

Abstract
Astrocytes, the third element of the tripartite synapse, are active players in neurotransmission. Up to now, their involvement in neuronal functions has primarily been investigated at the single cell level. However, a key property of astrocytes is that they communicate via extensive networks formed by gap junction channels. Recently, we have shown that this networking modulates the moment to moment basal synaptic transmission and plasticity via the regulation of extracellular potassium and glutamate levels. Here we show that astroglial gap junctional communication also regulates neuronal network activity. We discuss these findings and their implications for brain information processing.
Keywords: astrocytes, gap junctions, connexins, neuroglial interactions, neuronal network activity, hippocampus
Introduction
Astrocytes, as part of the tripartite synapse, can integrate neuronal activity through activation of their channels, receptors, and transporters, and modulate in turn synaptic transmission and plasticity, via the release of gliotransmitters or uptake of neuroactive substances.1 Individual astrocytes can contact up to 140 000 synapses.2 However, since astrocytes are interconnected via gap junctions, which allow the intercellular trafficking and redistribution of neuroactive substances, they can integrate and modulate large neuronal ensembles.3-6 We have recently shown that astroglial gap junctional networks provide the basis for precise neuronal communication, because they enable fast removal of extracellular potassium and glutamate, and regulate efficiently the activity-dependent changes in extracellular space volume.7 Thus dysfunction of astroglial networks severely affects extracellular neurotransmitter and ion homeostasis, which could, especially in highly recurrent connected neuronal networks, result in hyperexcitability. We therefore investigated whether astroglial network communication is important for the temporal and spatial precision of neuronal information processing, and thereby limits neuronal network activity.
Results and Discussion
We previously discovered that intercellular astroglial network communication controls synaptic transmission by limiting neuronal excitability, release probability and insertion of postsynaptic AMPA receptors, contributing to synapse silencing.7 Importantly, this has a major impact for synaptic plasticity, because converting functional synapses into silent synapses strongly shifts the threshold balance between long-term potentiation and long-term depression, favoring synaptic potentiation.7 Interestingly, imbalanced excitatory and inhibitory strength, as well as enhanced neuronal excitability are mainly the cause of pathological network activity, typical of epilepsy. We thus investigated whether astroglial gap junctional communication limits neuronal network activity, using mice with disconnected astrocytes, in which both astroglial gap junction forming proteins, connexin 30 (Cx30) and connexin 43 (Cx43), are knocked out (Cx30-/-Cx43-/- mice).4,7,8 We perform our study in the hippocampus, since this structure is implicated in the formation of physiological and pathological network activity, such as ripples and epileptic seizures, respectively. To study whether astroglial networks alter CA1 neuronal network activity in response to enhanced synaptic activation, we stimulated Schaffer collaterals with increasing stimulation strength (0.1 and 0.5 ms pulse width). Synchronous recordings of CA1 field excitatory postsynaptic potentials (fEPSP) and astrocytic membrane depolarizations (Fig. 1A–D), evoked by similar Schaffer collateral stimulation in wild type and Cx30-/-Cx43-/- hippocampal slices, as assessed by comparable fiber volley amplitudes from fEPSPs (Fig. 1C), allowed us to directly investigate alterations in neuronal activity and astroglial responses in both genotypes.

Figure 1. Astroglial gap junctional communication prevents amplification of neuronal activity and reduces neuronal network activity. Synchronous recordings of hippocampal CA1 extracellular field excitatory postsynaptic potentials (fEPSP) (A) and astroglial membrane depolarizations (C) were performed and representative traces are illustrated. (B) Increasing the stimulation length from 0.1 to 0.5 ms resulted in comparable presynaptic activity (fiber volley), but enhanced postsynaptic activity (charge transfer, p < 0.05) in Cx30−/−Cx43−/− mice (n = 6), as compared with the wildtype mice (n = 7). Scale bar 0.2 mV, 25 ms. (C, D) The extracellular potassium accumulation is increased (amplitude p < 0.05) and prolonged (decay, p < 0.001 for 0.1ms pulse width and p < 0.005 for 0.5 ms pulse width) in slices from Cx30−/−Cx43−/− mice (n = 6) compared with wildtype mice (n = 7), as measured by the astroglial membrane depolarization. Scale bar, 1 mV, 1 sec. (E, F) Spontaneous bursts, induced by inhibition of GABAergic transmission (100 µM picrotoxin) and removal of extracellular Mg2+, and recorded extracellularly in the hippocampal CA1 area, occur more frequently in Cx30−/−Cx43−/− mice (p < 0.001, n = 16) than in wildtype mice (n = 16). Scale bar, 0.2 mV, 30 sec. However, amplitude (p < 0.001), as well as charge transfer (p < 0.001), are reduced in Cx30−/−Cx43−/− mice (n = 16, WT n = 16). High magnifications of representative traces are shown on the right. Scale bar, 0.2 mV, 1s.
