

Abstract
Superoxide and its derivatives have been implicated as secondary messenger molecules that influence signaling cascades in non-phagocytes. B lymphocytes produce superoxide after BCR ligation. We found that these reactive oxygen species (ROS) regulate B cell signaling and entry into the cell cycle. B cells from mice deficient in the gp91phox subunit of the NADPH oxidase complex are unable to generate ROS after BCR ligation. However, after BCR stimulation, more gp91phox KO B cells enter the G1 stage of the cell cycle and proliferate than WT B cells. BCR ligation leads to a more rapid decrease in p27Kip1 levels in gp91phox KO B cells. Gp91phox KO mice display enhanced TI-2, but normal T-dependent antibody responses. ROS-dependent regulation of BCR-induced proliferation may help modulate the size of the humoral response to T cell-independent type 2 (TI-2) Ag immunization.
Keywords: ROS, B cell Ag receptor, cell cycle
Introduction
Signaling through the BCR controls a variety of important cell fate decisions [1–3]. Depending on a B cell’s differentiation state and the context of the signal, BCR ligation can lead to cell survival, apoptosis, proliferation or differentiation [1, 3–7]. The protein tyrosine kinases (PTK) Syk and Lyn, a Src-family kinase, are essential for signal initiation [1]. However, outcomes after BCR ligation are determined not only by which enzymatic cascades are triggered, but how they are modulated and attenuated [3]. Signal propagation relies on the balance between protein phosphorylation and dephosphorylation. Thus, activation of protein tyrosine phosphatases (PTP), which counter the activity of PTK, is also important [8].
Recent findings have demonstrated that modulation of PTP activity may have important consequences for BCR signaling, and PTP activity may be inhibited by oxidation [9–11]. Hydrogen peroxide, a type of reactive oxygen species (ROS), has long been used as a positive control in B lymphocyte signaling studies, as treatment of B cells with hydrogen peroxide (H2O2), similar to BCR ligation, results in strong upregulation of PTK activity and protein tyrosine phosphorylation [11]. However, H2O2 is likely to act by inhibition of PTP activity rather than by direct activation of PTK. Many non-phagocytic cells produce ROS after mitogenic stimulation, and these ROS modulate signaling pathways via inhibition of PTP [9, 12, 13]. For example, TCR-dependent induction of ROS inhibits the PTP SHP-2; inhibition of SHP-2 helps promote signaling to integrins and thus, T cell adhesion [13]. Receptor-induced generation of ROS has been demonstrated to augment enzymatic cascades downstream of platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) [14, 15]. Non-phagocytic cells make much smaller amounts of endogenous ROS compared to phagocytes; however, the enzyme utilized by non-phagocytic cells is often the same enzyme that phagocytes employ during their oxidative burst.
Many non-phagocytic cells, including B cells, express the NADPH oxidase (NOX) [16, 17, 17–19]. This multi-component enzyme inducibly generates superoxide after cell stimulation. In a resting cell, the NOX components are divided into two groups: the membrane-bound gp91phox catalytic subunit and p22phox subunit and the cytosolic p47phox, p67phox, and p40phox regulatory subunits [16, 17]. Upon stimulation, the cytosolic components translocate to the membrane, where, in conjunction with gp91phox, p22phox and activated Rac, they form the active NOX complex [16, 17]. In B cells, BCR ligation is a stimulus that leads to superoxide generation.
While the response of B cells to H2O2 suggested a role for ROS in B cell signaling, this role has not been fully examined. Singh et al. reported that ROS play a role in regulating the activity of Lyn in a mouse B cell line [9, 10]. Their data using inhibitors also suggested a link between ROS generation and calcium signaling; because these studies used a continuously dividing cell line, investigating a role for ROS in regulating B cell entry into the cell cycle was not examined. For this reason and because the outcome of signaling can differ between normal B cells and B cell lines, we decided to investigate the role for ROS in B cell signaling using B cells from mice deficient in the catalytic component of the NOX complex, gp91phox [20]. We found that the absence of BCR-generated superoxide specifically impacts BCR-dependent signaling outcomes. gp91phox KO B cells show increases in BCR-induced cell cycle entry and proliferation. This in vitro defect was accompanied by dysregulation in antibody responses of gp91phox KO mice to T cell-independent type 2 (TI-2) but not T cell-dependent (TD) Ag.
