

Abstract
We previously demonstrated that the ATP/PKA‑dependent activation of the human intermediate conductance, Ca2+‑activated K+ channel, hIK1, is dependent upon a C‑terminal motif. The NH2‑terminus of hIK1 contains a multi‑basic 13RRRKR17 motif, known to be important in the trafficking and function of ion channels. While individual mutations within this domain have no effect on channel function, the triple mutation (15RKR17/AAA), as well as additional double mutations, result in a near complete loss of functional channels, as assessed by whole‑cell patch‑clamp. However, cell‑surface -immunoprecipitation studies confirmed expression of these mutated channels at the plasma membrane. To elucidate the functional consequences of the 15RKR17/AAA mutation we performed inside‑out patch clamp recordings where we observed no difference in Ca2+ affinity between the wild‑type and mutated channels. However, in contrast to wild‑type hIK1, channels expressing the 15RKR17/AAA mutation exhibited rundown, which could not be reversed by the addition of ATP. Wild-type hIK1 channel activity was reduced by alkaline phosphatase both in the presence and absence of ATP, indicative of a phosphorylation event, whereas the 15RKR17/AAA mutation eliminated this effect of alkaline phosphatase. Further, single channel analysis demonstrated that the 15RKR17/AAA mutation resulted in a four‑fold lower channel open probability (Po), in the presence of saturating Ca2+ and ATP, compared to wild‑type hIK1. In conclusion, these results represent the first demonstration for a role of the NH2‑terminus in the second messenger‑dependent regulation of hIK1 and, in -combination with our previous findings, suggest that this regulation is dependent upon a close NH2/C‑terminal association.
Keywords : hIK1, KCa3.1, regulation, potassium channel, trafficking
Introduction
The human intermediate conductance, Ca2+‑activated K+ channel (hIK1) is known to play crucial roles in a wide array of physiological processes.1‑6 To fully appreciate the role of these channels in eliciting these physiological effects requires an understanding of the regulatory pathways involved in channel activation and inhibition. Recent reports have highlighted the role of the cytoplasmic C‑terminus in both second messenger‑dependent regulation and trafficking of hIK1. Indeed, the calmodulin binding domain,7,8 ATP/PKA‑dependent regulatory domain9,10 and PKC‑dependent regulatory domain11 have all been mapped to the C‑terminus of hIK1. In addition to these regulatory domains, Joiner et al.,12 demonstrated that the binding of calmodulin to the proximal C‑terminal tail of hIK1 was required for the efficient assembly and trafficking of hIK1 to the plasma membrane. Recently, our laboratory demonstrated that a leucine zipper in the distal C‑terminal tail of hIK1 was required for correct trafficking of hIK1 to the cell surface and that this effect was likely due to the self‑assembly of the leucine zipper into a coiled‑coil domain.13
In contrast to these studies on the C‑terminus of hIK1, the role of the cytoplasmic NH2‑terminus in the regulation and trafficking of hIK1 has been little studied. We recently demonstrated that the NH2‑terminus contains a series of leucines critical to channel tetramerization and trafficking.14 In addition to this hydrophobic domain, the NH2‑terminus of hIK1 also includes a multi‑basic motif (13RRRKR17). Multi‑basic motifs have been shown to be important in ER retention/retrieval of KATP channels and the GABAB receptor15,16 as well as ER exit and basolateral sorting of the betaine -transporter in epithelial cells.17 Importantly, NH2‑terminal basic clusters have also been shown to be critical in controlling the gating of K+ channels, including ROMK18 and KATP.19 To date, no second messenger‑dependent regulatory domains have been mapped to the NH2‑terminus of hIK1.
In the present study we investigated the role of this NH2‑terminal RKR motif in the trafficking and function of hIK1. We -demonstrate that alanine mutations within the RKR domain do not -abrogate cell surface expression of hIK1, indicating this multi‑basic motif is not required for trafficking of the channel to the plasma membrane. In contrast, we demonstrate, using whole‑cell and inside‑out patch‑clamp techniques, that the ATP‑dependent -regulation of hIK1 is abolished following mutation of the NH2‑terminal RKR motif with no change in apparent Ca2+ affinity. This abolition of ATP -dependence results in the mutated channel having a -dramatically reduced open -probability (Po) compared to wild-type hIK1. These results represent the first demonstration for a role of the NH2‑terminus in the regulation of hIK1.
Experimental Procedures
Molecular biology.
pBF plasmid containing the cDNA for full‑length hIK1 cDNA was kindly provided by J.P. Adelman (Vollum Institute, Oregon Health Sciences University). hIK1 was subcloned into pcDNA3.1(+) (Invitrogen, Carlsbad, CA) using the EcoRI and XhoI restriction sites. A hemagglutinin (HA; YPYDVPDYA) epitope was inserted into hIK1 (HA‑hIK1) between G132 and A133, i.e., the extracellular loop between transmembrane domains S3 and S4, by sequential overlap extension PCR, as described previously.13 An HA tag was appended to the C‑terminus of hIK1 (HA‑C‑Term) in a single‑step PCR. hIK1 was tagged with a C‑terminal myc‑epitope (EQKLISEEDL) through PCR amplification as described.13 All mutations in hIK1 (R15A/K16A/R17A (RKR/AAA), R13A, R14A, R15A, K16A, R17A, R13A/R14A, R15A/K16A and R15A/R17A) were generated using the Stratagene QuikChangeä site‑directed mutagenesis strategy (Stratagene, La Jolla, CA). The fidelity of all constructs utilized in this study was confirmed by sequencing (ABI PRISM 377 automated sequencer, University of Pittsburgh) and subsequent sequence alignment (NCBI BLAST) with hIK1 (GenBankTM accession number AF022150).
Cell culture.
Human embryonic kidney (HEK293) cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum and 1% -penicillin‑streptomycin in a humidified 5% CO2/95% O2 -incubator at 37°C. Cells were transfected using LipofectAMINE 2000 (Invitrogen) following the manufacturer’s instructions. Stable cell lines were generated for all constructs by subjecting cells to -antibiotic selection (1 mg/ml G418). Note that clonal cell lines were not -subsequently selected from this stable population in order to avoid clonal -variation.
Electrophysiology.
During whole‑cell patch‑clamp experiments the bath contained (in mM) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES (pH adjusted to 7.4 with NaOH). The pipette solution contained (in mM) 130 KCl, 5 NaCl, 0.12 CaCl2, 4 MgCl2, 10 HEPES and 0.2 EGTA (pH adjusted to 7.2 with KOH) this gives a calculated free‑Ca2+ concentration of 200 nM. All experiments were performed at room temperature (22°C) using stably transfected HEK293 cells plated the previous day. Electrodes were fabricated from thin‑walled borosilicate glass (World Precision Instruments, Sarasota, FL), pulled on a vertical puller (Narishige, Long Island, NY). Currents were recorded using an Axon Instruments 200B amplifier (Axon Instruments, Foster City, CA) interfaced to a computer using a Digidata 1322A (Axon Instruments). Following establishment of the whole‑cell -configuration, voltage steps (250 ms duration) were applied between ‑100 to +80 mV from a holding potential of ‑60 mV in 20 mV -increments to generate a current‑voltage (I-V) -relationship using pCLAMP 8.2 software (Axon Instruments). Current was sampled at steady‑state (100 msec) for the purpose of evaluating current density. Current density (pA/pF) at zero mV was -calculated by dividing the current by the whole cell capacitance. Similar I‑Vs were generated following stimulation with 10 mM DCEBIO (5,6‑dichloro‑1‑ethyl‑1,3--dihydro‑2H‑benzimidazol‑2‑one;20 and inhibition with the hIK1 blocker, clotrimazole (CLT; 3 mM). The current -densities reported throughout the -manuscript are the -clotrimazole‑inhibited currents. This ensures that if any baseline current is due to hIK1 it will be accounted for in the measurements reported.
Inside‑out patch‑clamp experiments.
The effects of Ca2+, DCEBIO, ATP and alkaline phosphatase on hIK1 were assessed with excised, inside‑out patch‑clamp experiments as a functional assay. During patch‑clamp experiments, the bath solution contained 145 mM K‑gluconate, 5 mM KCl, 2 mM MgCl2, 10 mM HEPES and 1 mM EGTA (pH adjusted to 7.2 with KOH). Sufficient CaCl2 was added to obtain the desired free Ca2+ concentration (program kindly provided by Dr. Dave Dawson, Oregon Health Sciences University). To obtain a 0 Ca2+ bath solution EGTA (1 mM) was added without CaCl2 (estimated free Ca2+ < 10 nM). The pipette solution was 140 mM K‑gluconate, 5 mM KCl, 1 mM MgCl2, 10 mM HEPES and 1 mM CaCl2 (pH adjusted to 7.2 with KOH). All -experiments were performed at room temperature. All patches were held at a holding potential of ‑100 mV. The voltage is -referenced to the extracellular compartment, as is the standard method for membrane potentials. Inward currents are defined as the -movement of -positive charge from the extracellular compartment to the -intracellular -compartment and are presented as downward -deflections from the baseline in all -recordings. Immediate bath solution changes were achieved using Automate Scientifics’ (San Francisco, CA) pressurized -perfusion system and recordings were continuous to allow for the most rapid changes in current to be recorded. Single channel analysis was performed on records after low pass filtering at 400 Hz and sampling at 1 KHz. Total channel current was determined using Biopatch -software (version 3.3, Bio‑Logic) or Clampfit 8.2 (Axon Instruments).