In mice with intact astroglial networks, increasing the stimulus length by 5-fold enhanced the presynaptic input by ~7-fold, as measured by the fiber volley amplitude (n = 7, Fig. 1B), and also resulted in synaptically-induced firing, detected as population spike (Fig. 1A).
However, in the absence of functional astroglial networks, even though the presynaptic presynaptic activity was comparable to the wildtype response (fiber volley amplitude, Fig. 1C), the postsynaptic activity was dramatically amplified (n = 6, Fig. 1B), and resulted mostly from the massive increase in synaptically-evoked firing, as measured by the charge of the evoked response, consisting of the field fEPSP and predominantly of the population spike [p < 0.05; Cx30-/-Cx43-/-, n = 6, wildtype (WT), n = 7, Fig. 1A]. Therefore dysfunction of astroglial network communication results in a distortion of neuronal information processing, due to the amplification of the postsynaptic response.
Because postsynaptic receptor activation is the main source (~80%) of extracellular potassium [(K+)o] during neuronal activity,8,9 persistent postsynaptic activity in the absence of functional astroglial networks should result in an accumulation of extracellular potassium due to impaired buffering.6 We indeed found that the synaptically-evoked astroglial membrane depolarization, a sensitive measure of extracellular potassium levels,10 was massively enhanced (peak amplitude, p < 0.005 for 0.1 and 0.5 ms pulse width, Fig. 1C-D) and sustained (decay, p < 0.001 for 0.1 ms pulse width and p < 0.005 for 0.5 ms pulse width, Fig. 1C-D) in Cx30-/-Cx43-/- mice (n = 6, WT n = 7), for both stimulation paradigms.
Interestingly in adult mice, absence of gap junction coupling between astrocytes and oligodendrocytes, also mediated by Cx43 and Cx30,11 may also lead to altered potassium buffering by oligodendrocytes, and further increase [K+]o.
Amplification and prolongation of the fEPSP most likely result from the combination of: 1) enhanced and prolonged extracellular potassium levels, due to altered potassium buffering by disconnected astrocytes, which in turn result in persistent spine depolarization facilitating receptor activation; 2) extracellular glutamate accumulation caused by decreased astroglial glutamate clearance rate in Cx30-/-Cx43-/- mice,7 which furthermore results in enhanced spillover of glutamate, activating neighboring synapses, as we recently found7; and 3) a severely decreased extracellular space volume during enhanced activity in Cx30-/-Cx43-/- mice, due to the increased swelling and delayed deswelling of disconnected astrocytes, as we observed during a train of stimulation (10 Hz, 10s),7 which should also further augment extracellular potassium and glutamate concentrations, and lead to an accumulation of diffusion barriers slowing down extracellular diffusion. Altogether these results indicate that astroglial network communication precisely tunes pre-to-postsynaptic signaling and prevents amplification of neuronal activity.
Such uncontrolled amplification of neuronal activity, especially in the highly recurrently connected hippocampal formation, could result in hypersynchronous firing, a hallmark of epilepsy. Indeed a previous study already reported enhanced seizure susceptibility of Cx30-/-Cx43-/- mice in response to low-frequency stimulation, as well as during persistent NMDA receptor activity.6 However a detailed analysis of the epileptiform activity and of the underlying mechanisms was not performed. Since our previous results indicate that in the absence of functional astroglial networks, both, excitatory, as well as inhibitory transmission increased,7 we first investigated whether the observed alterations in the excitatory drive alone could result in the enhanced seizure susceptibility.