Results
BCR-Generated Superoxide is Downstream of Calcium Flux
We first tested whether gp91phox KO B cells were incapable of producing superoxide after BCR crosslinking. As expected, loss of gp91phox completely abrogated superoxide generation after BCR ligation (Fig. 1A). The data generated by Singh et al. suggested that gp91phox KO B cells could have defects early in the BCR-dependent signaling pathway [9]. They found that incubating A20 B cells with antioxidants prior to BCR ligation resulted in reduced BCR-dependent calcium responses, suggesting ROS may regulate BCR-dependent calcium mobilization. Therefore, we compared BCR-induced intracellular calcium release in WT vs. gp91phox KO B cells (Fig. 1B). The data revealed that absence of BCR-dependent superoxide does not impact calcium mobilization in normal B cells, whether peak/base ratios or percent responding cells were compared. Furthermore, when we incubated WT and gp91phox KO B cells with the antioxidant NAC, BCR-dependent calcium release was reduced comparably in B cells from both genotypes (Fig. 1F). Thus, NAC affects B cells whether or not they produced superoxide, and NOX-derived superoxide is not required for BCR-induced calcium release.
Fig. 1.

BCR-induced superoxide production is downstream of calcium signaling. In (A, C–E) Diogenes chemiluminescent reagent was used to detect superoxide, shown in relative light units (RLU). (A) WT (triangles) and gp91phox KO B cells (circles) were left unstimulated (open), or stimulated with 10 μg/mL (black) anti-IgM sera. Arrow shows time of addition of anti-IgM. (B) Release of intracellular calcium was measured using Indo-1-labeled WT (grey bars) or gp91phox KO (black bars) B cells stimulated with graded doses of anti-IgM or ionomycin. (C) WT (square) or Syk-KO(triangle), Btk-KO (circle), or PLCγ2-KO (hatched diamond) DT40 chicken B cells were stimulated with anti-IgM. WT DT40 B cells were left unstimulated (black diamond) for comparison. (D) WT DT40 B cells were pre-incubated with DMSO only (square) or 10 μM (triangle), 20 μM (hatched diamond) or 50 μM (circle) BAPTA/AM before stimulation with anti-IgM. DMSO-only cells were left unstimulated (black diamond) as a control. (E) Primary WT B cells were preincubated with (circle) or without (square) 3 mM EGTA, then stimulated with 10 μg/mL anti-IgM or left unstimulated (diamond). (F) WT (grey) or gp91phox KO (black) B cells were loaded with Indo-1, then pre-incubated with or without 25 mM NAC. Calcium mobilization was measured after BCR ligation as above, using 30 μg/mL anti-IgM as stimulus. All data are representative of three independent experiments.
The signals required for NADPH oxidase activation have been examined primarily in neutrophils, either in whole cells or cell-free systems [21–23, 23, 24]. While NOX-derived ROS does not regulate BCR-dependent calcium flux (Fig. 1B), it remained unclear what signaling pathways are required for BCR-dependent NOX activation. Thus, using the chicken DT40 B cell system, we compared superoxide generation in WT cells with cells missing PTK known to be activated during BCR signaling. Cells missing either the Syk or Btk PTK or PLCγ2 did not produce superoxide after BCR ligation (Fig. 1C). The fact that Syk, Btk and PLCγ2 are required for BCR-induced mobilization of intracellular calcium (calcium flux) suggested that BCR-dependent calcium mobilization may be required for NOX activity in B cells. To test this possibility we utilized BAPTA/AM to cage calcium in DT40 B cells (Fig. 1D); graded doses of BAPTA/AM inhibited BCR-induced superoxide production, suggesting a dose-dependent reliance on calcium flux for BCR-dependent NOX activity. It was not possible to reproduce these findings with BAPTA/AM in normal mouse B cells. The DMSO vehicle abrogated the superoxide reaction at concentrations comparable to the necessary dose of BATPA/AM (data not shown). However, treatment of EGTA to reduce Ca2+ entry to B cells consistently reduced BCR-induced NOX-activity (Fig. 1E).
We also compared other early BCR-dependent processes in WT vs. gp91phox KO B cells including both BCR-dependent and constitutive internalization of the BCR (Fig. 2A). Neither of these processes was disrupted in gp91phox KO B cells. Global protein tyrosine phosphorylation appeared normal after BCR ligation (data not shown). Likewise, BCR capping in gp91phox KO B cells was not statistically different than capping in WT B cells (data not shown). Finally, we examined whether loss of an active NOX resulted in developmental defects in B cell subsets in vivo. WT and gp91phox KO had no significant differences in absolute numbers of splenic B cell subsets, and percentages of peritoneal B cell subsets were normal in KO mice (Fig. 2B) as well.
Fig. 2.