Variance analysis.
Excised patch‑clamp recordings that were subject to variance analysis were filtered at 10kHz and digitized at 20kHz. For these studies, current recordings were initiated -immediately following patch excision into a bath containing 10 mM free Ca2+, but lacking ATP. Following channel rundown the channels were reactivated with ATP (300 mM) and subsequently DCEBIO (10 mM). The total current record was divided into 250 episodes and mean current (I) and variance (s2) were calculated for each episode using Channelab Software (Synaptosoft Inc., Decatur, GA, USA). The number of channels (N) in the patch and single channel -amplitude (i) are obtained by fitting the variance (s2) against mean current (I) -distribution to Equation 1:
 |
(1) |
The maximum open probability (Pomax) can then be calculated using Equation 2:
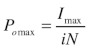 |
(2) |
where, Imax is the maximum current observed and i and N are the mean values for single channel amplitude and number of channels calculated from Equation 1. The product of i and N would give the theoretical current at Po = 1, hence, channel Po can be calculated at any given current as a proportion of Pomax.21‑24
Single channel analysis.
Amplitude histograms were compiled from 2 to 3 minutes of current trace binned in 0.1 pA intervals using pClamp8.0. To calculate Po, we initially calculated the area under each peak in the amplitude histogram (i.e., the total time spent at a current level) using Equation 3:
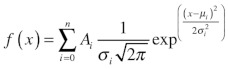 |
(3) |
where: i = 0, 1, 2, 3,……..n, are the individual current levels, A is the area under peak i, m is the mean of the Gaussian peak i and s is the Gaussian standard deviation. The area under each peak is normalized to the total area under the amplitude histogram, giving the probability of appearing in level i (Pi), where i = 0, 1, 2, 3…..n. The channel Po is calculated as NPo/n, where NPo is:
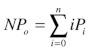 |
(4) |
where n is the predicted number of channels in the patch. We -estimated n from the highest peak observed in the amplitude -histogram under stimulation with DCEBIO. We are confident in our calculation of the total number of channels in a patch (n) as each individual level probability (Pi) is in very good agreement with a binomial distribution (see Equation 5) for the appropriate number of channels:
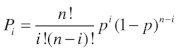 |
(5) |
where, n is the number of independent channels in the patch and i is the integer number of the amplitude histogram level (where the closed state is now level 0) and the equation is solved for p, the open channel probability.
Antibodies.
To detect hIK1 in immunoprecipitation (IP) and immunoblotting (IB) experiments, antibodies were obtained from the following sources (dilutions used are indicated): polyclonal HA (1:150) and monoclonal (1:1,000) HA (HA.11, Covance, Richmond, CA), c‑myc (clone 9E10, 1:1,000; Roche Molecular Biochemicals, Indianapolis, IN), HRP‑conjugated goat anti‑mouse IgG (1:2,000; KPL, Gaithersburg, MD) and HRP‑conjugated goat anti‑rabbit IgG (1:2,000; Transduction Laboratories, San Diego, CA).
Immunoprecipitation (IP).
For cell‑surface IP (CS‑IP), cells were grown to confluence in a 100 mm dish, washed in ice‑cold PBS, blocked in 1% BSA/PBS and labeled with polyclonal HA.11 Ab (1:500) for 90 mins at 4°C. Unbound Ab was removed by -extensive washing in 1% BSA followed by washes in PBS. All steps were performed at 4°C to prevent endocytosis of the channel and/or Ab. The cells were then lysed in IP buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1% v/v Triton X‑100, 1 mM EDTA containing CompleteTM EDTA‑free protease inhibitor cocktail mix, Roche). Protein concentrations were determined and normalized to achieve equivalent loading. Crude lysates were pre-cleared with protein A‑sepharose beads (Sigma‑Aldrich, St. Louis, MO) and incubated with rabbit polyclonal anti‑HA antibody. Immune complexes were precipitated with protein A‑sepharose beads, followed by sequential washes in IP buffer containing 500 mM, 300 mM and 150 mM (2x) NaCl, supplemented with 1x radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris‑HCl pH 7.5, 150 mM NaCl, 1% v/v Triton X‑100, 1% w/v sodium deoxycholate and 0.1% w/v SDS). After the final wash, the pellet was resuspended in Laemmli sample buffer, proteins resolved by SDS‑PAGE (12% gel) and transferred to nitrocellulose for immunoblot analysis using monoclonal HA Ab (1:1,000) as detailed below. As a control for intracellular labeling we utilized a channel in which the HA epitope was added to the -cytoplasmic C‑terminus. While this channel is expressed at high levels at the cell surface it can not be labeled in these studies unless the HA Ab has access to an intracellular compartment.
Immunoblot analysis.
HEK293 cells were grown to -confluence, lysed with IP buffer, separated by SDS‑PAGE electrophoresis, and transferred to nitrocellulose. Blots were blocked for one hour at room temperature using TBS‑blocking solution containing 5% w/v milk powder, 0.1% (v/v) Tween‑20, 0.005% (v/v) Antifoam A. Subsequently, blots were incubated in 1° Ab (mouse monoclonal HA, 1:1,000) for one hour at room temperature, extensively washed (TBS‑blocking solution deficient in milk powder) followed by -incubation in 2° Ab (1:2,000; HRP‑conjugated goat anti‑mouse IgG). The blot was then extensively washed and detection performed using West Pico Chemiluminescent Substrate (Pierce, Rockford, IL).
Chemicals.
All chemicals were obtained from Sigma‑Aldrich, unless otherwise stated. DCEBIO was synthesized in the laboratory of R.J. Bridges (University of Pittsburgh), as previously described.20 Both DCEBIO and clotrimazole were made as 10,000‑fold stock solutions in DMSO. Complete EDTA‑free protease inhibitor -cocktail mix was obtained from Roche Molecular Biochemicals.
Statistics.
All data are presented as means ± SEM, where n -indicates the number of experiments. Statistical analysis was performed using a Student’s t test. A value of p < 0.05 is considered statistically significant and is reported. All protein biochemical -experiments were carried out minimally three times on each construct to ensure the voracity of our results.
Results
An NH2‑terminal multi‑sasic RKR motif is required for the functional expression of hIK1.
While the predicted NH2‑terminus of hIK1 is only 26 amino acids long it contains a series of potentially important molecular motifs required for proper channel assembly, trafficking and function. We previously demonstrated that the NH2‑terminus of hIK1 contains a series of leucine residues required for both tetramerization as well as trafficking to the cell surface.14 In addition to these hydrophobic amino acids, the NH2‑terminus of hIK1 contains a multi‑basic 13RRRKR17 motif; a motif -previously shown to be important in ER retention,15,16 ER exit17 and channel gating.18,19 To evaluate the role of this multi‑basic motif in hIK1 we engineered a series of single, double and triple alanine-substituted mutations and determined both cell surface expression and function. Functional expression was evaluated by determining the DCEBIO‑stimulated, clotrimazole‑inhibited whole‑cell current density (pA/pF), whereas cell surface expression was determined by CS‑IP, as previously described.13,14 In wild‑type hIK1 expressing cells, DCEBIO (10 mM) increased the whole‑cell current as shown in Figure 1A. This current was subsequently inhibited by -clotrimazole (3 mM) to near baseline. For all whole‑cell recordings an average clotrimazole‑inhibited current density of 149 ± 22 pA/pF (n = 16, Fig. 1C) was recorded for wild-type hIK1 channels, which r-epresents a 610 ± 90% increase in current by DCEBIO. As shown in Figure 1C, individually mutating each of the 13RRRKR17 residues to alanine failed to affect the DCEBIO‑stimulated, clotrimazole-inhibited current density (R13A, 114 ± 23 pA/pF, n = 14; R14A, 146 ± 25 pA/pF, n = 12; R15A, 147 ± 30 pA/pF, n = 12; K16A, 162 ± 34 pA/pF, n = 14; R17A, 120 ± 17 pA/pF, n = 14). Similarly, the double mutation, R13A/R14A had no effect on total clotrimazole‑inhibited current density (104 ± 23 pA/pF, n = 14). In contrast to these results, a triple mutation of the 15RKR17 motif to alanines (RKR/AAA) resulted in a greatly diminished DCEBIO‑stimulated, clotrimazole‑inhibited response when compared to the wild-type channel (Fig. 1B); having an average current density of 1.3 ± 0.4 pA/pF (n = 12; Fig. 1C), which represents only a 20 ± 20% increase in current by DCEBIO. Similar reductions in clotrimazole‑inhibited current densities were seen with the double mutations R15A/K16A (2.0 ± 1.3 pA/pF, n = 6; p < 0.05) and R15A/R17A (6.2 ± 4.9 pA/pF, n = 6; p < 0.05). These results demonstrate a clear role for this RKR motif in the functional expression of hIK1.
Figure 1.