We found that astroglial network communication reduces bursting activity in an acute pharmacological model of epileptic-like activity (0 Mg2+, picrotoxin). Indeed, the frequency of interictal events was enhanced by nearly 3-fold in the absence of astroglial networks (Cx30-/-Cx43-/- mice p < 0.001; 4.5 ± 0.1 min−1, n = 16) compared with wildtype mice (1.7 ± 0.05 min−1; n = 16, Fig. 1E and 1F). However, detailed analysis of individual events revealed smaller burst amplitudes and reduced charge transfer (Fig. 1F) in slices from Cx30-/-Cx43-/- mice (n = 16) compared with wildtype (n = 16).
The increase in frequency of epileptiform events in hippocampal slices from Cx30-/-Cx43-/- mice may result from accumulation of extracellular potassium.7 Indeed an increase in extracellular potassium from 5 to 10 mM results in a 5-fold increase in the frequency of interictal events.12 Furthermore the enhanced postsynaptic AMPA receptor density, as well as the pronounced glutamate spillover, activating neighboring inactive synapses, should also contribute to synchronous firing and the increase in burst frequency. The reduced amplitude of epileptiform events in Cx30-/-Cx43-/- mice (Fig. 1F) might result from stronger depolarizations of CA1 pyramidal cells due to the enhanced extracellular potassium levels, leading to decreased excitatory postsynaptic activity. Alternatively, the decreased amplitude may come from a reduced number of neurons firing during an individual event, or from an altered energy supply, which is partially mediated by astroglial networks.4,13
The enhanced neuronal firing should also further elevate extracellular potassium levels. This might contribute to inappropriate information processing due to altered membrane depolarizations and repolarizations, resulting in dysfunction of voltage-gated ion channels, necessary for the generation of action potentials. The altered dynamics of extracellular potassium in Cx30-/-Cx43-/- mice should therefore prevent fast and adequate membrane depolarizations and hyperpolarizations, necessary for temporally and spatially-restricted channel activity.
In addition to the disturbed extracellular potassium homeostasis in Cx30-/-Cx43-/- mice, we also found insufficient astroglial glutamate clearance, measured by decreased glutamate transporter (GLT) clearance rate during basal evoked synaptic transmission (single stimulation of Schaffer collaterals).7 This results most likely from disturbance in K+ and Na+ gradients driving glutamate uptake. Indeed, a reduced electrogenic drive may be due to the altered intracellular ion homeostasis caused by the lack of intercellular coupling, and/or to alterations in astroglial membrane potential dynamics, resulting from enhanced extracellular potassium levels during neuronal activity.14 Interestingly, besides the well known involvement of gap junctions in potassium buffering, an activity-dependent Na+ spread through gap junction networks was shown in a recent study.13 We found that GLT dysfunction results in accumulation of released glutamate at synaptic, as well as at extrasynaptic sites, which increased postsynaptic AMPA receptor responses.7 However for higher regimes of activity, such as the bursting activity we induced acutely, further accumulation of glutamate in Cx30-/-Cx43-/- mice may lead to desensitization of AMPA receptors, as well as to activation of extrasynaptic metabotropic glutamate receptors, thereby limiting the development of uncontrolled excessive activity. It is furthermore possible that the impaired astroglial metabolic supply to neurons in Cx30-/-Cx43-/- mice,4 important during persistent neuronal activity, also contributes to limit the aberrant bursting activity.
Inter-astroglial connectivity provides individual astrocytes with large uptake and buffering capacities for potassium and glutamate, since intracellular accumulation can be prevented due to the fast redistribution via the network. Interestingly, our previous work showed that disconnection of astrocytes is still associated with a large uptake capacity,7 suggesting that disconnected astrocytes manage to redistribute and release the uptaken substances locally within their own microdomains. Nevertheless, we previously found that the glutamate clearance time course is decelerated,7 demonstrating a reduced functionality of GLT in Cx30-/-Cx43-/- astrocytes. We therefore investigated which strategies uncoupled astrocytes use to compensate for the lack in network redistribution capacity.