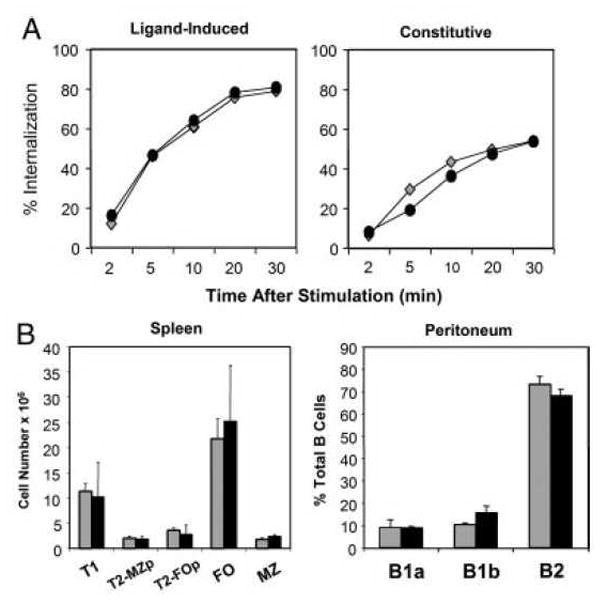
Early BCR-dependent signaling and B cell development are normal in gp91phox KO mice. (A) BCR internalization was analyzed using flow cytometry. To examine BCR internalization, WT (diamonds) or gp91phox KO (circles) B cells were incubated with F(ab′)2 anti-IgM to analyze ligand-induced internalization; or Fab anti-IgM to measure constitutive internalization. Data are representative of three experiments. (B) Splenic B subsets cells were analyzed as follows: Follicular (FO) B cells: IgMintIgDhiCD21lo; Transitional 1 (T1): IgDloIgMhiCD21lo. Transitional 2 Follicular Precursor (T2-FOp): IgDhiIgMhiCD21lo. Transitional 2 MZ precursor (T2-MZp): IgDhiIgMhiCD21hi. Marginal Zone (MZ): IgDloIgMhiCD21hi. WT (grey bars) and gp91phox KO (black bars). Peritoneal B cell subsets were analyzed using CD11b, CD5 and IgM as markers. B1a cells: CD11bhi, IgMlo, CD5hi. B1b cells: CD11bhi, IgMlo, CD5neg. B2: CD11bneg, IgMhi, CD5neg. Data mean + SD (n=3 spleens) and are representative of four independent experiments.
Loss of Superoxide Results in Enhanced BCR-Induced Proliferation
To explore the effect of NOX deficiency on B cell activation, we first labeled purified splenic B cells from WT or gp91phox KO mice with CFSE and then stimulated these cells with graded doses of anti-IgM. More gp91phox KO B cells proliferated at lower doses of anti-IgM than WT B cells (Fig. 3A, B) after 48 or 72 h. The enhanced proliferation of gp91phox KO B cells, while evident after BCR ligation, was not detected after stimulation with LPS (Fig. 3C) or after CD40 ligation (Fig. 3D). The selective elevated response of gp91phox KO B cells to BCR ligation did not appear to be related to differences in B cell populations, since B cell subset numbers were similar between WT and gp91phox KO mice (Fig. 2B). Furthermore, we compared the number of B cells in G0 or G1 of splenic WT and gp91phox KO B cells ex situ, and unlike with Caspase-6 KO B cells [25] there were no differences between WT and gp91phox KO B cells (data not shown).
Fig. 3.

gp91phox KO B cells display enhanced BCR-induced proliferation. Purified WT (grey bars) or gp91phox KO (black bars) B cells were loaded with CFSE and stimulated with graded doses of anti-mouse IgM (A, B), LPS (C), or anti-CD40 (D). Proliferation was measured by CFSE dilution at 48 or 72 h. ****p<0.006, ***p<0.02, **p<0.03, *p< 0.04, n.s., not significant; Student’s t-test. Data mean + SD (n≥3 spleens) and are representative of four independent experiments.
It was unclear where the dysregulation in the cell cycle of gp91phox KO B cells lay. After mitogenic stimulation, cells exit the quiescent stage of the cell cycle (G0), and enter the G1-phase [26, 27]. At this point they upregulate RNA and protein levels in preparation for DNA replication. Proteins which act to inhibit cell cycle progression, such as p27Kip1, are downregulated, and those which are necessary to promote cell cycle progression are upregulated. To examine early cell cycle events, we first compared changes in RNA levels at 24 h (Fig. 4A). More gp91phox KO B cells than WT B cells upregulated RNA 24 h after BCR stimulation. This was the case whether we used unfractionated B cells or dense B cells prepared as described [25]. To investigate potential mechanisms accounting for observed early cell cycle dysregulation, we measured the decrease of p27Kip1, an inhibitor of G1, and the increase of cyclin A, which is upregulated in S-phase (Fig. 4B). In gp91phox KO B cells, downregulation of p27Kip1 was enhanced, but cyclin A levels increased normally (Fig. 4B). The dysregulation of RNA and p27Kip1 levels suggested dysregulation early in the cell cycle. This suggests that ROS may modulate cell cycle entry by regulating a BCR-dependent pathway that maintains p27Kip1 levels.
Fig. 4.

