

Abstract
Human DNA polymerase δ (Pol δ) is involved in various DNA damage responses in addition to its central role in DNA replication. The Pol δ4 holoenzyme consists of four subunits, p125, p50, p68 and p12. It has been established that the p12 subunit is rapidly degraded in response to DNA damage by UV leading to the in vivo conversion of Pol δ4 to Pol δ3, a trimeric form lacking the p12 subunit. We provide the first analysis of the time-dependent recruitment of the individual Pol δ subunits to sites of DNA damage produced by UV irradiation through 5 μm polycarbonate filters by immunofluorescence microscopy and laser scanning cytometry (LSC). Quantitative analysis demonstrates that the recruitments of the three large subunits was near complete by 2 h and did not change significantly up to 4 h after UV exposure. However, the recruitment of p12 was incomplete even at 4 h, with about 70% of the Pol δ lacking the p12 subunit. ChIP analysis of Pol δ after global UV irradiation further demonstrates that only p125, p50 and p68 were present. Thus, Pol δ3 is the predominant form of Pol δ at sites of UV damage as a result of p12 degradation. Using LSC, we have further confirmed that Pol δ was recruited to CPD damage sites in all phases of the cell cycle. Collectively, our results show that Pol δ at the DNA damage site is the Pol δ trimer lacking p12 regardless of the cell cycle phase.
Keywords: DNA damage foci, DNA replication, POLD1, POLD4, UV damage, cyclobutane pyrimidine dimer, polymerase δ
Introduction
DNA polymerase δ (Pol δ) is a central enzyme in the replication of the eukaryotic genome. It functions in concert with PCNA, its DNA sliding clamp,1 which acts as a processivity factor. In addition, PCNA acts as a platform for coordinating the actions of other replication and repair components,2 including proteins involved in Okazaki fragment maturation and translesion synthesis, such as Fen1,3 ligase and translesion polymerases when PCNA is monoubiquitinated.4-7 In human and other mammalian cells, Pol δ is a heterotetramer consisting of p125 (the catalytic subunit), p50, p68 and p12.8-11 There is extensive conservation of both genes and mechanisms involved in DNA replication and repair from yeast to human,1,12 but significant differences do exist. Pol δ from Saccharomyces cerevisiae is a trimeric protein lacking the smallest subunit, p12.1 A homolog of p12 exists in Saccharomyces pombe13 but is non-essential for survival.14 In yeast, Pol δ and Pol ε have been assigned primary roles in lagging- and leading-strand DNA synthesis, respectively,15 but this division of labor has not been rigorously established for the replication of the much larger and more complex human genome.16
Pol δ has been implicated as an important gap-filling enzyme in DNA repair processes. Nucleotide excision repair (NER) is responsible for the removal of bulky adducts, such as those caused by UV, and involves two sub-pathways, global genomic NER and transcription-coupled NER.12 Human Pol δ activity has been shown to be involved in the gap-filling step in the late post-excision stage of repair.17-19 Pol δ has also been shown to be involved in the elongation of the invading strand in recombination processes of HR in yeast.20-22 In base excision repair (BER), Pol β is the primary DNA polymerase involved;23 however, there is evidence that Pol δ and Pol ε may participate in long patch BER.24
There has been enormous progress in understanding the cellular responses to DNA damage through elucidation of the events triggered by induction of double-stranded DNA breaks. The DNA damage response (DDR) involves the recruitment and assembly of large complexes of proteins that orchestrate and prioritize a network of responses that include DNA repair, activation of cell cycle checkpoints and the decision for apoptosis.25-28 Proteins involved in the cellular DNA damage responses as well as the DNA repair proteins are sequentially recruited to DNA damage sites to generate signaling complexes that involve both phosphorylation and ubiquitination reactions. The ATM kinase plays a central role as the apical protein kinase that initiates phosphorylation cascades in this signaling complex. In addition, a number of ubiquitination events play important roles in assembly of the complexes and in directing DNA repair.26-28
At the subnuclear level, individual DNA repair complexes are further assembled into DNA repair foci or DNA repair factories that are visible as small punctate areas under the fluorescence microscope by staining of DDR or repair factors.29,30 The analysis of the recruitment of DNA damage response and repair proteins to DNA damage foci has been an important experimental tool both for the identification of the protein factors and the determination of their ordering in the signaling cascades.
UV damage triggers the intra S-phase checkpoint that is regulated by the apical checkpoint kinase, ATR.31,32 The activation of the checkpoints leads to downregulation of DNA synthesis by inhibition of origin firing and DNA chain elongation.31,33,34 UV introduces bulky lesions such as CPDs (cyclopyrimidine butane dimers), which pose severe obstacles to DNA replication polymerases, including Pol δ. Failure to elongate past these lesions by replication polymerases leads to stalling of the replication forks, resulting in further DNA damage from collapsed replication forks, incomplete replication and ultimately cell death.6,35 Alternatively, DNA damage avoidance pathways allow these blockages to be bypassed by translesion polymerases that include Pol η, ι, κ and Rev1 and Pol ζ.36-39 Pol η is primarily responsible for the bypass of UV-induced lesions. Translesion bypass is triggered by the monoubiquitination of PCNA,40 which leads to recruitment of Pol η and switching with Pol δ. Pol δ is switched back once the bypass is accomplished, allowing replication to proceed.7,41,42
While the role of Pol δ activity in the response to DNA damage may be considered to be a passive one in that it is a late step in the repair processes, work from our laboratory has established that the Pol δ holoenzyme responds to DNA damage. The p12 subunit is degraded in response to genotoxic challenges by UV, alkylating agents and replication stress induced by hydroxyurea.43 This results in the in vivo alteration in the subunit structure of Pol δ, which is converted from the heterotetramer to a heterotrimer, Pol δ3. The process is dependent on an intact ubiquitination system and ATR but not ATM at low-dose UV.43 The properties of reconstituted human Pol δ3 provide insights as to how its formation could function as a response to DNA damage. Pol δ3 exhibits altered properties from Pol δ4 in that it exhibits a greater discrimination against bypass synthesis across DNA lesions, a greater discrimination against the extension of a mismatched primer terminus and greater proofreading activity.44 Comparison of the kinetic constants by pre-steady-state enzyme kinetics showed that Pol δ3 exhibited a decrease in kpol and an increase in kpol-exo, the rate of switching of the primer terminus from the polymerase active site to the exonuclease active site.45 These changes would endow Pol δ3 with a greater fidelity for DNA synthesis than its progenitor45 and support the concept that Pol δ3 could function as a novel defense mechanism that assists the cell in maintaining genomic integrity under DNA damage stress.
However, many aspects of the cellular behavior of Pol δ under conditions of genotoxic challenge remain to be elucidated. These include questions of whether p12 degradation takes place in all phases of the cell cycle, or whether Pol δ3 is formed and recruited to sites of DNA damage while CPDs are still present and in the process of repair. In this study, we have performed an analysis of the recruitment of all four subunits of Pol δ to sites of locally induced DNA damage using co-staining with CPDs. These results establish the physical presence of Pol δ3 at sites of UV damage. In addition, we demonstrate that Pol δ3 is the primary form of Pol δ associated with CPD damage by ChIP analysis.
Results
Subcellular localization of all four Pol δ subunits
In order to study the recruitment of the endogenous Pol δ complex, the antibodies for each subunit were optimized and characterized for use in indirect immunofluorescence (Materials and Methods). We chose indirect immunofluorescence rather than ectopic expression of tagged Pol δ subunits to avoid potential artifacts arising from the use of modified proteins or from artificially high levels of expression. Representative indirect fluorescence images of the localization of the four Pol δ subunits in A549 lung pulmonary cells are shown in Figure 1. All four Pol δ subunits exhibited a clear nuclear localization. Some variability in staining intensities was observed, with some cells exhibiting an intense, diffuse stain, while others display a less intense but more distinct punctate staining pattern (Fig. 1). However, the smallest subunit, p12, displayed a staining pattern different from the other three subunits in that a subpopulation of the cells appeared to have very little or no p12 content (Fig. 1, white arrows).