Since hippocampal excitatory transmission was enhanced in juvenile Cx30-/-Cx43-/- mice7 due in part to increased synaptic glutamate release, and expression of glutamate transporters is thought to be activity-dependent,15,16 we first analyzed whether hippocampal astrocytes exhibit expression changes in GLT-1 and GLAST, the two astroglial glutamate transporter subtypes. However protein levels of these two transporters, mainly expressed by glial cells, were indistinguishable between Cx30-/-Cx43-/- and wildtype mice (Fig. 2A). Thus in juvenile mice (P16–25), we did not observe the increase in GLT-1 and GLAST protein levels recently reported in the cortex and hippocampus of adult Cx30-/-Cx43-/- mice.17 This increase in adult Cx30-/-Cx43-/- mice might reflect a compensatory upregulation to counteract the enhanced excitatory transmission and decreased glutamate clearance rate,7 although a functional upregulation of glial glutamate uptake was not yet shown in adult Cx30-/-Cx43-/- mice.
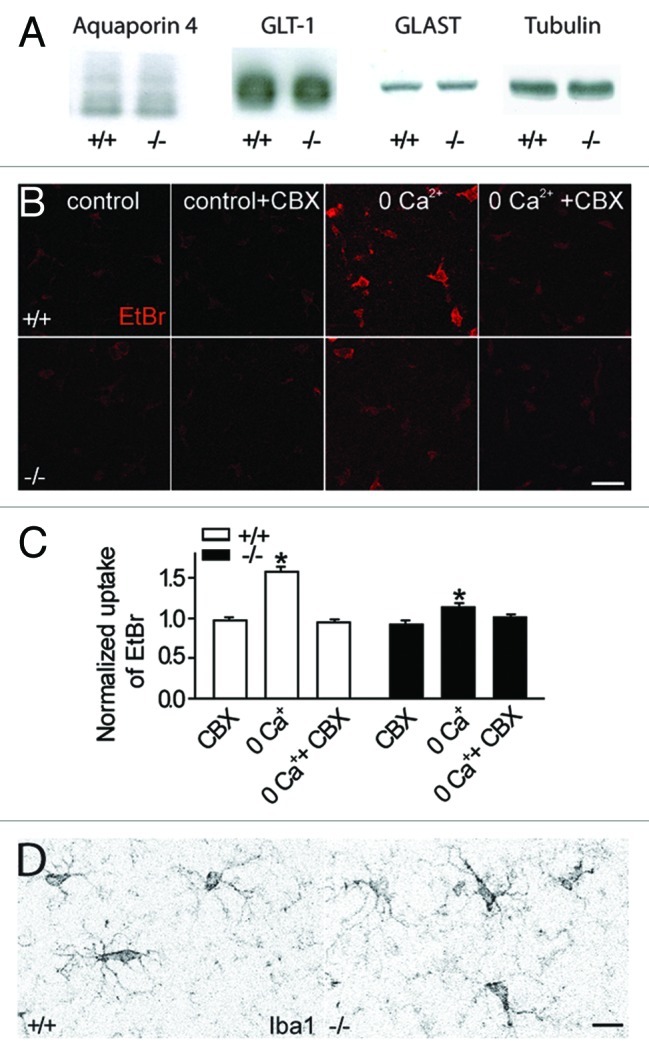
Figure 2. Absence of compensatory upregulation of water channels, glutamate transporters, opening of pannexin hemichannels or reactive microglia in Cx30-/-Cx43-/- mice. (A) Quantitative immunoblot analysis of hippocampal extracts from Cx30-/-Cx43-/- mice (n = 3) and wild type (n = 3) revealed no difference in protein expression for the water channel aquaporin 4 (~30 kD) and the glial glutamate transporters GLT-1 (~70 kD) and GLAST (~60 kD). Tubulin was used as a loading control. (B, C) Under control conditions, no enhanced ethidium bromide (EtBr) uptake was detectable in astrocytes from Cx30-/-Cx43-/- mice. Preincubation with carbenoxolone (CBX, 200 µM) for 15 min, a connexin and pannexin hemichannel blocker, has no effect on basal astroglial ethidium bromide uptake in wildtype (n = 50) and Cx30-/-Cx43-/- mice (n = 30). Removal of extracellular calcium (0 Ca2+) induced a carbenoxolone sensitive EtBr uptake in astrocytes from wildtype mice (n = 50), and to a much smaller extend in Cx30-/-Cx43-/- mice (n = 36). Scale bar, 25 µm. (D) Immunostaining of hippocampal slices with the microglia marker Iba-1 revealed no sign of microglia activation in slices from Cx30-/-Cx43-/- mice (n = 3; wild type n = 3). Scale bar, 10 µm.