More gp91phox KO B cells enter the cell cycle after BCR ligation. (A) 24 hr after stimulation via the BCR, WT and gp91phox KO B cells were incubated with 2 μg/mL Pyronin Y. Pyronin Y staining was measured by flow cytometry. WT (grey bars) and gp91phox KO (black bars). (B) Purified WT and gp91phox KO B cells were stimulated with 5 μg/mL anti-IgM F(ab′)2. Cells were cultured for the indicated time periods, collected and lysed. Western blots of cell lysates with anti-p27Kip1 or anti-cyclin A. All data are representative of three independent experiments.
Gp91phox KO Mice Display Dysregulated TI-2 Responses and IgG1 Production
While in vitro data from our lab and others support the hypothesis that ROS modulate BCR-dependent signaling pathways and cell fate, the in vivo role for superoxide in B cell signaling remained unexplored. To determine whether ROS regulate B cell responses in vivo, we first compared serum antibody levels in WT and gp91phox KO mice. Non-immunized gp91phox KO mice showed significantly enhanced serum titers of total IgG1 (Fig. 5A), which remained elevated as the mice aged, while titers of IgM, IgG3, IgG2a and IgG2b were normal (Fig. 5A, data not shown).
Fig. 5.

TI-2 antibody responses are enhanced in gp91phox KO mice. (A) Sera were collected from non-immunized WT (open circles) and gp91phox KO (closed circles) mice and specific isotype levels measured by ELISA. Horizontal bars show the mean. (B) WT (diamonds) and gp91phox KO mice (circles) mice were injected intra-peritoneally with 10 μg DNP-Ficoll. Sera were collected prior to immunization, then every week for two weeks. Ag-specific antibody titers were measured by ELISA. ****p<0.0001, ***p<0.002, **p< 0.005, *p<0.03; Student’s t-test, mean + SD, n= 5 mice per group.
To investigate whether the dysregulated proliferation in gp91phox KO B cells might translate into dysregulated Ag-specific in vivo antibody responses, we immunized WT and gp91phox KO mice with a TI-2 Ag, DNP-Ficoll. As shown in Fig. 5B, gp91phox KO mice displayed an enhanced Ag-specific antibody response after TI-2 Ag immunization. Both Ag-specific IgM and IgG3 were significantly elevated compared to levels in WT mice after injection of DNP-Ficoll. Gp91phox KO mice displayed significant increases in anti-DNP IgG1 as well.
IgG1 is mainly produced during TD responses. To determine whether gp91phox KO B cells display increased sensitivity to TD stimuli, first we compared WT and gp91phox KO B cell in vitro antibody responses to anti-CD40 plus IL-4 (Fig. 6A). While the gp91phox KO B cells produced more IgM at a low dose of anti-CD40, we detected no discernible differences in IgM production at higher doses. As the dose of anti-CD40 increased, the KO B cells produced less IgG1 compared to WT B cells. The in vivo Ag-specific IgG1 responses of gp91phox KO mice were normal at both doses of DNP-KLH tested (Fig. 6B). Thus, our data demonstrate that gp91phox KO mice generate enhanced responses to TI-2 immunization but not a TD antigen in vivo.
Fig. 6.

Reduced IgG1 production by gp91phox KO B cells in vitro does not correlate with dysregulated in vivo T-dependent antibody production by gp91phox KO mice. (A) WT (diamonds) or gp91phox KO (circles) B cells were cultured with medium alone (0) or 0.3, 1.0 or 3.0 μg/mL anti-CD40 plus 10 ng/mL IL-4 plus. After seven days, supernatants were collected and analyzed by ELISA. ***p≤0.0007, **p≤0.008, *p≤ 0.02; Student’s t-test, three mice per group. (B) WT (diamonds) and gp91phox KO (circles) mice were injected intra-peritoneally with either 5 μg (open) or 50 μg (closed) alum-precipitated DNP-KLH. Sera were collected prior to immunization and then every week for three weeks. Ag-specific antibody titers were measured by ELISA. n.s., not significant, student’s t-test. Data show mean + SD, n= 5 mice per group.
Discussion
Our data demonstrate the impact of ROS on cell fate decisions in normal B lymphocytes. ROS inhibit BCR-induced proliferation, regulating entry into the cell cycle. Loss of NOX results in enhanced BCR-mediated proliferation and dysregulated entry into the cell cycle in vitro, likely through diminished BCR-induced ROS production. Furthermore, loss of NOX results in increased Ag-specific TI-2 antibody responses in vivo.
Recent work has expanded our understanding of the role of ROS second messengers in non-phagocytic cells [10–13]. In many pathways, ROS inhibit the activity of PTP, promoting the amplification of PTK-dependent signaling. This activity may account for the efficacy of utilizing hydrogen peroxide as a positive control for BCR-dependent signaling studies. Previous studies indicating that ROS regulate enzymatic activity in B cells used pharmacologic ROS inhibitors in B cell lines [9]. We sought to extend these studies by investigating the role of NOX in normal B cells. Thus, we examined the role of ROS in normal mouse B cells by analyzing the impact of ROS on proliferation, differentiation, and immune responses. Because B cells employ the same NOX as phagocytes [17, 28], we chose the gp91phox KO strain [20].