Mutation of the NH2‑terminal RKR motif compromises function of hIK1. Representative whole--cell current traces from (A) HA‑hIK1 or (B) RKR/AAA stably transfected in HEK293 cells in response to 10 mM DCEBIO and 3 mM clotrimazole. (C) Average DCEBIO‑stimulated (10 mM), clotrimazole‑sensitive (3 mM) current densities (pA/pF) plotted for each construct at ‑20 mV. The number of experiments is indicated in parenthesis. The asterisks indicate -statistical significance (p < 0.05). (D) Cell Surface Immunoprecipitation (CS‑IP) of HA‑hIK1 channel constructs. (Top panel) CS‑IP confirms expression of HA‑hIK1, R13A/R14A, R15A, K16A, R17A, RKR/AAA, R15A/K16A and R15A/R17A at the cell surface of HEK293 cells. (Bottom panel) Immunoblot (20 mg total protein) indicating the cellular levels of IK1 channel expression for all constructs. The immunoblots shown are representative of three separate experiments.
While the above results demonstrate a lack of functional -expression following mutation of the NH2‑terminal RKR motif, they do not distinguish between the possibility that hIK1 is expressed at the cell surface and is non-functional versus hIK1 fails to traffic to the cell surface following mutation of this RKR motif. We previously -demonstrated that insertion of an HA epitope into the second -extracellular domain of hIK1 allows us to monitor cell surface -expression by CS‑IP while having no effect on the biophysical or regulatory properties of the channel.13 Thus, we performed CS‑IP to evaluate the possibility that the RKR/AAA, R15A/K16A and R15A/R17A mutations fail to express at the cell surface. As shown in Figure 1D, HA‑hIK1 is expressed at the cell surface (lane 1). In contrast to our functional data, all of the alanine‑substituted mutations expressed at the cell surface at levels similar to wild-type, except the RKR/AAA mutation (lane 6), which was reduced to an average of 21% of wild-type (n = 4), as determined by densitometry. Thus, this 4.7 ± 2.2‑fold decrease in cell surface expression cannot explain the 30‑fold decrease in stimulated current observed between wild‑type and RKR/AAA expressing cells. Also, a similar decrease in current density was observed for the R15A/K16A and R15A/R17A -mutations, channels that exhibit a cell surface expression level similar to wild-type hIK1.
Given this reduction in cell surface expression of the RKR/AAA mutation, we determined whether incubating the cells at reduced temperature (27°C), in butyrate (5 mM) or both could increase expression. We13 and others25‑27 have previously shown that incubating cells at 27°C or in the presence of butyrate can increase expression of trafficking compromised channels. As shown in Figure 2A, reducing the incubation temperature to 27°C in the -presence or absence of butyrate increased the cell surface expression of the RKR/AAA channel. As a control for this assay, we demonstrate that a hIK1 channel tagged with a HA epitope at the cytoplasmic C‑terminus (HA‑C‑term) was not detected by cell surface IP, as would be expected for an intracellular localized epitope. However, as shown in the immunoblot (Fig. 2A), HA‑C‑term is highly expressed in these cells. Indeed, even though 10‑fold less total protein was loaded (2 mg vs. 20 mg) we detected a similar signal on IB for HA‑C‑term compared to HA‑hIK1. Whether this difference is due to a true 10‑fold difference in expression or a difference in epitope recognition it clearly demonstrates the selective nature of our CS‑IP experiments for an extracellular epitope. Despite this increased expression at the cell surface, the RKR/AAA channels failed to express functionally, as assessed by DCEBIO‑stimulated, -clotrimazole‑sensitive current density measurements (Fig. 2B; 27°C, 0.5 ± 0.4 pA/pF, n = 6; 27°C + butyrate (But), 2.0 ± 1.1 pA/pF, n = 15). In total, our results with the RKR/AAA as well as R15A/K16A and R15A/R17A -channels -demonstrate that the lack of functional expression cannot be explained by an inability of these mutated -channels to traffic to the cell surface.
Figure 2.
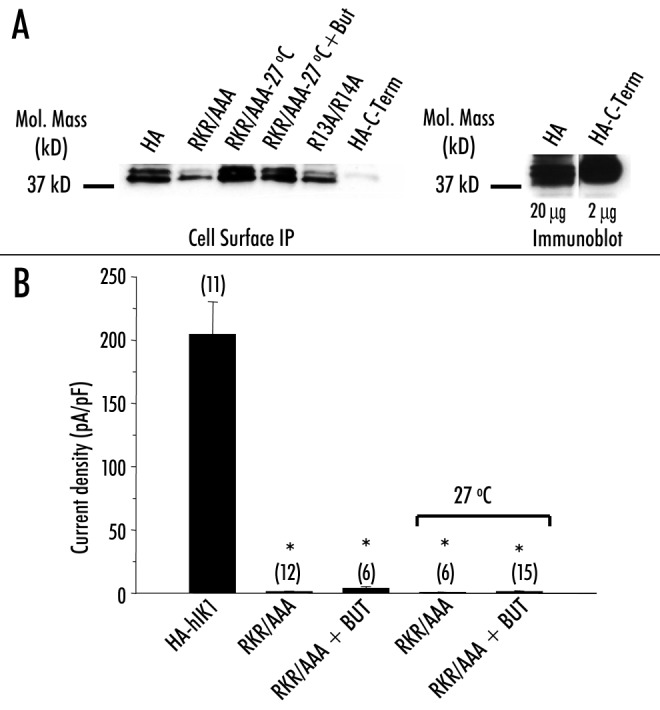
Reduced incubation temperature increases expression but not -function of RKR/AAA‑HA‑hIK1. (A, left panel) Cell surface -immunoprecipitation of HA‑hIK1 (lane 1), RKR/AAA grown at 37°C (lane 2), RKR/AAA grown at 27°C (lane 3), RKR/AAA grown at 27°C in the presence of 5 mM -butyrate (lane 4), R13A/R14A (lane 5) and a C‑terminal HA‑tagged -construct (lane 6). (A, right panel) Immunoblot of HA‑hIK1 (20 mg total protein) and HA‑C‑Term (2 mg total protein). Reduced incubation temperature (27°C) increases cell surface expression of RKR/AAA‑HA‑hIK1. HA‑C‑Term was not detected during cell surface IP confirming that only cell surface epitope is recognized. Similar results were obtained in three separate experiments. (B) Average DCEBIO‑stimulated, clotrimazole‑sensitive current density (pA/pF) for each construct at ‑20 mV. The number of experiments is indicated in parenthesis. Asterisks indicate statistical significance (p < 0.05).
Increasing Ca2+ activates RKR/AAA‑expressing hIK1 channels.
We demonstrate that expression of the RKR/AAA mutation results in a dramatically reduced DCEBIO‑stimulated, clotrimazole‑inhibited current density compared to wild‑type channels (Fig. 1B and C). This altered response could be caused by (a) an inability of the channel to respond to DCEBIO, (b) an inability of the channel to respond to Ca2+ and hence DCEBIO (DCEBIO activation requires Ca2+;1,28,29) or (c) another regulator of channel function is dependent upon this RKR domain. To begin to evaluate the Ca2+ dependence of the RKR/AAA expressing channels we stimulated the cells with the Ca2+ ionophore, ionomycin during whole‑cell patch‑clamp -recordings. Figure 3A shows the whole‑cell current response to ionomycin (2 mM) for a wild-type hIK1 transfected cell. Ionomycin induced a sustained increase in whole‑cell current, which was further increased by the addition of DCEBIO (10 mM). Likewise, in the RKR/AAA expressing channels ionomycin induced a sustained current response, which was further augmented by DCEBIO (Fig. 3B). In both wild‑type and RKR/AAA‑expressing cells DCEBIO induced an initial transient increase in current followed by a sustained plateau. Ionomycin (2 mM) increased the current densities of wild‑type and RKR/AAA to a similar extent (Fig. 3C, filled bars; WT, 31.4 ± 5.3 pA/pF, n = 15; RKR/AAA, 26.2 ± 7.9 pA/pF, n = 16). Importantly, after stimulating with ionomycin, DCEBIO (10 mM) induced a similar additional increase in the steady‑state current density in both wild-type‑ and RKR/AAA‑expressing cells (Fig. 3C, open bars; WT, 33.4 ± 8.4 pA/pF, n = 14; RKR/AAA, 28.2 ± 4.4 pA/pF, n = 15). These results clearly demonstrate that the RKR/AAA‑expressing hIK1 channels are capable of responding to both Ca2+ and DCEBIO.
Figure 3.