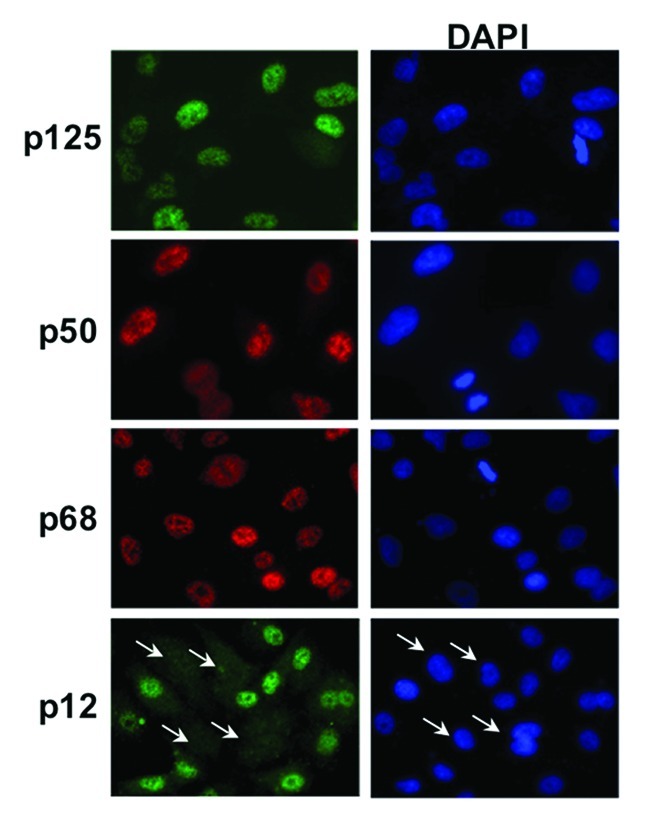
Figure 1. Indirect immunofluorescence imaging of the four Pol δ subunits. A549 cells were fixed and the subcellular localization of p125, p50, p68 and p12 subunits was determined by indirect immunofluorescence (Materials and Methods). p125 and p12 were stained with mouse monoclonal antibodies and Alexafluor488 conjugated anti-mouse IgG (green). p50 and p68 were stained with rabbit polyclonal antibodies and Rhodamine-X conjugated anti-rabbit IgG (red). The cells were co-stained with DAPI to reveal the nuclei. In the bottom panel the arrows indicate cells in which p12 was absent.
Spatiotemporal dynamics of the recruitment of Pol δ subunits to UV-damaged DNA
A major goal of this study was to determine the temporal dynamics of the UV-induced recruitment of Pol δ to sites of DNA damage. In addition, we sought to establish that Pol δ3 was present at the sites of DNA damage and therefore was involved in DNA repair by examining the behavior of each individual subunit. Cyclobutane pyrimidine dimers represent the major form of DNA damage caused by UV irradiation.46 We therefore utilized co-localization with CPDs for studying the recruitment of Pol δ subunits and other proteins to DNA damage as a direct and unambiguous indication of their recruitment to the primary DNA damage sites caused by UV irradiation.
In order to unambiguously visualize the recruitment of Pol δ to sites of DNA damage, we utilized the technique of irradiation through UV-opaque polycarbonate filters with 5 μm pores initially developed to study the recruitment of NER factors to UV damage foci.47 This method produces areas of local UV irradiation, to which recruitment of proteins can be readily visualized at low magnifications (40×).
Representative images for the co-staining of p125, p68, p50 and p12, respectively, with CPDs are shown for A549 cells 4 hours after UV (254 nm) irradiation (Fig. 2A). The recruitment of Pol δ subunits to local areas of DNA damage was readily observed by an intense staining to fairly uniform and well delineated foci (Fig. 2A). For convenience, we will refer to these areas as “macro-foci.” The three large subunits (p125, p68 and p50) are co-localized essentially to all the CPD containing macro-foci. On the other hand, the p12 subunit only exhibited partial co-localization with CPDs (Fig. 2A).
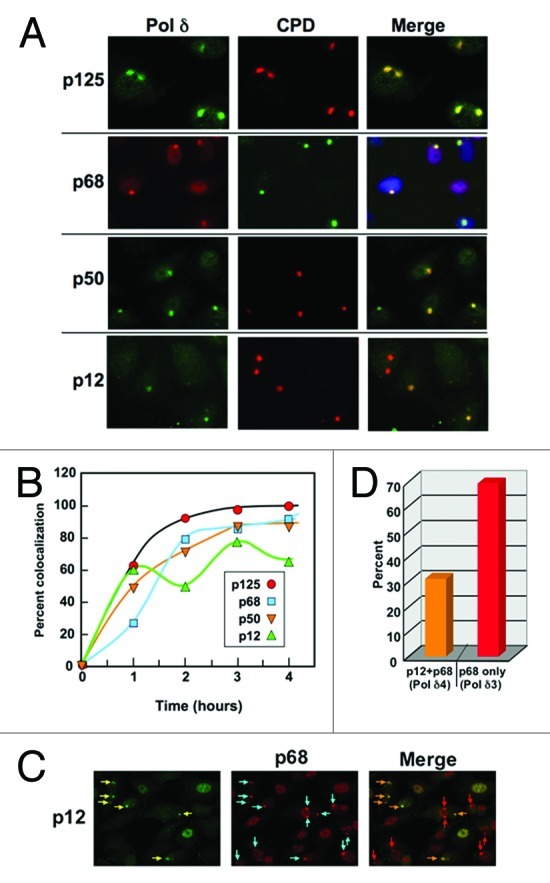
Figure 2. Recruitment of Pol δ subunits to local areas of UV-induced DNA damage. (A) A549 cells were irradiated with 75 J/m2 UV through 5 μm pore polycarbonate filters and co-stained for p125, p68, p50 and p12 with CPDs four hours after irradiation. Representative images of the co-localization of each subunit with CPDs are shown. Images for p125, p50 and p12 are shown by green fluorescence, and p68 by red fluorescence. The staining for CPDs is shown in the second column as red fluorescence, and with green in the case of p68. The merged panel for p68 also includes nuclear DAPI staining. (Images containing more cells from which the panels were taken are shown in Fig. S3). (B) A549 cells were irradiated as described above and stained after 1, 2, 3, and 4 h after UV treatment. Co-localization was performed by visual scoring of > 100 cells containing CPD foci for the percentage co-localization (p125, red circles; p68, blue squares; p50, inverted orange triangles; p12, green triangles). Representative panels that were used for counting are shown in Fig. S3). (C) p12 was co-localized with p68 in order to gain a measure of which CPDs contained all four subunits, and which lacked p12. Cells were fixed and stained four hours after irradiation as in panel A. Yellow arrows show macro-foci containing p12, cyan arrows show the presence of p68. In the merge panel, those foci containing p12 are shown with orange arrows and those without p12 are shown with red arrows. (D) The co-localization of p12 and p68 was scored for the experiment shown in panel (B). Orange bar, percentage of foci containing both p12 and p68, which was taken as an indication of the presence of Pol δ4; red bar, percentage of foci containing only p68, which was taken as an indication of the presence of Pol δ3.
The well-defined nature of the macro-foci provided a facile means for the quantitation of the recruitment of the subunits to CPD damage. This was performed by counting the co-localization of each subunit with CPDs by examining > 100 cells exhibiting the presence of macro-foci. We expressed the results as a percentage of co-localization with CPDs. This method was used to examine the recruitment of all four subunits of Pol δ at times of 0–4 h after irradiation through membrane filters at a UV fluence of 75 J/m2 (Fig. 2B). p125 was recruited to ca. 100% of the CPDs by 2 h after exposure to UV. [We have also examined the recruitment of p125 with γH2AX as a marker and found very similar results for A549 cells (Fig. S1)]. The p68 and p50 subunits exhibited similar kinetics, and they also exhibited near-complete co-localization with CPDs. The p12 subunit also exhibited similar recruitment kinetics but peaked at a lower level with an average (2, 3, 4 h) co-localization of ca. 60%, i.e., about 40% of the Pol δ appeared to be lacking p12. We have examined the staining of p12 using global irradiation of A549 cells by UV at a fluence of 20/Jm2 (Fig. S2). The nuclear staining for p12 is ablated in almost all the cells, eliminating an antibody specificity issue. The co-localization of p12 with p68 4 hours after irradiation with UV at a fluence of 75/J/m2 was examined in order to assess the relative amounts of Pol δ3 and Pol δ4 (Fig. 2C). Only a subset of the macro-foci that stained for p68 also stained for p12 (Fig. 2C). We can reasonably assume that the three large Pol δ subunits are present in all the macro-foci, so these images confirm the data of Figure 2A regarding the partial removal of p12. Quantitative scoring of the immunofluorescence data show that 30% of the macro-foci contained both p12 and p68, while 70% contained only p68 (Fig. 2D), i.e., only Pol δ3 is present in 70% of the DNA damage foci. This is higher than the value of ca. 40% obtained by co-localization with CPDs and confirms that p12 is not completely depleted. This was indeed surprising, since one might have expected that p12 would be completely degraded by 4 hours as in cells undergoing global irradiation (Fig. S2). A likely explanation is that degradation of p12 occurs only at the sites of damage, and that the observed level of p12 remains steady due to the free exchange of Pol δ4 and Pol δ3 with the nuclear pool of Pol δ (see Discussion). Nevertheless, the data provide the first direct physical evidence for the presence of Pol δ3 at sites of UV damage.