We hypothesized that the absence of superoxide, and the theoretical gain of PTP activity, would result in a hyporesponsive phenotype. Surprisingly, we found that gp91phox KO B cells were hyperresponsive to BCR stimulation. Compared with controls, more gp91phox KO B cells proliferated after BCR ligation. In gp91phox KO B cells, BCR ligation led to a more rapid reduction of the cell cycle inhibitor p27Kip1 (Fig. 4B). We did not find dysregulation in the downregulation of another important cell cycle inhibitor p21Cip1 (data not shown). Furthermore, Cyclin D2, a key promoter of cell cycle progression in B cells [27], was also upregulated normally after BCR stimulation in gp91phox KO B cells (data not shown). These observations suggest that, through maintenance of p27Kip1 levels, ROS act as a brake on the G0 to G1 transition; and loss of this brake resulted in enhanced cell cycle entry and ultimately more proliferation of NOX-deficient B cells. Rather than preventing the upregulation of factors such as cyclin D2 which promote cell cycle progression, ROS appear to help sustain negative regulation of the cell cycle. Additionally, ROS appear to regulate Ag-specific functions of B cells, as gp91phox KO B cells stimulated with either LPS or anti-CD40 proliferated at normal levels (Fig. 3C, D).
Proliferation is an important facet of the effector B cell’s response to Ag. Immunization with TI-2 Ag results in a proliferative burst before Ag-specific B cells differentiate into plasma cells and begin producing immunoglobulin [29]. T-independent type 2 Ags are so named because little or no T cell help is required to generate an Ag-specific antibody response to immunization with them [30]. We hypothesized that an increased proliferative burst would generate a larger pool of possible plasma cells after TI-2 immunization, and lead to increased antibody production. Thus, we immunized the NOX-deficient mice with DNP-Ficoll to test whether increased proliferation in vitro would correlate with an increased TI-2 response. Indeed, the Ag-specific immunoglobulin response of the gp91phox KO mice was increased compared to WT mice (Fig. 5B). This suggests that a NOX-dependent pathway in B cells influences the quantity of the TI-2 response, perhaps by regulating the size of the proliferative burst prior to plasma cell formation. Currently, it is unclear what function NOX plays in selectively regulating BCR-dependent proliferation. As shown in Fig. 3B, loss of NOX more strongly impacts B cells exposed to lower doses of anti-IgM, for shorter periods. It may be that NOX regulates a pathway that sets the threshold for B cell activation after exposure to Ag.
NOX may also regulate pathways which modulate the quality of the immunoglobulin response. Unimmunized gp91phox KO mice had elevated levels of total serum IgG1 (Fig. 5A), which remained elevated as the mice aged. In addition to increases in anti-DNP IgM, we saw enhanced anti-DNP IgG1 titers in NOX deficient mice immunized with DNP-Ficoll (Fig. 5B). IgG1 is considered a classic T-dependent isotype, and normally is not produced in large quantities in TI-2 responses. We hypothesized that gp91phox KO B cells would display dysregulated responses to T-dependent signals. Thus, we stimulated purified B cells with IL-4 and graded doses of anti-CD40. At low doses of anti-CD40, NOX-deficient B cells produced slightly higher amounts of IgG1 than WT B cells. But at the highest dose of anti-CD40 tested, the gp91phox KO B cell produced significantly less IgG1 than WT B cells (Fig. 6A). However, when we immunized WT and gp91phox KO mice with a T-dependent Ag, DNP-KLH, gp91phox KO mice generated comparable Ag-specific IgG1 responses (Fig. 6B). The increased IgG1 titers seen in unimmunized gp91phox KO mice may be due to their sensitivity to TI-2 signals. As mentioned previously, lower doses of anti-IgM resulted in stronger comparative proliferation in gp91phox KO B cells (Fig. 3A, B). It may be that at low levels of ongoing TI-2 stimulation, the NOX-deficient B cells generate a stronger antibody response. However, it is unclear at this time why gp91phox KO B cells demonstrate a slight dysregulation to T-dependent signals in vitro, yet generate normal TD responses. Stimulation with a T-dependent-type signal (anti-CD40) plus BCR ligation promotes comparable proliferation between WT and NOX-deficient B cells (Fig. 3B). It may be that BCR signals in addition to T-dependent help allow WT B cells to proliferate and differentiate at the same rate as gp91phox KO B cells.