We also found that disconnected astrocytes display several morphological changes, including a prominent swelling, a larger domain area, as well as enhanced GFAP and vimentin levels, reflecting reactive gliosis.7 Interestingly, a preferential swelling of astrocytic processes,18 known to cover synaptic compartments,19 may well occur, as shown during hypotonic stress.18 In addition, process volume changes depend on the expression of GFAP,18 which is increased in Cx30-/-Cx43-/- mice.7
Intracellular accumulation of potassium and glutamate in individual disconnected astrocytes should result in enhanced co-entry of water to equilibrate intracellular osmolarity, likely responsible for the swelling of Cx30-/-Cx43-/- astrocytes. Remarkably, a functional relationship has been proposed in astrocytes between connexin 43 gap junctions and aquaporin-4 water channels, prominently expressed in astrocytes.20 Indeed aquaporin-4 knockdown leads to a downregulation of Cx43 and associated cell coupling, as well as to an alteration in cell morphology.21 However our data, as well as previous results,11 suggest that the function of aquaporin-4 channels is unaltered in Cx43-/-Cx30-/- astrocytes. Indeed aquaporin-4 protein levels are unchanged (Fig. 2A). Furthermore disconnected astrocytes display larger domain area and important swelling, changes opposite to what is reported when aquaporin-4 expression is reduced.21
Finally uncoupled astrocytes, to compensate for the intracellular accumulation of potassium and glutamate, might open pannexin-mediated hemichannels to release potassium and/or glutamate at sites of lower concentration, and thereby balance intracellular osmolarity. Previous work revealed the presence of pannexin-1 hemichannels in astrocytes,22 enabling the release of gliotransmitters,23 and the opening of such channels by membrane depolarization.24 However carbenoxolone (CBX, 200 μM), known to inhibit connexin, as well as pannexin channels,25 did not reveal any additional functionality of these hemichannels in astrocytes, as assessed by ethidium bromide uptake assay in slices from Cx30-/-Cx43-/- mice (n = 26) or from wildtype mice (n = 37) in the presence of normal calcium concentration, as used throughout our experiments (Fig. 2B, C). Nevertheless, in the absence of extracellular calcium, we detected a prominent opening of CBX sensitive-hemichannels in wildtype mice (n = 49, in CBX n = 38), while in Cx30-/-Cx43-/- mice, a small uptake activity (~10%) was likely mediated by pannexin hemichannels (n = 33), since it was sensitive to CBX (n = 40, Fig. 2C). Hence we could not detect a compensatory opening of pannexin hemichannels in Cx30-/-Cx43-/- astrocytes in control conditions.
Since we reported that uncoupled astrocytes had enhanced GFAP and vimentin levels, reflecting reactive gliosis,7 we investigated whether microglial cells were reactive in Cx30-/-Cx43-/- mice, and thus could also possibly contribute to the alterations we reported in neuronal activity. Indeed, previous work revealed that elevation of extracellular potassium can activate microglial cells,26 which can release cytokines such as TNFα, known to increase the density of postsynaptic AMPA receptors enhancing synaptic strength,27 decrease glutamate clearance28 and promote excitotoxicity.29,30 However we did not observe morphological characteristics of reactive microglia in Cx30-/-Cx43-/- mice, such as increased soma size (Cx30-/-Cx43-/-: 30.1 ± 1.1µm2, n = 13; wt: 33.7 ± 2.1µm2, n = 14) or Iba1 reactivity (mean fluorescence intensity (arbitrary units): Cx30-/-Cx43-/-: 18.2 ± 0.8, n = 13; wt: 23.8 ± 1.4, n = 14), or signs of process retraction in hippocampal slices from Cx30-/-Cx43-/- mice stained by the Iba-1 marker (Fig. 2D). Although mild activation not affecting microglia morphology could occur, previous work showed that high levels of extracellular potassium are necessary to cause microglia activation.26 Therefore more detailed analysis of microglial properties in Cx30-/-Cx43-/- mice would be necessary to fully exclude an involvement of these cells in the observed alterations.