Pol δ3 is the primary form of Pol δ directly bound to damaged DNA with CPDs
In order to confirm that there is a strong association of Pol δ to damaged DNA, we examined the localization of p125 to UV damaged DNA after Triton X-100 treatment of the cells prior to fixing (Materials and Methods). The Triton X-100 pretreatment will remove proteins not directly associated with chromatin.48 In untreated cells, we observed some cells lack or contain very little amounts of p125 after in situ extraction (Fig. 3A, middle column, white arrows); these would represent cells that are not actively replicating DNA or undergoing DNA repair. In the irradiated cells, the p125 staining exhibits a clear punctate pattern throughout the nucleus and co-localizes with CPDs, which are also characterized by punctate dots that represent the repair foci (Fig. 3A, last column, blue arrows).
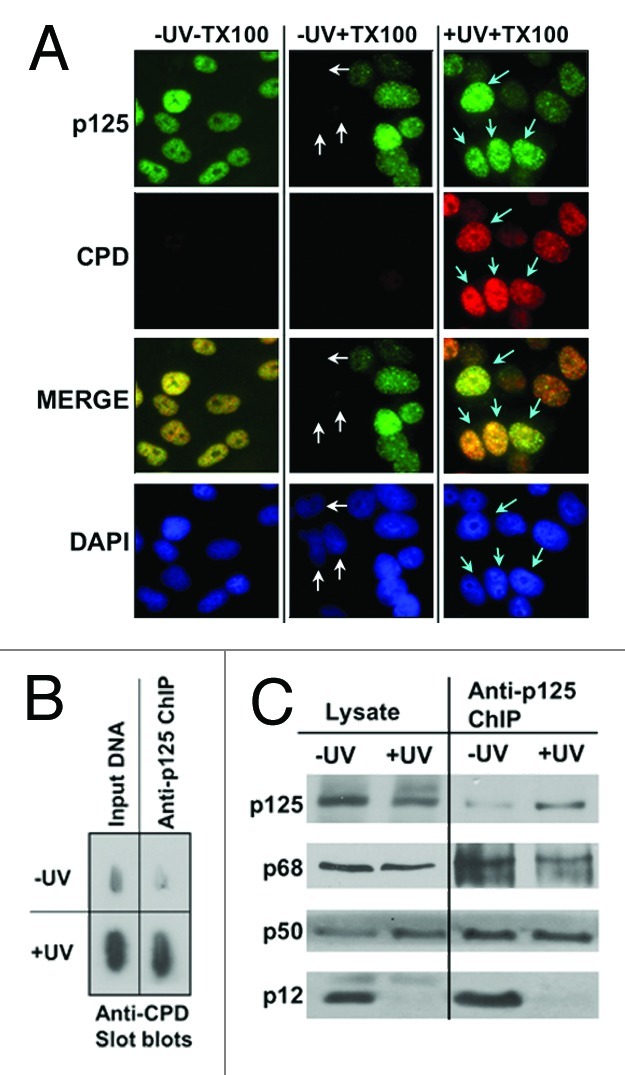
Figure 3. Pol δ is bound to CPD-damage containing DNA in UV irradiated cells and is present as Pol δ3. (A) A549 cells were globally irradiated with 20 J/m2 of UVC and extracted with a Triton-X100 based buffer in situ prior to fixing and staining for p125 (green AlexaFluor488 fluorescence) and for CPDs (Texas Red fluorescence). Cells were visualized at a magnification of 100X. The first column shows cells that were not extracted with Triton-X100 before fixing and staining, the second column cells that were pre-extracted, and the third column cells that were pre-extracted and treated with UVC. White arrows indicate a cluster of cells that show little to no nuclear p125 content in non-irradiated pre-extracted cells (second column). Aqua arrows indicate cells showing the presence of co-localization of p125 with CPDs at punctate DNA repair foci (third column). (B) A549 cells were irradiated with 20 J/m2 UVC, allowed to recover for four hours and analyzed by ChIP analysis. The panel shows slot blots of the ChIP immunoprecipitated DNA with anti-p125 for CPDs. The input DNA slots contained 10 ng DNA, and the ChIP slots contained the DNA from a similar amount of DNA immunoprecipitated with anti-p125. The blots were stained with anti-CPD. (C) western blots of the immunopreciptates for Pol δ subunits.
Chromatin immunoprecipitation (ChIP) analysis of DNA isolated from untreated and UV-treated A549 cells was used to confirm that Pol δ is in direct contact with UV-damaged DNA and to define the subunit composition of the Pol δ molecules that are in association with CPDs. The fragmented DNA containing cross-linked proteins was immunoprecipitated with antibodies against p125. A significant fraction of the CPDs was associated with the anti-125 immunoprecipitate, as shown by slot blots with anti-CPD antibody. The comparisons are from the same initial amount of fragmented DNA (Fig. 3B). Analyses of the immunoprecipitate by western blotting after release of the proteins from DNA showed that all four Pol δ subunits were present in the ChIP fraction derived from control cells, whereas only p125, p68 and p50 were present in that from UV-treated cells, with no evidence of p12 (Fig. 3C). Thus, those data provide direct confirmation that Pol δ3 alone is associated with UV-damaged DNA in UV-treated cells, underscoring the point that Pol δ3 is the only form of Pol δ present and available for DNA repair of the CPDs.
Recruitment of Pol δ to sites of DNA damage in relation to the recruitment of ATR, Pol η and Rad51
The temporal dynamics of the recruitment of p125 to the UV induced macro-foci was compared with proteins that are involved in DNA damage response and repair. These were ATR, the apical protein kinase that initiates checkpoint signaling in the intra-S phase checkpoint,31,33 Pol η, which is involved in translesion bypass of CPDs36 and Rad51, which is involved in homologous recombination.49 The recruitment of p125 was essentially complete by 2 h (Fig. 4). In comparison, ATR is rapidly recruited to the macro-foci, and reaches a maximum of ca. 100% within 30 min after exposure to UV (Fig. 4). Its rapid recruitment is expected, since ATR recruitment to DNA damage is signaled by the generation of ssDNA at stalled replication forks in the intra-S phase checkpoint.31 However, it is surprising that close to 100% recruitment is attained, since this implies that ATR is recruited to sites of UV damage in all phases of the cell cycle, even in G1 where DNA replication is not occurring. However, the UV fluence used was high, and under these conditions, multiple DNA damage pathways may be activated. In the case of Pol η, its recruitment peaks within the first hour, to a maximum of ca. 80% (Fig. 4). This suggests that Pol η recruitment to sites of UV damage is not restricted to cells active in DNA replication, since S-phase cells comprise much less than 80% of the cell population. Similar observations regarding the recruitment of Pol η have been made in G1 cells.50 Our data show that both Pol δ and Pol η recruitment is completed in a similar time frame (Fig. 4). In the case of Rad51, recruitment peaked at about 30% at 2 h after exposure to UV (Fig. 4). Since Rad51 is associated with HR, this may be due to the formation of DSBs in a subset of the cells.51
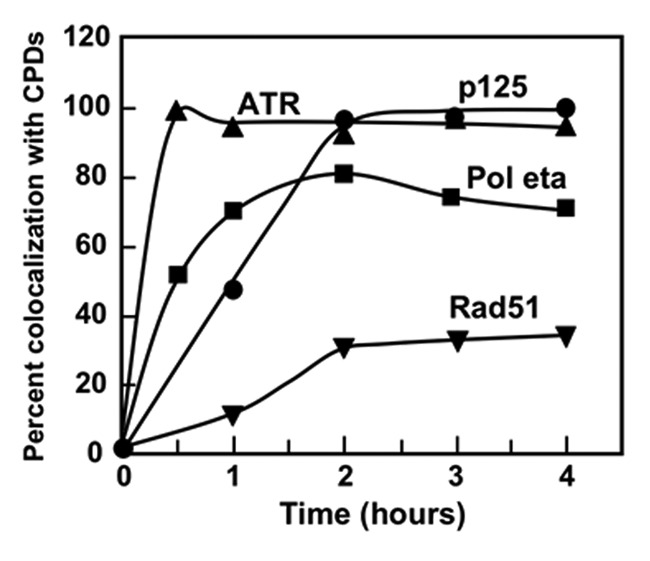
Figure 4. Dynamics of the recruitment of p125, ATR, Pol η and Rad51 to UV damage. A549 cells were irradiated with UVC through 5 μm pore polycarbonate filters and analyzed at 1, 2, 3, and 4 h after irradiation. Cells were co-stained for the indicated proteins with CPDs, and scored for the percentage co-localization with CPDs [ATR (triangles), p125 (circles), Pol η (squares) and Rad51 (inverted triangles)].