Previous studies investigating the signals which promote NOX complex assembly in neutrophils have implicated a variety of kinases, including protein kinase C family members, in this process [31]. However, investigations into the kinases important for NOX activation in B cells have lagged. Given the probable role of PKC in NOX assembly, we tested whether NOX activity depended on calcium mobilization in normal naive B cells. Our findings place BCR-generated NOX activity downstream of intracellular calcium release in normal B cells (Fig. 1B). Indeed, our data indicate that [Ca2+]i is required for BCR-induced ROS production (Fig. 1C–E). BCR-dependent NOX activity relies on Syk, crucial for signal initiation at the BCR complex, as well as Btk and PLCγ2, which regulate calcium mobilization downstream of BCR ligation.
Previous work by Singh and coworkers [9] implicated ROS in modulating BCR-induced calcium mobilization; their data were generated with cell lines and relied on inhibitors, and they identified a calcium-dependent dual oxidase (DUOX) as the ROS-generating enzyme in the A20 cell line [9]. However, normal naive B cells apparently do not rely on DUOX for BCR-dependent ROS induction, as our data demonstrated a complete absence of BCR-dependent superoxide production in cells lacking the catalytic subunit of NOX (Fig. 1A). Additionally, the deficit in BCR-dependent calcium mobilization seen in NOX-deficient cells after incubation with antioxidants was identical to that seen in WT B cells (Fig. 1F). This suggests that antioxidants may have off target effects, which influence signaling pathways independent of their ability to scavenge ROS generated by the NADPH oxidase. The differences between our findings and Singh may be due differences between B cell lines and normal B cells or because the use of inhibitors. Another possibility, but less likely in our view, is that since the A20 line is derived from an IgG+ B cell, memory and naive B cells may differ in how they regulate superoxide production. While we have identified gp91phox-dependent NOX as the enzyme responsible for BCR-induced superoxide generation in naive normal B cells (Fig. 1A), memory B cells, such as the A20 cell line, may rely on DUOX instead. Nevertheless, we can conclude that [Ca2+]i is upstream, and not downstream, of superoxide production in normal naive B cells.
ROS are thought to modulate signaling pathways by inhibiting PTP activity, helping to regulate the strength of signal. Despite their ability to inhibit a wide range of PTP, ROS have been demonstrated to act with selectivity [13]. Our own data investigating global BCR-induced tyrosine phosphorylation supports this; tyrosine phosphorylation appeared grossly normal in gp91phox KO B cells (data not shown). Currently, it is unclear which pathway downstream of calcium mobilization links NOX activity and regulation of p27Kip1 levels.
Many of the studies investigating superoxide’s role in modulating signaling pathways suggest that superoxide often enhances signaling pathways [12]. Indeed, if most PTP act to down-modulate signaling after mitogenic stimulation, a mechanism that inhibits this down-modulation would be expected to enhance those pathways. However, our data suggests that modulation of phosphorylation-dependent signaling pathways by superoxides may be much more complex. Our data demonstrates that the gp91phox-dependent NOX acts to inhibit pro-proliferative signaling after BCR stimulation. Indeed, phosphatases do not always counteract activation. Lyn, a Src family kinase, requires dephosphorylation at its negative regulatory site to achieve full activity.
In summary, the NAPDH oxidase in B cells appears to be involved specifically in a signaling pathway that negatively modulates BCR-induced proliferation. Loss of this enzyme activity results in dysregulated inhibition, leading to enhanced cell cycle entry and proliferation. Furthermore, this dysregulation in BCR-dependent proliferation likely impacts in vivo responses to immunization, resulting in enhanced TI-2 responses in gp91phox KO mice.
Materials and Methods
Mice
C57BL/6J Gp91phox KO mice were purchased from Jackson Laboratory [20]. WT C57BL6/Jmice (Jackson) were used as controls. Mice were housed under specific pathogen-free conditions, and all experiments were carried out in compliance with the University of Washington’s Institutional Animal Care and Use Committee (IACUC). Mice were utilized for experiments at 6 – 12 weeks of age.
B Cell Purification
B cells from mouse spleens were purified using the Easy Sep® Mouse B Cell Negative Purification kit from Stem Cell Technologies. Purification was performed as described in protocol, with B-cell enrichment exceeding 95%.
Detection of ROS and Intracellular Free Calcium Release
BCR-induced superoxide generation was detected using the Diogenes chemiluminescent probe (National Diagnostics). Cells were suspended at 2 × 107/mL in 50 μL HBSS and incubated in a 37°C water bath. 50 μL of Diogenes substrate was prior to stimulation with anti-mouse IgM F(ab′)2 (Jackson Immunoresearch). Superoxide generated at specific times was determined using a luminometer.
Intracellular calcium release was measured as in Craxton et al. [32]. In short, purified primary mouse B cells were incubated with Indo-1 in normal mouse media at 37°C for 45 minutes. Cells were then washed and maintained at room temperature in normal media. Prior to stimulation with anti-IgM, cells were incubated in a 37°C water bath with or without 25 mM NAC. A BD LSRII was used to measure calcium flux, and data analyzed using FlowJo software.