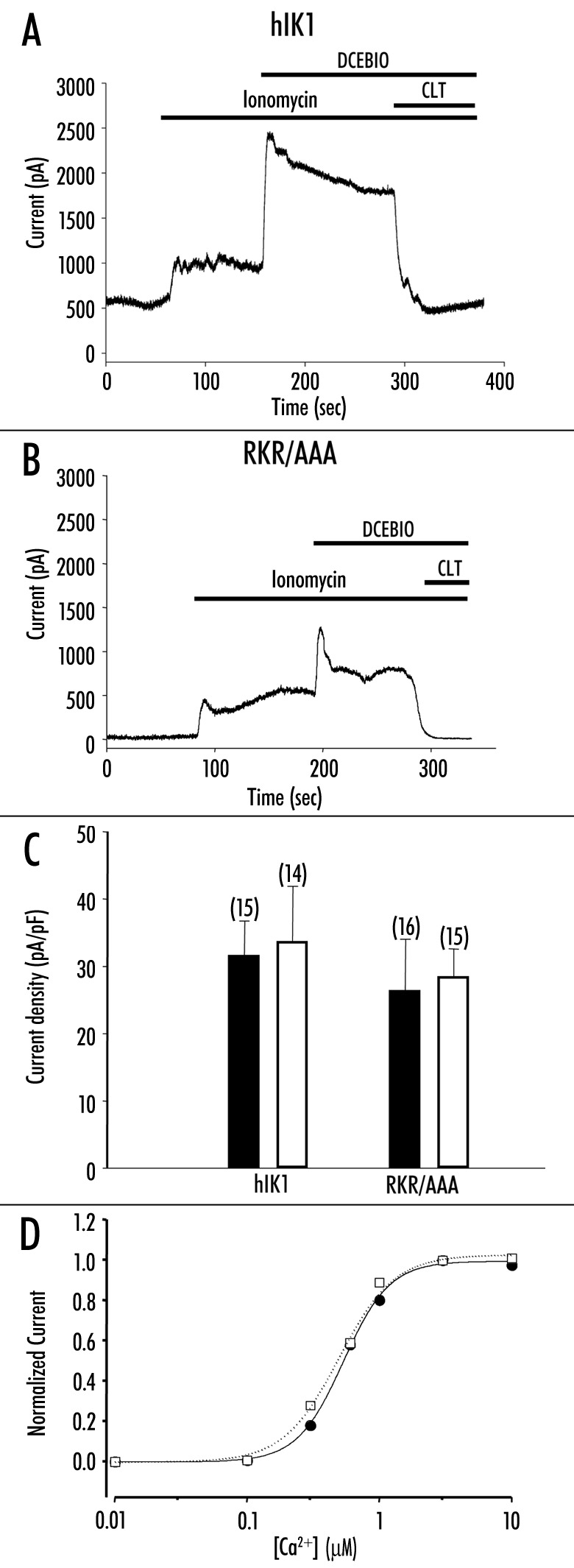
Increased Ca2+ activates RKR/AAA‑expressing channels. Representative whole-cell current traces from (A) HA‑hIK1 or (B) RKR/AAA -stably transfected HEK293 in response to 2 mM ionomycin, 10 mM DCEBIO and 3 mM clotrimazole. (C) Summary bar graph showing that ionomycin (2 mM) increases current density in both wild‑type (HA‑hIK1) and RKR/AAA‑expressing cells (solid bars). Subsequent to ionomycin, DCEBIO (10 mM) induces an equivalent increase in current density in wild‑type and RKR/AAA‑expressing cells (open bars). The numbers of experiments are indicated in parenthesis. The values are not statistically different between HA‑hIK1 and RKR/AAA‑expressing cells. (D) Average Ca2+ -concentration‑response curves for wild‑type hIK1 (filled circles, solid line, n = 12) and RKR/AAA‑expressing (open squares, dashed line, n = 11) cells. Error bars have been omitted for clarity. The lines show the best fit to the data using Hill equation (I = [Ca2+]n/(K0.5n + [Ca2+]n)). The apparent -affinity (K0.5) and Hill coefficient were not different for wild‑type and RKR/AAA hIK1 (WT, K0.5 = 512 ± 7nM, Hill -coefficient = 2.7; RKR/AAA, K0.5 = 527 ± 29 nM, Hill coefficient = 2.7).
The apparent Ca2+ affinity of hIK1 is not altered by the RKR/AAA mutation.
While the above results demonstrate that increasing levels of Ca2+ activate the RKR/AAA‑expressing channels it is important to determine whether there is an altered apparent Ca2+ affinity induced by this mutation. For these studies, we utilized the inside‑out patch‑clamp technique and, following rundown of the channels to a new steady‑state, exposed the patch to increasing levels of Ca2+. These data were then fit to the Hill equation to determine the half‑maximal Ca2+ concentration (K0.5) and Hill coefficient for Ca2+‑dependent activation. As shown in Figure 3D, the apparent K0.5 and Hill coefficient for Ca2+ was not different between wild‑type (K0.5 = 512 ± 7 nM, Hill coefficient = 2.7 ± 0.3; n = 12) and RKR/AAA‑expressing (K0.5 = 527 ± 29 nM, Hill coefficient = 2.7 ± 0.3; n = 11) channels, indicating this mutation does not alter the Ca2+‑dependent gating of hIK1.
The RKR/AAA mutation abolishes the ATP‑dependent -activation of hIK1.
During the course of our studies designed to elucidate the Ca2+‑dependence of hIK1 activation we observed a rundown of the RKR/AAA channel activity, despite the continued presence of ATP (300 mM), following patch excision. It should be noted that this rundown is highly variable (compare Figs. 4A, 6B and 8C). As we have previously shown that this rundown can be precluded by the addition of ATP in a Mg2+‑dependent fashion, we have -hypothesized that this is due to the existence of a protein -phosphatase in the patch.10 Based on the variability of this response we have not made any attempt to quantify this rate of current decline. This rundown, in the continued presence of ATP, is -illustrated in Figure 4A for a single patch excised into saturating levels of Ca2+ (10 mM). In five patches the current averaged ‑13.3 ± 4.9 pA immediately following patch excision and this ran down to ‑1.5 ± 0.5 pA in the continued -presence of ATP. Consistent with our whole‑cell patch‑clamp -recordings, in the presence of high Ca2+ (10 mM), DCEBIO (10 mM) increased the mean current to ‑18.5 ± 9.8 pA. These results confirm our whole‑cell patch‑clamp data by demonstrating that the RKR/AAA mutation does not preclude these channels from responding to the pharmacological opener, DCEBIO. However, we were surprised by the magnitude of the rundown in current observed following patch excision in the presence of ATP. We10 previously -demonstrated that ATP greatly diminishes this rundown and reactivates the channel following rundown in wild‑type hIK1 channels. This activation of wild‑type hIK1 by ATP is shown for a single recording in Figure 4B. In four patches, ATP increased the mean current an average of 4.1 ± 0.9‑fold; similar to the 3.5‑fold increase in current we -previously reported for hIK1.9 We next -determined whether ATP (1 mM) could -reactivate RKR/AAA‑expressing channels following rundown, similar to wild‑type channels. A representative current trace is shown in Figure 4C. For these experiments the channels were excised into 10 mM Ca2+ in the absence of ATP. Following rundown of current, ATP was perfused onto the patch. As is apparent, ATP failed to activate the RKR/AAA‑expressing channels, although the subsequent -addition of DCEBIO (10 mM) resulted in rapid activation. In eight patches the current averaged ‑27.0 ± 2.6 pA immediately following patch -excision and this decreased to ‑9.0 ± 4.6 pA in the continued -presence of 10 mM Ca2+. The subsequent- -addition of ATP (1 mM) failed to increase channel activity (‑7.5 ± 4.7 pA), whereas DCEBIO (10 mM) dramatically increased mean current to ‑75.4 ± 33.6 pA. These results demonstrate that the NH2‑terminal RKR domain is required for the ATP‑dependent activation of hIK1. As ATP is known to alter the Ca2+‑dependent gating of hIK19 this likely explains the difference observed between our whole‑cell recordings (100 nM free Ca2+) and our excised patch‑clamp recordings (10 mM free Ca2+; see Discussion).
Figure 4.

ATP fails to activate RKR/AAA‑expressing hIK1 channels. (A) Following patch excision into 10 mM free Ca2+ plus 1 mM ATP, RKR/AAA‑expressing channels exhibit a rapid decrease in current, so‑called “rundown.” Subsequent addition of DCEBIO (10 mM) increases current which is reversed upon -washout. (B) Following patch excision into 10 mM free Ca2+, in the absence of ATP, wild‑type hIK1 channels exhibit a slow rundown. Removal of Ca2+ from the bath results in an immediate inhibition of channel activity, which is restored upon re-addition of Ca2+. The subsequent addition of ATP (1 mM) results in a large increase in channel activity, which is completely inhibited by returning to zero Ca2+. (C) Excising RKR/AAA‑expressing channels into 10 mM free Ca2+, in the absence of ATP, results in a rapid rundown of channel activity. Addition of ATP (1 mM) does not increase channel activity, although the subsequent addition of DCEBIO (10 mM) results in a large increase in channel activity, which is reversed following washout of DCEBIO.
Figure 6.

Patches from HEK293 cells stably transfected with either (A) HA‑hIK1 or (B) RKR/AAA cells are excised into a bath containing 10 mM free Ca2+. The -channels are further activated with 10 mM DCEBIO. The bath is then switched to 200 nM Ca2+ and subsequently exposed to 10 mM DCEBIO. (B) The RKR/AAA channels cannot reactivate at low Ca2+. (C) DCEBIO‑stimulated increase in current for wild-type (n = 17) and RKR/AAA (n = 7) hIK1 in the -presence of either 10 mM (blue bars) or 200 nM (green bars) free Ca2+.
Figure 8.