Distribution of Pol δ subunits in cells during cell cycle progression
Laser scanning cytometry (LSC) was utilized to obtain information regarding the individual cells within the cell populations and to correlate recruitment of Pol δ after DNA damage with the cell cycle phase. Since Pol δ is confined to the nucleus, the DAPI fluorochrome of DNA was used to set the primary nuclear contour enabling the examination of the content of p125, p50, p68 and p12 within the nucleus. Intensity of DAPI fluorescence revealed the cellular DNA content and thus the cell cycle phase, while multiparameter analysis of these data made it possible to correlate content (expression) of these subunits with the cell cycle phase. The bivariate distributions (scatterplots) showing total nuclear p125, p50, p68 or p12 content vs. the DNA content in the untreated A549 cells are shown in Figure 5. As is evident, expression of p125 correlates well with the cell cycle progression, as the S-phase cells have a distinctly higher level of expression than G1 cells, and G2/M cells a still higher level than S cells. A similar pattern of expression vis-à-vis cell cycle phase is shown by p68. In fact, the ratio of p125 or p68 content per unit of DNA content remains nearly constant (0.90–1.06) across the cell cycle (Table 1).
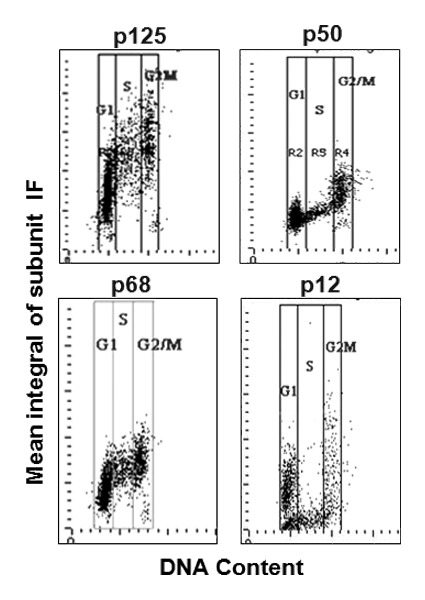
Figure 5. Analysis of Pol δ subunit content in A549 cells by laser scanning cytometry (LSC). A549 cells were fixed and stained for each subunit as in Figure 1 with modifications as described in Materials and Methods . The expression of p125, p50 p68 and p12 (integral values of IF intensity per nucleus) was DAPI (DNA content) as shown. For each panel the vertical lines show the gating analysis thresholds (“gates”) to identify subpopulations of cells in G1, S and G2/M. The mean values of the subunits IF in each population are presented in Table 1.
Table 1. Levels of Pol δ subunits in different cell cycle phases.
| Cell cycle phase | Mean integral values (×10−6) | |||
|---|---|---|---|---|
| |
p125 |
p68 |
p50 |
p12 |
| G1 |
3.4a (1.00)b |
3.0a (1.00)b |
0.80a (1.00)b |
1.5a (1.00)b |
| S |
5.4 (1.06) |
4.7 (1.03) |
0.92 (0.76) |
0.84 (0.37) |
| G2/M | 6.4 (0.94) | 5.4 (0.90) | 1.0 (0.98) | 2.0 (0.67) |
a Determined from LSC data (Fig. 5). bMean integral values divided by mean integral values for DAPI (DNA) and normalized to the values for G1 set as 1.00.
The p50 and p12 subunits reveal different patterns of expression from p125 and p68. Specifically, expression of p50 and p12 is reduced in S-phase cells. This is reflected by the fact that p50 and p12 exhibit the lowest ratios of content per unit of DNA in S-phase cells (0.76 and 0.37, respectively) as compared with G1 (1.0 and 1.0) or G2M (0.98 and 0.67) cells (Table 1). In fact, the expression of p12 in the S phase is at low levels that fall in the range below the minimal expression of this subunit in G1 or G2M cells (Fig. 5, p12 panel). These data show that the subunit composition of Pol δ within the cell is dynamic during the cell cycle, particularly in the case of the p12 subunit which is so markedly diminished in S-phase cells.
The recruitment of p125 to UV damage takes place in all phases of the cell cycle
One of the striking aspects of the recruitment of Pol δ that we have demonstrated thus far is the completeness of its recruitment to sites of CPD damage (Figs. 2 and 4), which indicates that this process occurs throughout the cell cycle. For this reason, we sought direct evidence for the recruitment of Pol δ to CPD damage by cell cytometric analysis using LSC.
A549 cells were immuno-stained for p125 and CPDs. For multiparameter analysis by LSC, the nucleus identified by DAPI staining served as the primary contour, while the DNA damaged areas (CPDs) produced by UV irradiation through polycarbonate filters served as the sub-contours. The cytometric data illustrating the kinetics of the recruitment of Pol δ to DNA damage sites is presented in the form of DNA content frequency histograms (Fig. 6A) and bivariate distribution plots (scatterplots; Fig. 6B and C). In the multivariate “paint-a-gate” analysis, the cells in which p125 was recruited into DNA damage foci (represented in the sub-contours defined by CPDs) were electronically colored red; the cells with CPDs contours with no p125 were marked blue. We can visualize the kinetics of recruitment of p125 to DNA damage sites by following the rate of “replacement” of the “blue” by the “red” cells on the scatterplots. The increase in the number of cells exhibiting p125 recruitment to damaged DNA with time is clearly demonstrated. The data also shows that Pol δ is recruited in all phases of the cell cycle. The progression of recruitment is consistent with those expected from the analysis shown in Figure 2, i.e., recruitment is nearly complete by 2 h after UV treatment.