B Cell Subsets and BCR Internalization
B cell subsets were quantified as described in Loder et al. [33] using antibodies to sIgM (BD Pharmingen), sIgD (eBioscience) and CD21 (BD Pharmingen) to phenotype splenic B cells.
For B cell internalization, purified B cells were incubated with either F(ab)′2 anti-mouse IgM (for ligand-induced internalization) or Fab anti-mouse IgM (Jackson Immunoresearch) (constitutive internalization) for forty minutes on ice, protected from light. Cells were then washed to remove excess antibody, and incubated for various times at 37°C. Internalization was halted by addition of ice-cold 1% PBSA. Secondary anti-mouse-PE antibodies (Jackson) were added to determine internalization of the BCR. Cells were run on a FACScan analyzer and data analyzed on CellQuest software. Internalization was determined using the formula
where t0 indicates time 0, and tn indicates a given time after beginning incubation.
Western Blotting
Lysates were prepared using 20 × 106 cells per sample; cells were stimulated, then lysed with RIPA buffer and sonicated briefly on ice. Proteins were resolved on 10% SDS-PAGE gel, then transferred to nitrocellulose in transfer buffer (25 mM Tris, 0.2 M glycine, 20% methanol, pH 8.5). Membranes were washed in 0.1% TBST, then incubated overnight at 4°C in blocking buffer (0.1% TBST plus 5% (w/v) nonfat dry milk). After blocking, membranes were probed with anti-p27Kip1 (Santa Cruz Biotechnology) or anti-cyclin A (Cell Signaling Technology) in TBST plus 5% (w/v) BSA. Blots were reprobed with anti-p38MAPK (Santa Cruz) to control for protein loading. Scanning densitometry software (NIH Image 1.63) was used to quantify bands.
Cell Cycle and Proliferation Assays
Purified B cells were labeled for 10 minutes at 37°C with 5 μM CFSE. Cells were resuspended at a concentration of 5 × 105 cells/mL and cultured in the presence or absence of anti-IgM, LPS (Sigma) or 1C10 (anti-CD40) mAb. Cells were collected at the indicated times. Prior to cell cycle analysis, cells were incubated in the presence of Pyronin-Y. CFSE dilution and cell cycle stage were analyzed by flow cytometry. Measurement of cells in dense B cells in G0 as described in Watanabe et al. [25].
Immunizations and ELISA
For T Independent Type-2 (TI-2) immunizations, mice were injected intraperitoneally with 10 μg DNP-Ficoll in 200 μL PBS. For T-Dependent (TD) immunizations, mice were injected intraperitoneally with either 5 or 50 μg DNP-KLH in PBS. Orbital eye bleeds were performed to obtain sera before injection and once a week for three weeks thereafter.
Total serum antibody levels were measured by ELISA. Briefly, plates were incubated overnight with either donkey anti-mouse IgG or IgM (Jackson), then blocked with PBS plus 1% BSA for 1 h at room temperature. After serial dilution, sera were allowed to bind to isotype-specific goat anti-mouse antisera (Jackson) and donkey anti-goat antisera conjugated to horseradish peroxidase (Jackson). Anti-DNP antibodies were detected using plates incubated overnight with DNP-BSA (Biosearch Technologies), then blocked overnight with 1% BSA. Sera were serially diluted and analyzed as before. Purified mouse isotypes (Zymed) were used as standards.
For measurement of in vitro antibody production, B cells purified by EasySep® were cultured as above with varying doses of anti-CD40 plus IL-4. Antibody production after seven days was determined by ELISA as described above.
Acknowledgments
These studies supported by a Cancer Research Institute Training Grant and NIH grants GM37905 and DE 16381. We thank Dr. Tomo Kurosaki for mutant DT40 cell lines and Dr. Hiroaki Niiro for help with initial experiments with DT40 lines. We also thank Geraldine Shu for performing tyroinse phosphorylation blots and technical assistance. The authors would like to thank Kevin Draves for technical help and Dr. Craig Chappell, Daphne Ma, Dr. Grant Hughes and Shinji Kasahara for helpful comments.