In conclusion, our work shows that astroglial network communication restricts neuronal activity temporally and spatially. Therefore gap junction-mediated glial networks, by contributing to extracellular neurotransmitter and ion homeostasis through regulation of astroglial clearance rate and extracellular space volume, enable precise synaptic information transfer, processing and storage. Indeed, astroglial gap junctions limits neuronal excitability, release probability, spillover, the number of functional synapses7 and the generation of neuronal network activity. Importantly, this has a major impact for synaptic plasticity, and favors synaptic potentiation.7
Interestingly, astroglial networks have also been proposed to participate to heterosynaptic depression, another form of hippocampal synaptic plasticity, by releasing ATP,5 suggesting that other mechanisms potentially involving coordinated release of gliotransmitters by astroglial networks, may also restrict and dampen synaptic transmission.
Such astroglial networks are also likely to be functionally crucial in pathological conditions associated to enhanced release of ions and neurotransmitters, and should therefore preserve synapse independence and restrict excitotoxicity. Thus our work reveals the cellular and molecular mechanisms underlying the key role of astroglial connexins in cognitive functions and possibly in psychopathology, as suggested by recent studies reporting altered expression of astroglial connexins in human brains of patients with psychiatric disorders including autism31 and major depression32 or of suicide completers.33 Since astroglial gap junctional communication is disrupted in many pathological conditions (neuroinflammation, excitotoxic injury, multiple sclerosis, epilepsy, oculo-dentodigital dysplasia, neurodegenerative diseases, psychopathologies), connexins should be a promising therapeutic target to restore normal brain information processing.
Materials and Methods
Animals
Experiments were performed according to the guidelines of the European Community Council Directives of November 24th 1986 (86/609/EEC). Experiments were performed on the hippocampus of wild type mice and Cx30-/-Cx43fl/fl:hGFAP-Cre mice (Cx30-/-Cx43-/-, double knockout) (provided by Pr. Willecke, University of Bonn, Germany), with conditional deletion of Cx43 in astrocytes34 and additional total deletion of Cx30,35 as previously described.6 For all analyses, mice of both genders and littermates were used (P16-P25).
Electrophysiology
Acute transverse hippocampal slices (300–400 µm) were prepared as previously described36 from 16–25 d-old wild-type and Cx30-/-Cx43-/- mice. Slices were maintained at room temperature in a submerged storage chamber containing an artificial cerebrospinal fluid (ACSF) (containing in mM: 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3 and 11 glucose, saturated with 95% O2 and 5% CO2). Slices were stored either in ACSF or, for generation of epileptiform activity in Mg-free ACSF with 100 µM picrotoxin for at least one hour prior to recording. Slices were transferred to a submerged recording chamber mounted on an Olympus BX51WI microscope equipped for infrared-differential interference (IR-DIC) microscopy and were perfused with ACSF at a rate of 1.5 ml/min at room temperature. All experiments were performed in the presence of picrotoxin (100 µM) and a cut was made between CA1 and CA3 to prevent the propagation of epileptiform activity, except for recordings of epileptic discharges. Extracellular field and whole-cell patch-clamp recordings were performed. Evoked postsynaptic or astrocytic responses were induced by stimulating Schaffer collaterals (0.1 Hz) with ACSF filled glass pipettes. Stratum radiatum astrocytes were identified by their small cell bodies, low input resistance (~20 MΩ), high resting potentials (~-80 mV) and linear IV curves. Field excitatory postsynaptic potentials (fEPSPs) were recorded with glass pipettes (2–5 MΩ) filled with ACSF and placed in stratum radiatum. Somatic whole-cell recordings were obtained from visually identified stratum radiatum astrocytes, using 5–10 MΩ glass pipettes filled with (in mM): 105 K-gluconate, 30 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Tris, 0.3 EGTA (pH 7.4, 280 mOsm); Astroglial whole-cell membrane depolarizations were evoked by stimulation of Schaffer collaterals and were recorded simultaneously with fEPSPs. The field recording pipette was placed 50–100 µm away from the recorded astrocyte. Epileptiform activity recordings were performed in Mg-free ACSF with the field recording electrode placed in the stratum radiatum region of the hippocampus.
Recordings were acquired with Axopatch-1D amplifiers (Molecular Devices), digitized at 10 kHz, filtered at 2 kHz, stored and analyzed on computer using Pclamp9 and Clampfit9 software (Molecular Devices). All data are expressed as mean ± SEM. Statistical significance for comparisons was determined by unpaired t-tests. picrotoxin and carbenoxolone were obtained from Sigma.