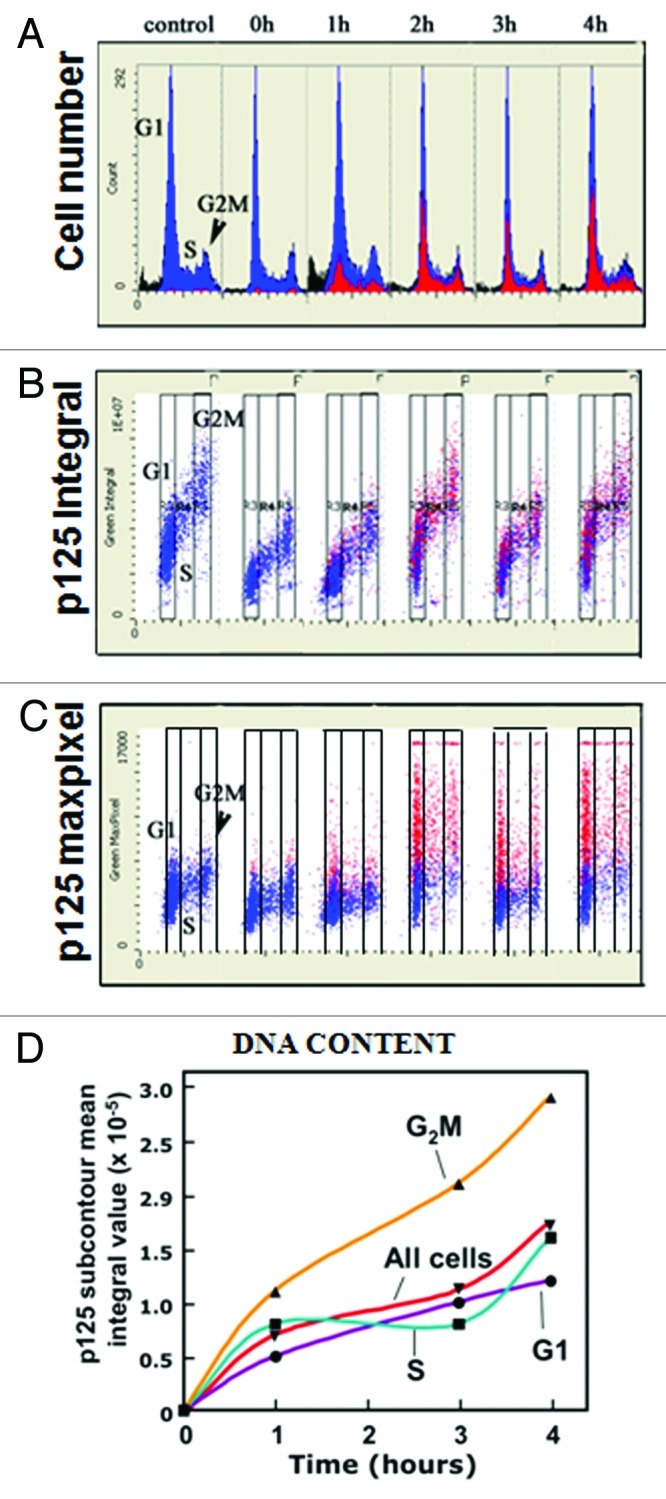
Figure 6. Analysis of the kinetics of recruitment of p125 to the UV induced CPD macro-foci. A549 cells were treated with 75 J/m2 UVC through a 5 μm polycarbonate filter and fixed after 0, 1, 2, 3 and 4 h and co-stained with different colors of IF for p125 and CPDs, respectively. DNA was stained with DAPI; fluorescence intensity [integral and maximal pixel (MaxPixel) value, per cell] was measured by LSC. (A) DNA content frequency histograms. Through the “paint-a-gate” multiparameter analysis the cells strongly expressing p125 within the macro-foci marking CPDs (detected by sub-contouring within the nucleus for areas of high intensity, as measured by maximal pixel of p125 IF) are colored red. (B) Bivariate distributions (scatterplots) representing integral IF of p125 in the nuclei plotted against DNA content. (C) Scatterplots of the maximal pixel of p125 IF vs. DNA content. In all plots (A, B and C) the cells with CPDs expressing strong intensity of p125 IF are colored red. (D) Plot of p125 IF mean integral values assessed for CPDs macro-foci (IF sub-contours) for G1, S, G2/M cells as well as for all cells shown as circles, squares, triangles and inverted triangles, respectively.
The scatterplots of the p125 integral of IF (immunofluorescence), which reflects the total p125 protein content per nucleus (Fig. 6B), show that the expression of this protein decreases somewhat at 0 and 1 h after irradiation but then returns to the level of the control. The maximal pixel values, however, reflecting the maximum p125 IF local intensity and hence its high “punctate” concentration at the sites of DNA damage, reveals quite dramatic increases between 1 and 2 h after UV irradiation and a rather constant level between 2 and 4 h. This increase is observed in cells in all phases of the cell cycle. This is consistent with the visually observed intensity of p125 fluorescence in the macro-foci over the background (e.g., Fig. 2A).
When the mean integral values within the sub-contours, reflecting the amounts of p125 in the macro-foci, are plotted against time, a time course of recruitment that reaches a near maximum by 2 h is found (Fig. 6D). The kinetics is consistent with the data obtained by visual quantitation of the co-localization of p125 with CPDs by fluorescence microscopy (Fig. 2B). However, we observed that between 2 and 4 h the content of p125 continues to increase, but at a slower rate. In the visual co-localization experiments (Fig. 2) we would be unable to distinguish this increase as the foci were simply scored as positive or negative.
Discussion
Previous studies, limited to the use of anti-p125 antibodies, have shown that Pol δ is recruited to UV-induced DNA damage foci together with the NER factor TFIIH18 as well as PCNA, Pol η, Rad1852 and p21.53 However, there have been no detailed studies of the spatiotemporal dynamics of the recruitment of Pol δ to DNA damage foci or direct evidence that Pol δ3 is present at DNA damage sites. In this study, we established that Pol δ3 is present at local areas of UV-induced DNA damage through subcellular co-localization of Pol δ subunits with CPDs, and that it represents the primary form of Pol δ associated with CPDs through biochemical evidence by ChIP analysis. CPDs are repaired more slowly in relation to the depletion of p12, which is completed by 4 h after UV treatment. We observed that CPD fluorescence persists as long as 18 h after UV treatment, consistent with earlier reports on the dynamics of removal of CPDs from cultured cells.54 Thus, Pol δ3 is present at a time when CPD repair must still be ongoing.
Our studies of the recruitment of Pol δ to CPDs in an asynchronous population shows that recruitment takes place in nearly all the CPD containing macro-foci by two hours post-irradiation (Figs. 2 and 4). The recruitment rate is not dependent on cell cycle phase and, when examined within each cell cycle phase population, occurs at similar rates to that of the entire asynchronous population (Fig. 6D). One scenario that could contribute to recruitment timing is related to the time it takes for the chromatin reorganization to make the damage accessible, particularly in G1 cells, given that the gap filling step is the penultimate event in DNA repair.