Abbreviations
- NOX
NADPH oxidase
- cdk
cyclin-dependent kinase
- NOX
NADPH oxidase
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- TI-2
T-independent type 2
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Cell. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Buhl AM, Cambier JC. Co-receptor and accessory regulation of B-cell antigen receptor signal transduction. Immunol Rev. 1997;160:127–138. doi: 10.1111/j.1600-065x.1997.tb01033.x. [DOI] [PubMed] [Google Scholar]
- 3.Richards S, Watanabe C, Santos L, Craxton A, Clark EA. Regulation of B-cell entry into the cell cycle. Immunol Rev. 2008;224:183–200. doi: 10.1111/j.1600-065X.2008.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Nagro CJ, Otero DC, Anzelon AN, Omori SA, Kolla RV, Rickert RC. CD19 function in central and peripheral B-cell development. Immunol Res. 2005;31:119–131. doi: 10.1385/IR:31:2:119. [DOI] [PubMed] [Google Scholar]
- 5.Callard R, Hodgkin P. Modeling T- and B-cell growth and differentiation. Immunol Rev. 2007;216:119–129. doi: 10.1111/j.1600-065X.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- 6.O’Rourke L, Tooze R, Fearon DT. Co-receptors of B lymphocytes. Curr Opin Immunol. 1997;9:324–329. doi: 10.1016/s0952-7915(97)80077-5. [DOI] [PubMed] [Google Scholar]
- 7.Reth M. The B-cell antigen receptor complex and co-receptors. Immunol Today. 1995;16:310–313. doi: 10.1016/0167-5699(95)80141-3. [DOI] [PubMed] [Google Scholar]
- 8.Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. 2005;5:43–57. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- 9.Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, Rao KV. The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell. 2005;121:281–293. doi: 10.1016/j.cell.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK. Redox redux: Revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 11.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 12.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 13.Kwon J, Qu C-K, Maeng J-S, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T cell adhesion through SLP-76-ADAP. EMBO. 2005;24:2331–2341. doi: 10.1038/sj.emboj.7600706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 15.Bae YS, Kang SW, Seo MS, Baines IC, Teckle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 16.Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338:677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 17.Morel F, Cohen Tanugi Cholley L, Brandolin G, Dianoux AC, Martel C, Champelovier P, Seigneurin JM, Francois P, Bost M, Vignais PV. The O2-generating oxidase of B lymphocytes: Epstein-barr virus-immortalized B lymphocytes as a tool for the identification of defective components of the oxidase in chronic granulomatous disease. Biochim Biophys Acta. 1993;1182:101–109. doi: 10.1016/0925-4439(93)90159-x. [DOI] [PubMed] [Google Scholar]
- 18.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 19.Grandvaux N, Elsen S, Vignais PV. Oxidant-dependent phosphorylation of p40phox in B lymphocytes. Biochem Biophys Res Commun. 2001;287:1009–1016. doi: 10.1006/bbrc.2001.5665. [DOI] [PubMed] [Google Scholar]
- 20.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 21.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 22.El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation an implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majumdar S, Kane LH, Rossi MW, Volpp BD, Nauseef WM, Korchak HM. Protein kinase C isotypes and signal transduction in human neutrophils: Selective substrate specificity calcium-dependent classical beta-PKC and novel calcium-independent nPKC. Biochim Biophys Acta. 1993;1176:276–286. doi: 10.1016/0167-4889(93)90056-u. [DOI] [PubMed] [Google Scholar]
- 24.Siow YL, Au-Yeung KK, Woo CWOK. Homocysteine stimulated phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase cbeta activation. Biochem J. 2006;398:73–82. doi: 10.1042/BJ20051810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe C, Shu GL, Zheng TS, Flavell RA, Clark EA. Caspase 6 regulates B cell activation and differentiation into plasma cells. J Immunol. 2008;181:6810–6819. doi: 10.4049/jimmunol.181.10.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arellano M, Moreno S. Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem Cell Biol. 1997;29:559–573. doi: 10.1016/s1357-2725(96)00178-1. [DOI] [PubMed] [Google Scholar]
- 27.Chiles TC. Regulation and function of cyclin D2 in B lymphocyte subsets. J Immunol. 2004;173:2901–2907. doi: 10.4049/jimmunol.173.5.2901. [DOI] [PubMed] [Google Scholar]
- 28.Paclet MH, Coleman AW, Burritt J, Morel F. NADPH oxidase of epstein-barr-virus immortalized B lymphocytes. Effect of cytochrome b(558) glycosylation. Eur J Biochem. 2001;268:5197–5208. doi: 10.1046/j.0014-2956.2001.02455.x. [DOI] [PubMed] [Google Scholar]
- 29.Garcia de Vinuesa C, O’Leary P, Sze DM, Toellner KM, MacLennan IC. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur J Immunol. 1999;29:1314–1323. doi: 10.1002/(SICI)1521-4141(199904)29:04<1314::AID-IMMU1314>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 31.Waki K, Inanami O, Yamamori T, Nagahata H, Kuwabara M. Involvement of protein kinase Cdelta in the activation of NADPH oxidase and the phagocytosis of neutrophils. Free Radic Res. 2006;40:359–367. doi: 10.1080/10715760500539121. [DOI] [PubMed] [Google Scholar]
- 32.Craxton A, Draves KE, Clark EA. Bim regulates BCR-induced entry of B cells into the cell cycle. Eur J Immunol. 2007;37:2715–2722. doi: 10.1002/eji.200737327. [DOI] [PubMed] [Google Scholar]
- 33.Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]