Antibodies
The following primary antibodies were used: GLT-1 and GLAST rabbit polyclonal antibodies (Frontier Science Co); Aquaporin 4 rabbit monoclonal antibody, Tubulin mouse monoclonal antibody (Sigma) and Iba-1 rabbit ployclonal antibody (Wako chemichals). The following HRP conjugated secondary antibodies were used: Donkey anti rabbit IgG (Amersham Biosciences) and Goat anti-mouse IgG (Santa-Cruz). The fluorescent conjugated secondary antibody was used in appropriate combinations: Goat anti-mouse IgG Alexa 488 conjugated and Goat anti-rabbit IgG Alexa 488 conjugated (Molecular probes).
Immunohistochemistry
Mice were anesthetized, perfused with PBS and their brains rapidly removed and frozen in isopentane cooled at -30°C. Coronal sections (20 µm) were cut on a cryostat, collected on slides and fixed with 4% paraformaldehyde in PBS for 30min at 4°C. Coronal sections or fixed hippocampal slices were permeabilized and immuno-blocked with PBS, containing 0.2% gelatin and 0.2% Triton-X100, for 1h and processed for immunostaining by overnight incubation at 4°C with primary antibodies diluted in PBS. After three washes, sections were incubated for 2 h at room temperature with appropriate secondary antibodies. After several washes, slices were mounted in Fluoromount (Southern Biotechnology) and examined with a confocal laser-scanning microscope (Leica TBCS SP2, SP5), equipped with 16, 40 and 63 x objectives. Stacks of consecutive confocal images taken at 0.5 µm intervals were acquired sequentially with two lasers (argon 488 nm) and Z projections were reconstructed using Leica Confocal Software. Cell soma size and immunoreactivity were measured in Image J on overlaid projections of several consecutive images.
Immunoblotting
Hippocampi were frozen, pulverized and homogenized in 2% SDS with protease inhibitor cocktail, β-glycerophosphate (10 mM) and orthovanadate (1 mM). Equal amounts of protein were separated on 10% PAGE gel followed by transfer to nitrocellulose membranes. Proteins were detected by immunoblotting using the HRP-ECL kit from Perkin Elmer. Tubulin was used as loading control.
Dye uptake by hemichannels
The hemichannel permeable fluorescent tracer ethidium bromide (EtdBr, 314 Da) was included in either ACSF or ACSF/0Ca2+/5 mM EGTA solutions at a final concentration of 4 μM. Slices were incubated for 10 min in the solutions (equilibrated with 95% O2 - 5% CO2, at RT). In blocking experiments, slices were pre-incubated 15 min prior to and during EtdBr application with the GJ and connexin/pannexin hemichannel blocker carbenoxolone (CBX, 200 μM). Slices were then rinsed 15 min in ACSF, fixed for 2 h in 4% paraformaldehyde in 0.12 M buffer phosphate and mounted in Fluoromount. Labeled cells were examined in a confocal laser-scanning microscope (TCS SP2, Leica) with a 63x objective. Stacks of consecutive confocal images were taken at 300 nm intervals and acquired with a laser (561 nm); Z projections were reconstructed using the LAS AF software. At least three fields were selected in each slice. Fluorescence was digitized in arbitrary units (AU) with image J processing software. Dye uptake was expressed as the difference between the fluorescence measured in cells (5–10 per slice) and the background fluorescence measured where no labeled cells were detected. Values of fluorescence in different experimental conditions were normalized relative to the control level.