Variance analysis of wild-type and RKR/AAA hIK1. (A) Following patch excision of wild-type hIK1 into a bath containing 10 mM free Ca2+ current decreased over time until a new steady‑state was reached. The subsequent addition of both ATP (1 mM) and DCEBIO (10 mM) resulted in an increase in current flow. Current traces have been data reduced for -display. Spike artifacts during solution changes were removed prior to analysis as they -produce large deviations during variance analysis. (B) Plot of variance (s2) against mean -current (I) for the current record shown in (A). The data were fitted to equation 1 yielding an N of 1,907 ± 45 channels and a single channel amplitude (i) of 1.90 ± 0.03 pA. These data allowed us to calculate a PoMAX of 0.64 for this recording using equation 2. (C) Excised patch‑clamp recording of RKR/AAA hIK1 demonstrating a loss of ATP‑dependent activation. (D) Plot of variance (s2) against mean current (I) for the current record shown in (C). The data were fitted to equation 1 yielding an N of 136 ± 5 channels and a single channel amplitude (i) of 2.87 ± 0.05 pA. These data allowed us to calculate a PoMAX of 0.61 for this recording using equation 2.
The RKR/AAA mutation abolishes the alkaline -phosphatase‑ dependent inhibition of hIK1.
The above results suggest that the RKR/AAA mutation eliminates the kinase‑dependent regulation of hIK1 which we previously described,9,10 resulting in a channel with a marked reduction in Po. As we previously demonstrated that this ATP‑dependent activation of hIK1 could be reversed by alkaline phosphatase, indicative of a kinase‑mediated event,10 our present results would predict that the RKR/AAA‑expressing channels would be insensitive to alkaline phosphatase. Initially we confirmed the alkaline phosphatase‑dependence of wild-type hIK1. As shown in Figure 5A, following patch excision in the absence of ATP and the establishment of a steady‑state current, wild-type hIK1 activity was further decreased by the addition of alkaline phosphatase (5 --U/ml). In seven experiments, the steady‑state current averaged 118 ± 19 pA which was reduced to 53 ± 14 pA following addition of alkaline phosphatase. In a separate series of experiments (Fig. 5C), channel activity was increased by the addition of ATP (1 mM). The -subsequent -addition of alkaline phosphatase (10 U/ml) again dramatically reduced this current. In six experiments the steady‑state current averaged 113 ± 34 pA and this was increased to 146 ± 50 pA by the addition of ATP. Further addition of alkaline phosphatase (10 U/ml) reduced this current to 44 ± 9 pA, similar to what we -previously reported.10
Figure 5.

Patches from HEK293 cells stably transfected with either HA‑hIK1 (A and C) or RKR/AAA (B and D) are excised into a bath containing 10 mM free Ca2+. Note that the recordings shown in this figure were initiated subsequent to the rundown of channel activity. (A) Following the establishment of a steady‑state -current wild-type hIK1 channel activity was further decreased in the presence of alkaline phosphatase (5 U/ml). (B) Alkaline phosphatase (5 U/ml) has no effect on RKR/AAA channels, although 0 Ca2+ eliminates channel activity. (C) Wild-type hIK1 channels are activated by ATP (1 mM) in the -presence of -saturating Ca2+ and this effect is reversed by the subsequent addition of alkaline phosphatase (10 U/ml) in the continued presence of ATP. (D) RKR/AAA‑expressing channels are not activated by ATP (1 mM) and the subsequent addition of alkaline phosphatase (10 U/ml) has no effect on channel activity.
In contrast to the above results, following rundown of the RKR/AAA‑expressing channels the current was not increased by ATP and was not inhibited by alkaline phosphatase (Fig. 5B and D). In five experiments the current averaged 19 ± 3 pA and this was not altered by alkaline phosphatase (5 U/ml; 18 ± 2 pA). In an additional five experiments the current was not increased by ATP (control, 11 ± 2 pA; ATP, 11 ± 2 pA) nor reduced by the subsequent addition of alkaline phosphatase (10 U/ml; 11 ± 2 pA). These results clearly demonstrate that the RKR/AAA mutation eliminates the ATP‑ and alkaline phosphatase‑dependent regulation of hIK1.
The activation of RKR/AAA by DCEBIO is Ca2+ dependent.
We demonstrate that wild-type hIK1 expressing cells respond to DCEBIO in the presence of both low levels of intracellular Ca2+ (200 nM, Fig. 1) as well as in the presence of elevated Ca2+ (-ionomycin stimulated, Fig. 3). In contrast, RKR/AAA‑expressing cells only respond to DCEBIO in the presence of elevated Ca2+ (ionomycin, Figs. 1 and 3) during whole‑cell recording. To extend these observations, we utilized the inside‑out patch‑clamp technique where the DCEBIO dependence on Ca2+ levels can be more directly evaluated in wild-type‑ and RKR/AAA‑expressing cells. As shown in Figure 6A, excising wild-type hIK1 into -saturating Ca2+ (10 mM) results in actively gating channels, which can be further activated by DCEBIO, as previously reported.29 This -activation averaged 3.8 ± 1.8-fold in 17 patches. Following reduction in bath Ca2+ to 200 nM channel activity was significantly decreased. Once more, the addition of DCEBIO resulted in a large increase in channel activity, averaging 4.9 ± 0.8-fold (n = 17). As shown in Figure 6B, DCEBIO similarly activated RKR/AAA in the presence of saturating Ca2+ (10 mM), averaging 3.9 ± 1.4-fold in seven patches. However, following reduction of free Ca2+ to 200 nM, DCEBIO failed to increase RKR/AAA channel activity (1.1 ± 0.1‑fold, n = 7). These results are -consistent with our whole‑cell data and -demonstrate that the -activation of RKR/AAA by DCEBIO is -dependent upon elevated Ca2+.
The activation of hIK1 by DCEBIO is ATP dependent.
We demonstrate: (a) ATP fails to activate RKR/AAA (Fig. 4); (b) alkaline phosphatase does not alter RKR/AAA -function (Fig. 5), suggesting that RKR/AAA‑expressing channels are not phosphorylated; and (c) that the activation of RKR/AAA by DCEBIO is dependent upon the level of free Ca2+ in the bath (Fig. 6). This raises the question of whether these observations are directly linked. That is, we have previously shown that ATP activates hIK1 by increasing the channel open probability (Po) at all levels of Ca2+ while not shifting apparent Ca2+ affinity.10 Here we demonstrate that RKR/AAA fails to respond to ATP, i.e., it is behaving in a manner analogous to hIK1, which has not been exposed to ATP. Thus, RKR/AAA‑expressing -channels would have a lower Po at all levels of Ca2+ than wild-type hIK1 in the -presence of ATP. We previously demonstrated that the activation of hIK1 by EBIO is dependent upon the channel being actively gating in the presence of Ca2+, i.e., EBIO can not activate hIK1 in the absence of Ca2+.1 Thus, we would propose that following de‑-phosphorylation, hIK1 would have a reduced response to DCEBIO, analogous to the RKR/AAA channels. To test this hypothesis we determined the effect of alkaline phosphatase (5 U/ml) on hIK1 channel activity in the -presence of DCEBIO. As shown in Figure 7, decreasing free Ca2+ from 10 mM to 200 nM resulted in a large decrease in channel activity which was stimulated 4.8 ± 1.4‑fold by DCEBIO. The subsequent addition of alkaline phosphatase reduced channel activity such that the response above baseline was only 1.3 ± 0.6 fold (n = 9). This result is consistent with the notion that the RKR/AAA channels are analogous to a -de‑phosphorylated wild-type hIK1 channel such that their Po is dramatically reduced. This reduced Po results in a channel, which cannot respond to DCEBIO, as we have previously reported for wild‑type hIK1 in the absence of Ca2+.
Figure 7.

Alkaline phosphatase inhibits hIK1 response to DCEBIO. The patch is excised from a wild‑type HA‑hIK1 expressing HEK cell into a bath -containing 10 mM free Ca2+. The subsequent reduction in free Ca2+ to 200 nM decreased current, and this activity could be increased with the addition of DCEBIO (10 mM). The further addition of alkaline phosphatase (5 U/ml) results in a loss of channel activity (n = 9).
RKR/AAA channels have a reduced Po compared to wild‑type hIK1.
The above results suggest that the RKR/AAA channels will have a reduced Po compared to wild-type hIK1. Unfortunately, patches expressing wild-type hIK1, where the number of individual channels can be accurately determined are rarely observed in our cell line and so standard methods of estimating channel Po cannot be utilized. Thus, we used variance analysis to estimate channel Po as the channel was activated first with ATP and then DCEBIO (see Methods). A representative experiment for wild-type hIK1 is shown in Figure 8A. Following patch excision into 10 mM Ca2+, in the absence of ATP, channel activity decreased over time until it reached a steady state. Addition of ATP (300 mM) increased channel activity as previously described,9,10 and this was further increased by DCEBIO. The variance plot for this channel recording is shown in Figure 8B. As is apparent, the variance goes through a maximum such that the maximum Po (PoMAX) for this recording is 0.64 in the presence of DCEBIO. In 6 patches the PoMAX in the presence of DCEBIO -averaged 0.69 ± 0.02, with a single channel amplitude of 2.1 ± 0.1 pA. Based on this PoMAX, we are able to calculate a Po in the presence of 10 mM Ca2+ and ATP of 0.38 ± 0.03 and a Po in the presence of only 10 mM Ca2+of 0.10 ± 0.06.