Two major areas in which Pol δ has been shown to be involved in response to DNA damage are in the repair of CPDs by NER,17 which is regarded to operate mainly in G1 cells, and the operation of the DNA damage tolerance pathway in S phase, in which Pol η functions to perform largely error-free bypass of the CPD lesions.36 Previous examination of the dynamics of the recruitment of NER proteins to UV damage foci in human fibroblasts showed that TFIIH is recruited by 10 min, while Pol δ (p125 subunit) and PCNA are recruited by 30 min.18 In vitro analysis of the temporal dynamics of the recruitment of NER factors from HeLa cell extracts showed that the NER factors are recruited in a temporally sequential manner, with Pol δ, PCNA and RFC being recruited after TFIIH and RPA, with maximal recruitment after 2 h.18 Future studies on the re-synthesis step in NER using both Pol δ4 and Pol δ3 will be needed to establish and ascertain the molecular basis for the utilization of Pol δ3. Pol δ has been shown to be involved in HR in yeast by both genetic means20,55 and in vitro reconstituted systems, in a process that requires PCNA and RFC.21 Yeast Pol δ has been shown to be able to extend D-loop strand invasion intermediates involved in HR.22 Pol δ3 is likely to be the form that may be involved in translesion synthesis, where Pol δ and Pol η must switch places at the site of the CPD lesions.42,56 There are important questions regarding the mechanisms of switching that are raised by the involvement of Pol δ3 in translesion synthesis. Previously, the switching of Pol δ with Pol η has not been considered in regard to differences in yeast and human Pol δ, or in regard to the existence of Pol δ3. All four Pol δ subunits have been reported to bind PCNA, and p12 has been shown to contribute to the interaction of Pol δ4 with PCNA.57 One possibility is that the loss of p12, which contributes to the interaction of Pol δ4 to PCNA, facilitates the displacement of Pol δ by Pol η. It is enticing to suggest that Pol δ3 is the form which is involved in switching with Pol η and other TLS pols.4
Our findings, which support the existence of Pol δ3 at sites of DNA damage strengthen the concept that mutability of the subunit composition of Pol δ represents a physiologically relevant response to DNA damage. The question arises as to whether there are other conditions where Pol δ3 is formed, and if this mutability extends to the other subunits. We have observed in a number of experiments that there is a small but consistent fraction of the population of untreated cells with little or no p12 present (Fig. 1). Also, our cell cycle analysis data indicate that p12 may be lower in S phase, suggesting that Pol δ3 levels are increased in S phase. However, given that p12 levels respond to DNA damage, this could be reasonably explained since the cells are constantly responding to endogenous sources of DNA damage, such as deamination and oxidative damage.58,59 It should be noted, however, that whereas the S-phase cells are characterized by minimal expression of p12 subunit, distinctly below the minimal level of that of G1 and G2M cells, the expression of p50 is also somewhat lower during S phase, although not to such an extent as p12 (Fig. 5 and Table 1). However, a word of caution in interpretation of the immunocytochemical data in quantitative terms is needed. Namely, while the lowered expression of these subunits detected immunocytochemically most likely reflects their diminished content, it is possible, albeit less likely, that accessibility of epitope of these proteins to the respective Abs may be altered, if the epitope binds to other proteins or undergoes conformational changes. Nevertheless, the question of whether Pol δ3 has a role in normal chromosomal replication could be raised. This idea may not be far-fetched, since there are recent reports that suggest that translesion polymerases may participate in normal DNA replication. This could arise from the presence of sequences or structures that pose difficulties for Pol δ, where translesion polymerases may be switched in to perform bypass synthesis, e.g., in the replication of microsatellite sequences, where Pol κ has been shown to exhibit greater fidelity than the Pol δ holoenzyme.60 Thus, it would be of interest to examine the ability of Pol δ3 in this context, since our studies show that it exhibits greater fidelity than Pol δ4.44
A broader context of the mutability of Pol δ subunit composition comes from recent studies of the interaction between human Pol ζ and the Pol δ p50 subunit. Pol ζ is a member of the B-family of DNA polymerases and has a C-terminal domain (CTD) that is conserved with that of Pol δ. Using recombinant p50 complexed with the N-terminal fragment of the p68 subunit, it was demonstrated that the CTD of Pol ζ interacts with the p50-p68 complex. This led to the hypothesis that Pol ζ may switch places with the p125 catalytic subunit of Pol δ to form a complex of Pol ζ, p50 and p68.61 This adds yet another twist to the potential role of Pol δ3 in translesion polymerase switch, where loss of the p12 subunit is a trigger for the switching of the catalytic subunits. Finally, recent reports demonstrate that expressions of the POLD4 gene encoding p12 are altered in certain population of lung cancer tissues.62 siRNA depletion of p12 in cultured cells resulted in phenotypes exhibiting alterations in cell cycle progression, checkpoint activation and chromosomal aberrations as well as increased genomic instability.62,63
One of the unexpected observations we made was that p12 degradation within areas of local irradiation through membrane filters was incomplete, whereas it is completely degraded in globally irradiated cells exposed to UV. This phenomenon may provide insights into the mechanisms involved in p12 degradation, which are triggered by ubiquitination.43 A major theme in the assembly of DNA damage response complexes is the involvement of E3 ubiquitin ligases, which act only when they are recruited to the sites of DNA damage.27,28 Similarly, ubiquitination of PCNA by Rad18/Rad6 requires their recruitment to the site of the stalled replication, viz., PCNA is ubiquitinated in situ at the site of the UV lesion.40,52 The sequence of events that lead to p12 degradation would require the recruitment of Pol δ4 and the E3 ligase involved, followed by the degradation of p12. Our findings can be explained if there is an exchange of Pol δ3 and Pol δ4 to and from the nuclear pool to the damage sites. In the case of globally irradiated cells, the total amount of CPD damage throughout the nuclei would govern the conversion of the total nuclear pool of Pol δ4 to Pol δ3. The areas within the observed foci are small compared with that of the entire nucleus that are being irradiated and would have a smaller net capacity for degradation of p12. The area of the observed foci through local irradiation is roughly 1/40 of the total area of the entire nuclei, i.e., the total damage incurred within the foci was 40 times smaller than incurred when the whole nucleus was irradiation. On the assumption that DNA is equally distributed in the nucleus, and that the capacity for enzymatic ubiquitination of p12 is constrained in situ to the CPDs, then the overall capacity for degradation of p12 by a single macro-foci will be 1/40 that of the globally irradiated nucleus. This should not affect the rate at which p12 is observed to disappear, unless there is an exchange between Pol δ in the macro-foci and Pol δ in the rest of the nucleus. In the presence of exchange, this would slow the rate by a large factor. In other words, confinement of the DNA damage to a small concentrated area reduces the overall nuclear rate of conversion of Pol δ4 to Pol δ3. This reveals an unexpected limitation of the use of local areas of irradiation where in situ conversion of p12 or any given protein is being observed. While we currently have no information on the rates of exchange of Pol δ localized to CPDs and the nuclear pool of unbound Pol δ, we have examined the dynamics of recruitment of Pol δ3 to CPD macro-foci by prior siRNA depletion of p12 (Fig. S4). The time course of recruitment of Pol δ3 was the same as that of Pol δ4. Thus, we can reasonably state that Pol δ3 is readily recruited and bound to CPD damage at rates comparable to Pol δ4.
Materials and Methods
Cell lines and cell culture
A549 cells, which are adherent epithelial cells derived from human lung adenocarcinoma, were used for the majority of the experiments. Cells were maintained according to the protocols provided by the supplier.
Treatment of cells with UV irradiation and Triton X-100
Cells (24 h after seeding) were exposed to UVC (254 nm) irradiation for the indicated dosage using a UVLMS-38EL series 3 UV lamp with a flux rate of 2 J/s. The lamp was calibrated using a UV radiometer (UVP, Inc.). The medium was removed from the cells, and the cells were washed with PBS prior to irradiation, and fresh medium was replaced thereafter. The cells were returned to the incubator and allowed to recover for the indicated time periods before harvesting or fixing. For the experiments in which cells were locally irradiated, Millipore polycarbonate filters with 5 µm pores were laid on top of the cells before irradiating the cells with an incident UV dose of 75 J/m2. The medium was then replaced and the filter removed before cells were returned to the incubator for recovery.
For the pre-extraction of soluble proteins to examine chromatin bound proteins by immunofluorescence, cells were incubated on ice for 5 min with a 0.1% Triton X-100 buffer (10 mM HEPES PH 7.4, 100 mM KCl, 2 mM MgCl2, 1 mM EDTA and 0.1% Triton X-100) prior to fixing with paraformaldehyde.
SDS-PAGE and western blotting
Cells were harvested at approximately 60% confluency and collected by either centrifugation or scraping and lysed by the addition of a Tris-based lysis buffer. After 20 min rotation at 4°C, cells were sonicated, and lysates were centrifuged for 10 min at 10,000 × g at 4°C to pellet cellular debris. Protein concentration of the lysates was measured using the Bradford assay, and samples of 50–75 µg were used for separation and western blotting analysis with selected antibodies.
Indirect immunofluorescence microscopy
For p125, we used a mouse monoclonal antibody against human p125 at a final dilution of 1:4000 in 2% BSA/PBS. An incubation time of 1–2 h at room temperature was found to be sufficient for binding and subsequent staining. For p50 and p68, rabbit polyclonal antibodies were used. For p12, a mouse monoclonal anti-p12 was used. The specificity of this anti-p12 was previously tested using cell lysates with overexpressing FLAG-tagged p12.43 Antibodies against γH2AX (phosphorylated ser139, clone 2F3) was from BioLegend, ATR (c-20) and Pol η (B-7) were obtained from Santa Cruz. Anti-Rad51 was purchased from CalBiochem. Anti-CPD antibody (D194–1) was obtained from MBL Inc. Secondary antibodies used were: Alexafluor488 conjugated anti-mouse or Rhodamine-X conjugated anti-rabbit IgG (Molecular Probes). 4',6-diamidino-2-phenylindole (DAPI) was used to counterstain the cells for DNA.