Acknowledgments
We thank K. Willecke for providing the Cx30-/-Cx43fl/fl:hGFAP-Cre mice, and P. Ezan for his technical assistance. This work was supported by grants from the HFSPO (Career Development Award), ANR (Programme Jeunes chercheurs and Programme Blanc Neurosciences), FRC (Fédération pour la Recherche sur le Cerveau), INSERM and La Pitié Salpêtrière hospital (Translational research contract) to N.R., from French Research Ministry and Deutsche Forschungsgemeinschaft postdoc fellowships to U.P., and from FRM postdoc fellowships to M.D. and O.C.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/19410
References
- 1.Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–31. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–92. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–6. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 4.Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–5. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- 5.Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–82. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–47. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, et al. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 2011;108:8467–72. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol. 1987;58:404–16. doi: 10.1152/jn.1987.58.2.404. [DOI] [PubMed] [Google Scholar]
- 9.Pumain R, Heinemann U. Stimulus- and amino acid-induced calcium and potassium changes in rat neocortex. J Neurophysiol. 1985;53:1–16. doi: 10.1152/jn.1985.53.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22:1042–53. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743–52. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutecki PA, Lebeda FJ, Johnston D. Epileptiform activity induced by changes in extracellular potassium in hippocampus. J Neurophysiol. 1985;54:1363–74. doi: 10.1152/jn.1985.54.5.1363. [DOI] [PubMed] [Google Scholar]
- 13.Langer J, Stephan J, Theis M, Rose CR. Gap junctions mediate intercellular spread of sodium between hippocampal astrocytes in situ. Glia. 2012;60:239–52. doi: 10.1002/glia.21259. [DOI] [PubMed] [Google Scholar]
- 14.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 15.Genoud C, Quairiaux C, Steiner P, Hirling H, Welker E, Knott GW. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006;4:e343. doi: 10.1371/journal.pbio.0040343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perego C, Vanoni C, Bossi M, Massari S, Basudev H, Longhi R, et al. The GLT-1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem. 2000;75:1076–84. doi: 10.1046/j.1471-4159.2000.0751076.x. [DOI] [PubMed] [Google Scholar]
- 17.Unger T, Bette S, Zhang J, Theis M, Engele J. Connexin-deficiency affects expression levels of glial glutamate transporters within the cerebrum. Neurosci Lett. 2012;506:12–6. doi: 10.1016/j.neulet.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 18.Chvátal A, Anderová M, Hock M, Prajerová I, Neprasová H, Chvátal V, et al. Three-dimensional confocal morphometry reveals structural changes in astrocyte morphology in situ. J Neurosci Res. 2007;85:260–71. doi: 10.1002/jnr.21113. [DOI] [PubMed] [Google Scholar]
- 19.Hirrlinger J, Hülsmann S, Kirchhoff F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 2004;20:2235–9. doi: 10.1111/j.1460-9568.2004.03689.x. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicchia GP, Srinivas M, Li W, Brosnan CF, Frigeri A, Spray DC. New possible roles for aquaporin-4 in astrocytes: cell cytoskeleton and functional relationship with connexin43. FASEB J. 2005;19:1674–6. doi: 10.1096/fj.04-3281fje. [DOI] [PubMed] [Google Scholar]
- 22.Santiago MF, Veliskova J, Patel NK, Lutz SE, Caille D, Charollais A, et al. Targeting pannexin1 improves seizure outcome. PLoS One. 2011;6:e25178. doi: 10.1371/journal.pone.0025178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–96. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–8. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Spray DC, Ye ZC, Ransom BR. Functional connexin “hemichannels”: a critical appraisal. Glia. 2006;54:758–73. doi: 10.1002/glia.20429. [DOI] [PubMed] [Google Scholar]
- 26.Abrahám H, Losonczy A, Czéh G, Lázár G. Rapid activation of microglial cells by hypoxia, kainic acid, and potassium ions in slice preparations of the rat hippocampus. Brain Res. 2001;906:115–26. doi: 10.1016/S0006-8993(01)02569-0. [DOI] [PubMed] [Google Scholar]
- 27.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–5. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 28.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271:15303–6. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- 29.Balosso S, Ravizza T, Perego C, Peschon J, Campbell IL, De Simoni MG, et al. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann Neurol. 2005;57:804–12. doi: 10.1002/ana.20480. [DOI] [PubMed] [Google Scholar]
- 30.Balosso S, Ravizza T, Pierucci M, Calcagno E, Invernizzi R, Di Giovanni G, et al. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: implications for seizure susceptibility. Neuroscience. 2009;161:293–300. doi: 10.1016/j.neuroscience.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Fatemi SH, Folsom TD, Reutiman TJ, Lee S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008;62:501–7. doi: 10.1002/syn.20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2011;16:634–46. doi: 10.1038/mp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ernst C, Nagy C, Kim S, Yang JP, Deng X, Hellstrom IC, et al. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol Psychiatry. 2011;70:312–9. doi: 10.1016/j.biopsych.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 34.Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–76. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 36.Rouach N, Byrd K, Petralia RS, Elias GM, Adesnik H, Tomita S, et al. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–33. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]