In contrast to wild-type hIK1, ATP did not induce a -significant increase in current in RKR/AAA, in the presence of 10 mM Ca2+, whereas DCEBIO stimulated a large response (Fig. 8C). An -analysis of variance for this recording demonstrates a PoMAX of 0.61 (Fig. 8D). In four patches, the PoMAX for RKR/AAA -averaged 0.63 ± 0.05, which is not significantly different from wild-type hIK1. However, based on these measurements, the calculated Po for RKR/AAA in the presence of 10 mM Ca2+ and ATP is only 0.11 ± 0.03, a value significantly less than wild-type hIK1 (p < 0.001). This value is not different from that in the presence of 10 mM Ca2+ alone (Po = 0.09 ± 0.02). Indeed, this value is the same as that predicted for wild-type hIK1 in the absence of ATP (0.10 ± 0.06), further suggesting that the RKR/AAA -mutation behaves similarly to wild-type hIK1, which has not been exposed to ATP.
We were able to obtain three patches on RKR/AAA where we could accurately count the number of channels in the presence of DCEBIO and thus directly estimate Po. Figure 9 shows the results from one patch containing 12 channels. In the absence of DCEBIO only three channel levels were observed both in the channel record (Fig. 9A) and the amplitude histogram (Fig. 9B). Following DCEBIO addition (10 mM), channel activity was dramatically increased and 12 channel levels could be discerned in the -amplitude histogram. The dashed line shows the fits to the individual peaks in the amplitude histogram while the colored lines show the sum of these individual peaks for ATP (red) and DCEBIO (blue). We are confident that we are not underestimating the number of -channels in the patch (n) as each individual level probability (Pi) is in -excellent agreement with a binomial distribution (see Equation 5). Based on these data, we calculate a Po in the absence of DCEBIO (in the -presence of ATP) of 0.036, which is increased to 0.603 in the -presence of DCEBIO. In four patches the average Po increased from 0.044 ± 0.004 in the absence of DCEBIO to 0.658 ± 0.028 in the presence of DCEBIO. These values are not significantly different from those obtained using variance analysis, confirming the utility of this approach for multi‑channel patches expressing hIK1. These results further -demonstrate that the RKR/AAA mutation, in the presence of ATP, functions in a manner analogous to wild type hIK1 in the absence of ATP.
Figure 9.
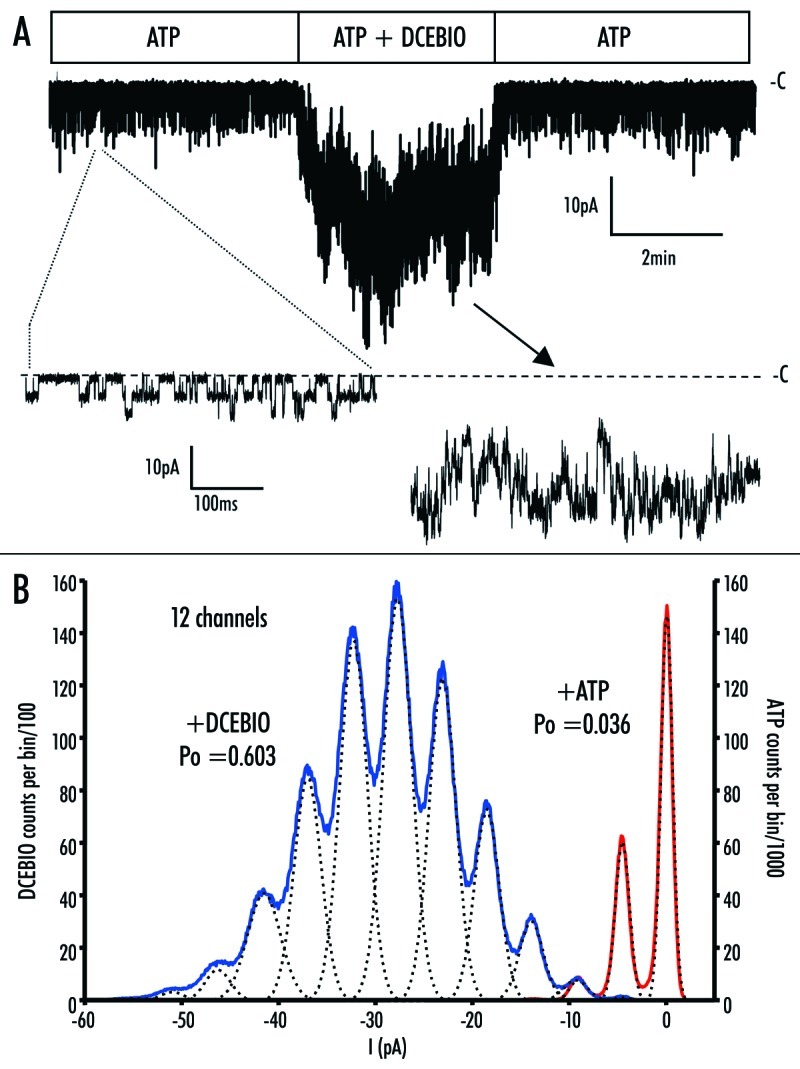
(A) Excised patch of RKR/AAA hIK1 showing activation by DCEBIO (10 mM). Expanded current traces in the presence of ATP (300 mM) and DCEBIO are shown where the dashed line indicates the closed state (‑C) of the channel. Changes in seal resistance were compensated for and removed by processing current trace with Biopatch software. Current spike artifacts during solution change have been removed. (B) Amplitude histogram of the recording shown in (A). A bin size of 0.1 pA was used and the data were fit as described in the Methods. The red trace shows the gaussian fit to the current in the presence of ATP, whereas the blue trace shows the gaussian fit to the current trace in the presence of DCEBIO. The dashed lines are the fits to the individual peaks in the amplitude histogram. Based on these fits we calculated open probabilities (Po) of 0.036 and 0.603 for RKR/AAA hIK1 in the presence of ATP and DCEBIO, respectively.
Discussion
While the NH2‑terminus of hIK1 is predicted to be only 26 amino acids long it contains numerous -well‑defined motifs, including a leucine zipper, -di‑leucine and multi‑basic motifs, known to play crucial roles in the assembly, regulation and -trafficking of a host of proteins. Indeed, we recently defined a role for both the NH2‑terminal leucine zipper and di--‑-leucine motifs in the assembly and trafficking of hIK1.14 While hIK1 possesses a -cytoplasmic NH2‑terminal multi‑basic motif (RRRKR) closely apposed to the predicted first transmembrane domain,30,31 the role of this domain has not -previously been evaluated. Multi‑basic motifs are known to play crucial roles in a wide array of cell biological processes, including ER exit,17,32 ER retention,15,16 acting as nuclear -localization signals33,34 as well as the gating of K+ channels.18,19 Thus, the role these multi‑basic motifs play in protein trafficking and function is dependent upon the context in which they are expressed. Here we demonstrate that the ATP‑dependent activation and -alkaline -phosphatase‑dependent inhibition of hIK1 is dependent upon a multi‑basic RKR motif. That is, mutating this motif results in a channel with a dramatically reduced Po such that it behaves like wild-type hIK1, which has not been exposed to ATP. As we have previously demonstrated a role for the C‑terminus in the ATP‑dependent -activation of hIK1, our results are consistent with the -hypothesis that the ATP‑dependent regulation of hIK1 is conferred by the expected close apposition of the NH2‑ and C‑termini, similar to what has been described for the SK channels.35
We initially demonstrate that double and triple mutations of the 15RKR17 domain of hIK1 result in a near complete loss of channel activity, as assessed by DCEBIO‑induced, -clotrimazole‑sensitive whole‑cell current density measurements (Fig. 1C). We further demonstrate that mutation of the two -arginines preceding this RKR motif (R13A/R14A) has no effect on channel function (Fig. 1) suggesting these additional basic amino acids are not part of the motif identified. The lack of a functional response to DCEBIO in these experiments could be the result of several possibilities: First, mutation of this RKR domain could result in the expression of a -trafficking defective channel such that the channel fails to express at the cell surface. However, as shown in Figure 1D, the R15A/R17A and R15A/K16A channels were expressed at the cell surface at wild‑type levels, although no response to DCEBIO was observed. Additionally, the 4.7‑fold decrease in cell surface channel expression, as assessed by cell surface IP, can not account for the 30‑fold decrease in current response observed for the RKR/AAA‑expressing cells. Finally, while the RKR/AAA mutation resulted in both diminished protein -expression as well as cell surface -expression this was completely restored by incubating the RKR/AAA expressing channels at 27°C in either the presence or absence of butyrate (Fig. 2A). Despite this increased cell surface expression, the RKR/AAA‑expressing channels failed to respond to DCEBIO during whole‑cell patch‑clamp recording (Fig. 2B). Together, these results indicate that the lack of a functional DCEBIO response during these whole‑cell -recordings is not the result of altered channel -trafficking or expression. Nevertheless, we cannot completely exclude the possibility that the decreased expression observed for the RKR/AAA mutation may in some way alter channel function.