Cells were seeded at about 60% confluence onto glass chamber slides 24 h before each experiment. After UV treatment and recovery, cells were fixed in 4% paraformaldehyde for 15 min at room temperature then placed in 70% ethanol overnight at -20°C. Cells were then permeabilized at room temperature for 10 min with 0.1% Triton-X-100 in PBS followed by blocking with 2% BSA in PBS for 1 h. If the cells were double labeled with antibodies from different hosts, both primary antibodies were added at the appropriate dilution usually for overnight incubation at 4°C. This was followed by three 10 min washes in 2% BSA and simultaneous incubation with the appropriate secondary antibodies at a dilution of 1:2000 for 1 h at room temperature. For the cells that were double labeled with p125 and CPD, a sequential method of staining was used due to the same host origin of the antibodies. Slides were washed 3× for 10 min with 2% BSA followed by incubation with AlexaFluor488 (green) conjugated goat anti-mouse at a dilution of 1:2000 in 2% BSA for 1 h at room temperature. Following another set of three washes with 2% BSA for 10 min each, cells were incubated with non-immune goat serum for 30 min to bind any excess secondary antibody. After another three washes with BSA, cells were incubated with anti-CPD at a dilution of 1:4000 for 30 min at room temperature, again followed by three washes with BSA. Cells were then incubated with Texas Red conjugated goat anti-mouse secondary antibody for 30 min at a dilution of 1:3000. Finally, cells were washed 3× with 1X PBS for 10 min each, then dried and mounted with ProLong Antifade reagent with DAPI (Invitrogen). Slides were visualized and imaged using a Zeiss Axiovert 200M with a black and white CCD AxioCam and pseudo-colored with Axiovision 4.6 software. Unless otherwise stated, most images were taken at a magnification of 40X.
Quantitative analysis of the time course of recruitment of Pol δ subunits to localized areas of damage
After UV irradiation through 5 μm polycarbonate filters, cells were allowed to recover and examined at different time periods, usually 1, 2, 3 and 4 h. Cells were stained for CPDs and the individual Pol δ subunits. Randomly selected fields containing 20–30 cells were scored by counting the total number of large CPD foci, and those that displayed co-localization with the specific Pol δ subunit or other proteins. A total of at least 100 cells were examined, and the data expressed as percentage of co-localization.
Cell analysis by laser scanning cytometry (LSC)
Cells to be analyzed using LSC were maintained on glass chamber slides using the same procedure as for indirect immunofluorescence (IF) labeling for microscopy, with some modifications. Secondary antibody labeling was done with the green wavelength emitting fluorochrome AlexaFluor488 and far-red AlexaFluor 647, at a 1:500 dilution. Cellular DNA was counterstained with DAPI at a concentration of 2.5 µg/ml in PBS for 15 min at room temperature following the set of three rinses after secondary antibody incubation. Staining was completed with another set of three washes at 10 min each with PBS followed by mounting with ProLong Anti-fade reagent. Cellular green and far-red immunofluorescence (IF) representing the binding of the respective Abs as well as the blue emission of DAPI-stained DNA was measured using an LSC (iCys Research Imaging Cytometer; CompuCyte) with standard filter settings; fluorescence was excited with helium-neon (633 nm) argon ion (488 nm) and violet (405 nm) lasers. For all experiments, the primary contour was set using DAPI to define the nucleus. For the experiments in which local UV irradiation was applied through the Millipore filters, a secondary contour was set using the far red stain for CPD as a positive marker for DNA damage. These parameters were then used to analyze the recruitment of p125 to the localized areas of DNA damage, which are represented by the changes in intensity of fluorescent staining. The intensities of maximal pixel and integrated fluorescence were measured and recorded for each cell. No electronic compensation for possible emission spectral overlap was applied. At least 3,000 cells were measured per sample. Gating analysis to obtain the mean values of IF for populations of cells in G1, S and G2M phases of the cell cycle was performed as shown in Figure 5. Other details of cells analysis by LSC are presented elsewhere.64
Chromatin immunoprecipitation (ChIP) analyses
A549 cells were seeded onto 4 × 15 cm plates at approximately 60% confluency 24 h before UV irradiation. Following 4 h of recovery after 20 J/m2 of UV irradiation, cells were cross-linked by adding 11% formaldehyde solution directly to the medium for a final concentration of 1% formaldehyde for 10 min at room temperature. The cross-linking reaction was neutralized by the addition glycine to a final concentration of 0.25 M for 5 min. The harvested cell pellet was then sonicated twice for 20 sec each. The ChiP assay was performed using a ChIP-IT Express Enzymatic Kit (Active Motif, Carlsbard, CA) following the supplied protocol. Lysate containing 1–2 mg of protein was aliquoted per immunoprecipitation (IP) reaction and diluted at least 1:1 with RIPA buffer. Anti-p125 (20 µg) or control mouse IgG was added to each reaction and allowed to rotate overnight at 4°C. Protein A/G beads (Santa Cruz) were added to each reaction and rotated for 1 h at 4°C to pull down the antibodies and precipitated proteins. Immunoprecipitated proteins were released with SDS sample buffer and heating at 100°C and analyzed with SDS-PAGE and western blotting.
For DNA analysis, immunoprecipitates were eluted twice by adding 50 µl of elution buffer to the beads and heating at 65°C each time. Eluates were combined and NaCl added to a final concentration of 0.1 M. Eluates were then incubated at 65°C overnight or for at least 6 h. DNA was isolated and purified via phenol/chloroform extraction. This was done as following: an equal volume of phenol/chloroform was added to each sample and vortexed before centrifuging for 5 min at 13,000 rpm in a microcentrifuge. Supernatants were transferred to a fresh tube and 3 M sodium acetate (pH 5.2) added to a final concentration of 0.3 M along with 500 µl of 100% ethanol. Samples were mixed thoroughly and left at -20°C overnight. Precipitated DNA was then collected by centrifuging at 13,000 rpm in a microcentrifuge for 10 min at 4°C. Supernatant was removed and 500 µl of 70% ethanol was added to the DNA pellet to wash and subsequently centrifuged for 5 min at 4°C at 13,000 rpm. After removal of the supernatant, the pellet was air-dried and resuspended in 30 μl of double distilled water (ddH2O).
A slot blot apparatus was used to apply the DNA samples to nitrocellulose before probing with anti-CPD. The nitrocellulose membrane was soaked in 20× SSC (3 M NaCl, 300 mM sodium citrate, pH 7.0) for 20 min prior to application of the sample by vacuum and each sample volume was brought up to 100 µl with ddH2O. Following sample application, DNA was denatured by adding 0.3 M NaOH to each slot, then neutralizing with 6× SSC and solutions were pulled through by vacuum suction. The membrane was allowed to dry, then baked for 2 h at 80°C and washed with TBST before the addition of anti-CPD (1:3000) and subsequent incubation for 1 h at room temperature. The membrane was then processed following the procedure for western blotting as detailed above.
siRNA depletion of p12 in A549 cells
Human P12 (Pol δ4) siGENOME SMARTpool and Non-target siRNA control were purchased from Dharmacon. Oligofectamine™ Reagent was used for transfection according to the supplied protocol. After 48–72 h post transfection, cells were examined for immunostaining after UV treatment and for western blotting for p12 without treatment.
