]
- Sawa T, et al. Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J Biol Chem. 2000;275:32467–32474. doi: 10.1074/jbc.M910169199. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Sellevold OF, et al. High performance liquid chromatography: a rapid isocratic method for determination of creatine compounds and adenine nucleotides in myocardial tissue. J Mol Cell Cardiol. 1986;18:517–527. doi: 10.1016/s0022-2828(86)80917-8. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Tieu K, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotti D, et al. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- Trova MP, et al. Superoxide dismutase mimetics. Part 2: synthesis and structure-activity relationship of glyoxylate- and glyoxamide-derived metalloporphyrins. Bioorg Med Chem. 2003;11:2695–2707. doi: 10.1016/s0968-0896(03)00272-4. [DOI] [PubMed] [Google Scholar]
- Waldbaum S, et al. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J Neurochem. 2010;115:1172–1182. doi: 10.1111/j.1471-4159.2010.07013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, et al. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological and biochemical characterization of a mitochondrial DNA disease. Cell. 1988;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- Williams PA, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.