A more likely scenario to explain the lack of a DCEBIO response, despite cell surface expression, is that these NH2‑terminal basic amino acids are necessary for the correct gating of hIK1. This could come about in at least three ways: First, the channels, although trafficking to the plasma membrane, may be functionally dead and therefore incapable of carrying current. However, we demonstrate in both whole‑cell (Fig. 3) and excised patch‑clamp (Figs. 3, 5 and 6) -recordings that the RKR/AAA‑expressing channels respond to changes in Ca2+, indicative of an actively gating channel. Second, although the Ca2+‑dependent gating of these -channels is conveyed via the binding of Ca2+ to calmodulin,36,37 the NH2‑terminus may modify this Ca2+‑dependent gating. This hypothesis is refuted by our -demonstration that the apparent K0.5 and Hill coefficient for Ca2+‑dependent gating are not different between wild‑type and RKR/AAA‑expressing -channels (Fig. 3D).
Third, as shown in Figures 4 and 8, the -dramatically reduced gating potential of RKR/AAA‑expressing hIK1 -channels is the result of an inability of these channels to correctly respond to ATP. This result raises the question of why a lack of an ATP response results in a channel which cannot respond to DCEBIO at low Ca2+ -concentrations (Figs. 1 and 6), but which does respond to DCEBIO at high levels of Ca2+ (Figs. 3, 4, 6 and 8)? We previously -demonstrated that (a) hIK1 exists in a phosphorylated state following patch -excision, (b) the open probability (Po) of hIK1 could be reduced approximately 50% by exogenous -phosphatases, and (c) ATP activates hIK1 via a kinase-‑dependent -mechanism.9,10 This ATP‑dependent activation of hIK1 involved an increase in Po at all levels of Ca2+ with no change in the apparent K0.5 or Hill -coefficient for Ca2+ activation.10 Here, we -demonstrate that the RKR/AAA mutation has no effect on the Ca2+‑dependence of channel gating (Fig. 3D). Thus, we interpret our data to -indicate that the RKR/AAA mutation is functionally equivalent to a dephosphorylated channel, i.e., current is dramatically reduced with no change in Ca2+‑dependent gating. We, -therefore, propose that the inability of the RKR/AAA channels to respond to DCEBIO during our whole‑cell patch‑clamp experiments is based upon the extremely low Po expected for hIK1 in 200 nM free Ca2+ (pipette solution) in a de‑phosphorylated (non-ATP) state. As Ca2+‑mediated channel activity is a prerequisite for activation by EBIO1,28,29 the -compromised response to ATP would be expected to impinge upon the DCEBIO‑dependent activation at these resting levels of Ca2+. This proposal is supported by a series of experiments. First, while wild-type hIK1 is activated by DCEBIO to a similar extent (3.8 ± 1.8 vs. 4.9 ± 0.8‑fold) in both low and high Ca2+ (Fig. 6) the response of the RKR/AAA mutation to DCEBIO is completely abrogated in low Ca2+ as channel activity is dramatically diminished at these levels of Ca2+. It is important to note that the response of the RKR/AAA mutation to DCEBIO in high Ca2+ is identical to wild-type hIK1 (3.8 ± 1.8 vs. 3.9 ± 1.4‑fold) indicating the response to DCEBIO is not compromised per se, but that the lack of response in low Ca2+ is due to the diminished channel activity exhibited by the RKR/AAA mutation. Second, if the RKR/AAA mutation behaves as a de‑phosphorylated channel then the response to DCEBIO on a wild-type channel should be reduced following the addition of alkaline phosphatase as this would mimic the RKR/AAA mutation. This is exactly what was observed (Fig. 7) and clearly demonstrates that reducing the Po of hIK1 via a de--phosphorylation mechanism will diminish the ability of DCEBIO to activate the channel. Finally, we utilized variance analysis to measure channel Po in 10 mM Ca2+, 10 mM Ca2+ plus ATP and 10 mM Ca2+ plus ATP and DCEBIO. We demonstrate that wild-type hIK1 has an Po in 10 mM Ca2+ alone that is identical to the Po of RKR/AAA in the presence of 10 mM Ca2+ plus ATP (0.10 vs. 0.11, -respectively). We confirmed this low Po for RKR/AAA directly in excised patches expressing relatively low numbers of channels (Po = 0.044), -demonstrating the utility of the variance analysis approach. Given the very low Po of RKR/AAA in saturating Ca2+ (Po = 0.04 to 0.1) this mutation would result in a completely inactive channel at resting levels of Ca2+, thus explaining the lack of response to DCEBIO in 200 nM free Ca2+ (Figs. 1 and 6). Given the -observations that (a) RKR/AAA has a Po in the presence of 10 mM Ca2+ plus ATP that is identical to wild-type hIK1 in 10 mM Ca2+ alone, (b) that RKR/AAA fails to respond to ATP or alkaline phosphatase and (c) the effect of the RKR/AAA mutation can be mimicked by de‑phosphorylating wild-type hIK1 we conclude that the multi‑basic RKR motif is required for ATP‑dependent -activation of hIK1.
It is interesting to note that Hirschberg et al.38 observed an Po of approximately 0.6 for SK2 channels in saturating Ca2+ (note however, that transitions to a low Po were spontaneously observed), whereas we observe a Po of only approximately 0.1 for hIK1 in saturating Ca2+ in the absence of additional modulators of channel activity. As Ca2+ is known to primarily alter the channel opening rate,38 this would suggest that while IK and SK channels are highly conserved throughout the pore and calmodulin binding domains the energetics involved in the Ca2+‑dependent transition between the closed and open states are markedly different. We also consistently observe a decrease in current flow across the patch following excision from the cell (present data and refs. 9 and 10), suggesting that hIK1 (or an alternate protein that modulates hIK1 function) is -phosphorylated cell‑attached. Indeed, in the phosphorylated state, hIK1 has an Po approaching that of the SK channels. Also, while the crystal structure for the calcified and apo‑calmodulin‑bound calmodulin‑binding domains have been solved,39,40 the mechanism whereby this results in channel opening, how this is distinct between hIK1 and rSK2 and how ATP influences this transition remain unclear.
It is also critical to note that the RKR/AAA mutation does not abrogate rundown of the channel following patch excision (Figs. 4, 6 and 8), although the ATP‑dependent activation and alkaline -phosphatase-dependent inhibition are lost (Figs. 4, 5 and 8). Thus, we must consider the possibility that there are two distinct regulatory events being observed in these -recordings. First, there is rundown of the channel and second there is an ATP‑dependent -activation, which we have previously shown to be kinase driven.9,10 One -possibility to explain the ATP‑independent run‑down observed in the RKR/AAA mutation (Figs. 4, 6 and 8), is that this is -dependent upon lipid mediators, such as PIP2. PIP2 is a well‑known -modulator of channel function41,42 and has been shown to depend upon positively charged amino acids in cytosolic domains.43‑45 However, we (unpublished observations) and Donald Hilgemann (personal communication) have found no effect of PIP2 on hIK1 channel function. Similarly, we found no effect of PI(3)P on hIK1 function in inside‑out patches (-unpublished -observations). Thus, these lipid mediators cannot explain the rundown observed in hIK1 that is ATP‑independent.
It is important to note that we previously reported that the ATP‑dependent activation of hIK1 was dependent upon a 14 amino acid region within the Ca2+‑dependent calmodulin‑binding domain of the C‑terminus.9 As this region of hIK1 contains no consensus phosphorylation sites, we speculated that a b‑subunit might be involved in the ATP‑dependent regulation observed. Our present results clearly demonstrate that the NH2‑terminus of hIK1 is involved in the ATP‑dependent activation of hIK1. Based on these results, we would modify our hypothesis to suggest that the ATP‑dependent activation of hIK1 involves a close association between the NH2‑ and C‑termini of the channel, although further experimentation is required to directly address this possibility.
In this regard, Skolnik and colleagues46 identified a novel b‑subunit of hIK1, MTMR6, which belongs to a family of -phosphatases that dephosphorylate the 3' position of -phosphatidylinositol 3‑phosphate (PI(3)P). Interestingly, MTMR6 was shown to interact with hIK1 via a C‑terminal leucine zipper,47 which we previously -demonstrated was required for the correct trafficking of hIK1 to the cell surface.13 Further, the MTMR6‑dependent regulation of hIK1 requires the identical 14 amino acid C‑terminal domain47 that we mapped as being critical for the ATP‑dependent activation of hIK1.9 More recently, these authors demonstrated that nucleoside diphosphate kinase B (NDK‑B) functions downstream of PI(3)P to directly -phosphorylate a histidine in this 14 amino acid C‑terminal domain.48 Thus, the ATP‑dependent regulation of hIK1 which we initially demonstrated9,10 may be due to the NDK‑B model described by Skolnik and colleagues.48 How the RKR domain described in the present work may impinge upon this novel -regulatory mechanism remains to be determined.
In conclusion, we demonstrate that mutation of an NH2‑terminal RKR domain in hIK1 results in a complete loss of ATP‑dependent activation. This results in a channel with a reduced Po such that the response to pharmacologic agents is also compromised; further highlighting the relationship between Ca2+, kinase (ATP) and -pharmacologic (DCEBIO) activators of hIK1. These results represent the first demonstration for a role of the NH2‑terminus in the second messenger‑dependent regulation of hIK1.