Supplementary Material
Acknowledgments
This work was supported by grants from the United States Public Health Service GM31973 and ES14737 to M.Y.W.T.L. and CA28704 to Z.D.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21280
References
- 1.Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005;40:115–28. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 2.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol. 2011;3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Z, Zhang S, Lin SH, Wang X, Wu L, Lee EY, et al. Structure of monoubiquitinated PCNA: Implications for DNA polymerase switching and Okazaki fragment maturation. Cell Cycle. 2012;11:2128–36. doi: 10.4161/cc.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirchmaier AL. Ub-family modifications at the replication fork: Regulating PCNA-interacting components. FEBS Lett. 2011;585:2920–8. doi: 10.1016/j.febslet.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18:162–73. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann AR. Ubiquitin-family modifications in the replication of DNA damage. FEBS Lett. 2011;585:2772–9. doi: 10.1016/j.febslet.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Xie B, Li H, Wang Q, Xie S, Rahmeh A, Dai W, et al. Further characterization of human DNA polymerase delta interacting protein 38. J Biol Chem. 2005;280:22375–84. doi: 10.1074/jbc.M414597200. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem. 2003;278:10041–7. doi: 10.1074/jbc.M208694200. [DOI] [PubMed] [Google Scholar]
- 10.Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MY. Reconstitution and characterization of the human DNA polymerase delta four-subunit holoenzyme. Biochemistry. 2002;41:13133–42. doi: 10.1021/bi0262707. [DOI] [PubMed] [Google Scholar]
- 11.Mo J, Liu L, Leon A, Mazloum N, Lee MY. Evidence that DNA polymerase delta isolated by immunoaffinity chromatography exhibits high-molecular weight characteristics and is associated with the KIAA0039 protein and RPA. Biochemistry. 2000;39:7245–54. doi: 10.1021/bi0000871. [DOI] [PubMed] [Google Scholar]
- 12.Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 13.Zuo S, Bermudez V, Zhang G, Kelman Z, Hurwitz J. Structure and activity associated with multiple forms of Schizosaccharomyces pombe DNA polymerase delta. J Biol Chem. 2000;275:5153–62. doi: 10.1074/jbc.275.7.5153. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds N, Watt A, Fantes PA, MacNeill SA. Cdm1, the smallest subunit of DNA polymerase d in the fission yeast Schizosaccharomyces pombe, is non-essential for growth and division. Curr Genet. 1998;34:250–8. doi: 10.1007/s002940050394. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–7. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rytkönen AK, Vaara M, Nethanel T, Kaufmann G, Sormunen R, Läärä E, et al. Distinctive activities of DNA polymerases during human DNA replication. FEBS J. 2006;273:2984–3001. doi: 10.1111/j.1742-4658.2006.05310.x. [DOI] [PubMed] [Google Scholar]
- 17.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37:714–27. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Mocquet V, Lainé JP, Riedl T, Yajin Z, Lee MY, Egly JM. Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J. 2008;27:155–67. doi: 10.1038/sj.emboj.7601948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overmeer RM, Gourdin AM, Giglia-Mari A, Kool H, Houtsmuller AB, Siegal G, et al. Replication factor C recruits DNA polymerase delta to sites of nucleotide excision repair but is not required for PCNA recruitment. Mol Cell Biol. 2010;30:4828–39. doi: 10.1128/MCB.00285-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maloisel L, Fabre F, Gangloff S. DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol. 2008;28:1373–82. doi: 10.1128/MCB.01651-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Stith CM, Burgers PM, Heyer WD. PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol Cell. 2009;36:704–13. doi: 10.1016/j.molcel.2009.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sebesta M, Burkovics P, Haracska L, Krejci L. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair (Amst) 2011;10:567–76. doi: 10.1016/j.dnarep.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–82. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 24.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hübscher U, et al. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene. 1998;17:835–43. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 25.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 26.Huen MS, Chen J. Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem Sci. 2010;35:101–8. doi: 10.1016/j.tibs.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA. 2007;104:20759–63. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Lisby M, Rothstein R. Localization of checkpoint and repair proteins in eukaryotes. Biochimie. 2005;87:579–89. doi: 10.1016/j.biochi.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 30.Yan J, Jetten AM. RAP80 and RNF8, key players in the recruitment of repair proteins to DNA damage sites. Cancer Lett. 2008;271:179–90. doi: 10.1016/j.canlet.2008.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufmann WK. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis. 2010;31:751–65. doi: 10.1093/carcin/bgp230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J. 2011;436:527–36. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol Cell Biol. 2007;27:5806–18. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle. 2007;6:2760–7. doi: 10.4161/cc.6.22.4932. [DOI] [PubMed] [Google Scholar]
- 35.Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25:7158–69. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13:141–52. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Bozza W, Zhuang Z. Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis. Cell Biochem Biophys. 2011;60:47–60. doi: 10.1007/s12013-011-9187-3. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Qin Z, Zhang X, Xiao W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS Lett. 2011;585:2786–94. doi: 10.1016/j.febslet.2011.04.044. [DOI] [PubMed] [Google Scholar]
- 39.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–54. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann AR. Translesion synthesis in mammalian cells. Exp Cell Res. 2006;312:2673–6. doi: 10.1016/j.yexcr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 43.Zhang S, Zhou Y, Trusa S, Meng X, Lee EY, Lee MY. A novel DNA damage response: rapid degradation of the p12 subunit of dna polymerase delta. J Biol Chem. 2007;282:15330–40. doi: 10.1074/jbc.M610356200. [DOI] [PubMed] [Google Scholar]
- 44.Meng X, Zhou Y, Zhang S, Lee EY, Frick DN, Lee MY. DNA damage alters DNA polymerase delta to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 2009;37:647–57. doi: 10.1093/nar/gkn1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meng X, Zhou Y, Lee EY, Lee MY, Frick DN. The p12 subunit of human polymerase delta modulates the rate and fidelity of DNA synthesis. Biochemistry. 2010;49:3545–54. doi: 10.1021/bi100042b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeifer GP. Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem Photobiol. 1997;65:270–83. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 47.Volker M, Moné MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, et al. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–24. doi: 10.1016/S1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 48.Okuno Y, McNairn AJ, den Elzen N, Pines J, Gilbert DM. Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J. 2001;20:4263–77. doi: 10.1093/emboj/20.15.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soria G, Belluscio L, van Cappellen WA, Kanaar R, Essers J, Gottifredi V. DNA damage induced Pol eta recruitment takes place independently of the cell cycle phase. Cell Cycle. 2009;8:3340–8. doi: 10.4161/cc.8.20.9836. [DOI] [PubMed] [Google Scholar]
- 51.Sinha RP, Häder DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1:225–36. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–96. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perucca P, Cazzalini O, Mortusewicz O, Necchi D, Savio M, Nardo T, et al. Spatiotemporal dynamics of p21CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J Cell Sci. 2006;119:1517–27. doi: 10.1242/jcs.02868. [DOI] [PubMed] [Google Scholar]
- 54.Mellon I, Bohr VA, Smith CA, Hanawalt PC. Preferential DNA repair of an active gene in human cells. Proc Natl Acad Sci USA. 1986;83:8878–82. doi: 10.1073/pnas.83.23.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brocas C, Charbonnier JB, Dhérin C, Gangloff S, Maloisel L. Stable interactions between DNA polymerase δ catalytic and structural subunits are essential for efficient DNA repair. DNA Repair (Amst) 2010;9:1098–111. doi: 10.1016/j.dnarep.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 56.Lehmann AR. Clubbing together on clamps: The key to translesion synthesis. DNA Repair (Amst) 2006;5:404–7. doi: 10.1016/j.dnarep.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Xie B, Zhou Y, Rahmeh A, Trusa S, Zhang S, et al. Functional roles of p12, the fourth subunit of human DNA polymerase delta. J Biol Chem. 2006;281:14748–55. doi: 10.1074/jbc.M600322200. [DOI] [PubMed] [Google Scholar]
- 58.Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, et al. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging (Albany NY) 2011;3:1028–38. doi: 10.18632/aging.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 60.Hile SE, Wang X, Lee MY, Eckert KA. Beyond translesion synthesis: polymerase κ fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012;40:1636–47. doi: 10.1093/nar/gkr889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baranovskiy AG, Lada AG, Siebler HM, Zhang Y, Pavlov YI, Tahirov TH. DNA Polymerase δ and ζ Switch by Sharing Accessory Subunits of DNA Polymerase δ. J Biol Chem. 2012;287:17281–7. doi: 10.1074/jbc.M112.351122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang QM, Tomida S, Masuda Y, Arima C, Cao K, Kasahara TA, et al. Regulation of DNA polymerase POLD4 influences genomic instability in lung cancer. Cancer Res. 2010;70:8407–16. doi: 10.1158/0008-5472.CAN-10-0784. [DOI] [PubMed] [Google Scholar]
- 63.Huang QM, Akashi T, Masuda Y, Kamiya K, Takahashi T, Suzuki M. Roles of POLD4, smallest subunit of DNA polymerase delta, in nuclear structures and genomic stability of human cells. Biochem Biophys Res Commun. 2010;391:542–6. doi: 10.1016/j.bbrc.2009.11.094. [DOI] [PubMed] [Google Scholar]
- 64.Zhao H, Rybak P, Dobrucki J, Traganos F, Darzynkiewicz Z. Relationship of DNA damage signaling to DNA replication following treatment with DNA topoisomerase inhibitors camptothecin/topotecan, mitoxantrone, or etoposide. Cytometry A. 2012;81:45–51. doi: 10.1002/cyto.a.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.