Acknowledgements
This work was supported by a National Institutes of Health grants (DK54941, HL083060) to Daniel C. Devor, a National Institute of Health T32 training grant (DK061296‑03) to Heather M. Jones, the University of Otago (Department of Physiology) for sabbatical support of Kirk L. Hamilton and a postdoctoral fellowship (AHA 0120544U) to Colin A. Syme.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/3999
References
- 1.Devor DC, Singh AK, Frizzell RA, Bridges RJ. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am J Physiol. 1996;271:L775–84. doi: 10.1152/ajplung.1996.271.5.L775. [DOI] [PubMed] [Google Scholar]
- 2.Devor DC, Singh AK, Lambert LC, DeLuca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J Gen Physiol. 1999;113:743–60. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards G, Gardener MJ, Feletou M, Brady G, Vanhoutte PM, Weston AH. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br J Pharmacol. 1999;128:1064–70. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, et al. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–49. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- 5.Köhler R, Degenhardt C, Kühn M, Runkel N, Paul M, Hoyer J. Expression and function of endothelial Ca(2+)-activated K(+) channels in human mesenteric artery: A single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ Res. 2000;87:496–503. doi: 10.1161/01.RES.87.6.496. [DOI] [PubMed] [Google Scholar]
- 6.Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, Adelman JP, et al. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J Biol Chem. 1998;273:21542–53. doi: 10.1074/jbc.273.34.21542. [DOI] [PubMed] [Google Scholar]
- 7.Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, et al. Domains responsible for constitutive and Ca(2+)-dependent interactions between calmodulin and small conductance Ca(2+)-activated potassium channels. J Neurosci. 1999;19:8830–8. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J Biol Chem. 1999;274:14838–49. doi: 10.1074/jbc.274.21.14838. [DOI] [PubMed] [Google Scholar]
- 9.Gerlach AC, Syme CA, Giltinan L, Adelman JP, Devor DC. ATP‑dependent activation of the intermediate conductance, Ca2+‑activated K+ channel, hIK1, is conferred by a C‑terminal domain. J Biol Chem. 2001;276:10963–70. doi: 10.1074/jbc.M007716200. [DOI] [PubMed] [Google Scholar]
- 10.Gerlach AC, Gangopadhyay NN, Devor DC. Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. J Biol Chem. 2000;275:585–98. doi: 10.1074/jbc.275.1.585. [DOI] [PubMed] [Google Scholar]
- 11.Wulf A, Schwab A. Regulation of a calcium-sensitive K+ channel (cIK1) by protein kinase C. J Membr Biol. 2002;187:71–9. doi: 10.1007/s00232-001-0149-3. [DOI] [PubMed] [Google Scholar]
- 12.Joiner WJ, Khanna R, Schlichter LC, Kaczmarek LK. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J Biol Chem. 2001;276:37980–5. doi: 10.1074/jbc.M104965200. [DOI] [PubMed] [Google Scholar]
- 13.Syme CA, Hamilton KL, Jones HM, Gerlach AC, Giltinan L, Papworth GD, et al. Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J Biol Chem. 2003;278:8476–86. doi: 10.1074/jbc.M210072200. [DOI] [PubMed] [Google Scholar]
- 14.Jones HM, Hamilton KL, Papworth GD, Syme CA, Watkins SC, Bradbury NA, et al. Role of the NH2 terminus in the assembly and trafficking of the intermediate conductance Ca2+-activated K+ channel hIK1. J Biol Chem. 2004;279:15531–40. doi: 10.1074/jbc.M400069200. [DOI] [PubMed] [Google Scholar]
- 15.Margeta-Mitrovic M, Mitrovic I, Riley RC, Jan LY, Basbaum AI. Immunohistochemical localization of GABA(B) receptors in the rat central nervous system. J Comp Neurol. 1999;405:299–321. doi: 10.1002/(SICI)1096-9861(19990315)405:3<299::AID-CNE2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22:537–48. doi: 10.1016/S0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 17.Perego C, Bulbarelli A, Longhi R, Caimi M, Villa A, Caplan MJ, et al. Sorting of two polytopic proteins, the gamma-aminobutyric acid and betaine transporters, in polarized epithelial cells. J Biol Chem. 1997;272:6584–92. doi: 10.1074/jbc.272.10.6584. [DOI] [PubMed] [Google Scholar]
- 18.Schulte U, Hahn H, Wiesinger H, Ruppersberg JP, Fakler B. pH-dependent gating of ROMK (Kir1.1) channels involves conformational changes in both N and C termini. J Biol Chem. 1998;273:34575–9. doi: 10.1074/jbc.273.51.34575. [DOI] [PubMed] [Google Scholar]
- 19.Cukras CA, Jeliazkova I, Nichols CG. The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels. J Gen Physiol. 2002;120:437–46. doi: 10.1085/jgp.20028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh S, Syme CA, Singh AK, Devor DC, Bridges RJ. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2001;296:600–11. [PubMed] [Google Scholar]
- 21.Alvarez O, Gonzalez C, Latorre R. Counting channels: a tutorial guide on ion channel fluctuation analysis. Adv Physiol Educ. 2002;26:327–41. doi: 10.1152/advan.00006.2002. [DOI] [PubMed] [Google Scholar]
- 22.Sigworth FJ. Sodium channels in nerve apparently have two conductance states. Nature. 1977;270:265–7. doi: 10.1038/270265a0. [DOI] [PubMed] [Google Scholar]
- 23.Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Traynelis SF, Jaramillo F. Getting the most out of noise in the central nervous system. Trends Neurosci. 1998;21:137–45. doi: 10.1016/S0166-2236(98)01238-7. [DOI] [PubMed] [Google Scholar]
- 25.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–65. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–4. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–6. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- 28.Singh AK, Devor DC, Gerlach AC, Gondor M, Pilewski JM, Bridges RJ. Stimulation of Cl(-) secretion by chlorzoxazone. J Pharmacol Exp Ther. 2000;292:778–87. [PubMed] [Google Scholar]
- 29.Syme CA, Gerlach AC, Singh AK, Devor DC. Pharmacological activation of cloned intermediate- and small-conductance Ca(2+)-activated K(+) channels. Am J Physiol Cell Physiol. 2000;278:C570–81. doi: 10.1152/ajpcell.2000.278.3.C570. [DOI] [PubMed] [Google Scholar]
- 30.Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci U S A. 1997;94:11651–6. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joiner WJ, Wang LY, Tang MD, Kaczmarek LK. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc Natl Acad Sci U S A. 1997;94:11013–8. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller SH, Lindstrom J, Ellisman M, Taylor P. Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-Subunit. J Biol Chem. 2001;276:18384–91. doi: 10.1074/jbc.M100691200. [DOI] [PubMed] [Google Scholar]
- 33.Munster AK, Weinhold B, Gotza B, Muhlenhoff M, Frosch M, Gerardy-Schahn R. Nuclear localization signal of murine CMP-Neu5Ac synthetase includes residues required for both nuclear targeting and enzymatic activity. J Biol Chem. 2002;277:19688–96. doi: 10.1074/jbc.M201093200. [DOI] [PubMed] [Google Scholar]
- 34.Fagerlund R, Mélen K, Kinnunen L, Julkunen I. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin alpha 5. J Biol Chem. 2002;277:30072–8. doi: 10.1074/jbc.M202943200. [DOI] [PubMed] [Google Scholar]
- 35.Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, et al. Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–58. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 36.Takahata T, Hayashi M, Ishikawa T. SK4/IK1-like channels mediate TEA-insensitive, Ca2+-activated K+ currents in bovine parotid acinar cells. Am J Physiol Cell Physiol. 2003;284:C127–44. doi: 10.1152/ajpcell.00250.2002. [DOI] [PubMed] [Google Scholar]
- 37.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–7. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 38.Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J Gen Physiol. 1998;111:565–81. doi: 10.1085/jgp.111.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher MA, Crum M, Miller MC. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure. 2004;12:849–60. doi: 10.1016/j.str.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 40.Schumacher MA, Rivard AF, Bächinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–4. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 41.Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–9. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- 42.Hilgemann DW. Cytoplasmic ATP-dependent regulation of ion transporters and channels: mechanisms and messengers. Annu Rev Physiol. 1997;59:193–220. doi: 10.1146/annurev.physiol.59.1.193. [DOI] [PubMed] [Google Scholar]
- 43.Dong K, Tang L, MacGregor GG, Hebert SC. Localization of the ATP/phosphatidylinositol 4,5 diphosphate-binding site to a 39-amino acid region of the carboxyl terminus of the ATP-regulated K+ channel Kir1.1. J Biol Chem. 2002;277:49366–73. doi: 10.1074/jbc.M208679200. [DOI] [PubMed] [Google Scholar]
- 44.Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–8. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- 45.Shyng SL, Cukras CA, Harwood J, Nichols CG. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J Gen Physiol. 2000;116:599–608. doi: 10.1085/jgp.116.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava S, Li Z, Lin L, Liu G, Ko K, Coetzee WA, et al. The phosphatidylinositol 3-phosphate phosphatase myotubularin- related protein 6 (MTMR6) is a negative regulator of the Ca2+-activated K+ channel KCa3.1. Mol Cell Biol. 2005;25:3630–8. doi: 10.1128/MCB.25.9.3630-3638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava S, Choudhury P, Li Z, Liu G, Nadkarni V, Ko K, et al. Phosphatidylinositol 3-phosphate indirectly activates KCa3.1 via 14 amino acids in the carboxy terminus of KCa3.1. Mol Biol Cell. 2006;17:146–54. doi: 10.1091/mbc.E05-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–75. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]