

Abstract
Dendritic cells (DCs), known to support immune activation during infections, may also regulate immune homeostasis in resting animals. Here we show that mice lacking A20 specifically in DCs spontaneously exhibited DC activation and expansion of activated T cells. DC-specific epistasis experiments using A20fl/fl Myd88fl/fl Cd11c-Cre compound mice revealed that A20 restricts both MyD88-independent signals, which drive DC and T cell activation, and MyD88-dependent signals, which drive T cell expansion. In addition, A20fl/fl Cd11c-Cre mice spontaneously developed lymphocyte-dependent colitis, sero-negative ankylosing arthritis and enthesitis, conditions stereotypical for human inflammatory bowel disease (IBD). These findings indicate that DCs require A20 to preserve immune quiescence and suggest that A20-dependent DC functions may underlie IBD and IBD-associated arthritides.
Introduction
Immune homeostasis in resting animals has long been considered a consequence of the absence of infectious or inflammatory stimulation. However, recent studies suggest that immunostimulatory ligands such as microbial molecules may be sensed in non-perturbed animals1–4. This notion has raised multiple questions regarding which ligands are delivered to host cells under basal conditions, which immune cells respond to such ligands and how these cells regulate their responses to these ligands.
Dendritic cells (DCs) are specialized sentinels that detect inflammation or microbial pathogens via mutiple receptors, such as tumor necrosis factor (TNF)-receptors and Toll-like receptors (TLR)5,6. During overt immunization or infection, these stimuli cause DCs to upregulate expression of major histocompatibility complex (MHC) molecules, T cell costimulatory molecules and cytokines, thereby recruiting and activating other immune cells5,7,8. As TLR ligands are expressed by commensal microbes and as host nucleic acids can trigger inflammatory pathways9,10, it is possible that host cells such as DCs may also encounter activating ligands during steady state conditions. In this context, the nature, duration and/or intensity of signals in DCs may determine whether and how DCs stimulate innate and adaptive immune cells6,11,12. Thus, intracellular proteins that regulate these signaling pathways could play critical roles in controlling DC activation and immune homeostasis during steady state conditions.
A20 is a potent anti-inflammatory protein that utilizes de-ubiquitinating, E3 ligase and ubiquitin binding functions to regulate NF-κB signaling13,14. Genetic and biochemical studies indicate that A20 restricts TNF, TLR, nucleotide-binding oligomerization domain protein (NOD) and CD40 activation signals by editing ubiquitin chains on key signaling proteins that transduce these signals14–18. A20-deficient mice spontaneously develop severe inflammatory disease and perinatal lethality, indicating that A20 preserves immune homeostasis14,19. In addition, single nucleotide polymorphisms (SNPs) in the human A20 locus (also known as TNFAIP3) have been associated with several human autoimmune diseases, including systemic lupus erythematosus20,21, rheumatoid arthritis 22,23, psoriasis24,25, and celiac disease26, suggesting that altered A20-dependent functions contribute to human autoimmunity. Given the critical roles of A20 in restricting diverse activating signals, and the potential roles such signals play in DC functions, we hypothesized that A20 expression in DCs may be important for immune homeostasis. To investigate the roles of A20 in regulating DC functions, we have generated mice in which A20 expression is deleted specifically in dendritic cells.
Results
A20 prevents spontaneous DC activation
To determine the relative expression of A20 in DCs during steady state conditions, we analyzed mRNA in conventional DCs (cDCs—CD11chi Ly6C− MHC-II+) and plasmacytoid DCs (pDCs—CD11clo CD11b− Ly6C+ B220+) purified from spleens of wild-type mice. While the relative expression of interleukin 1β mRNA was markedly disparate between cDCs and pDCs, A20 mRNA was similarly expressed by both cDCs and pDCs (Fig. 1a). To determine whether A20 regulates DC functionality, we generated mice carrying a loxP-flanked allele of A20 and bred these mice to Cd11c-Cre transgenic mice (Supplementary Fig. 1a)18,27. Consistent with prior reports of “floxed” (FL) alleles deleted by the Cd11c-Cre transgene, recombination and deletion of the loxP-flanked A20 exon occurred at high efficiency in cDCs (and slightly lower efficiency in pDCs) of A20fl/fl Cd11c-Cre mice (Supplementary Fig. 1b–e)27. In contrast to animals globally deficient in A20 (A20−/− mice)19, A20fl/fl Cd11c-Cre mice were grossly healthy until at least 6 months of age (data not shown). Thus, A20 deficiency in DCs does not cause the cachexia and perinatal lethality seen in A20−/− mice.
Figure 1. A20 prevents spontaneous activation of dendritic cells in A20FL/FL CD11c-Cre mice.

(a) Sorted populations of splenic cDCs and pDCs from wild-type (WT) mice were analyzed for RNA expression of Il1β (p = 0.004) and A20 (p = 0.03). Data is relative to HPRT expression and is pooled from three independent experiments including at least three mice each. (b) A20+/+ and A20fl/fl Cd11c-Cre mice were analyzed by flow cytometry to distinguish cDCs (CD11chigh, upper box) and CD11clowDCs (dashed box), which includes pDCs. Expansion of myeloid cells in obscures the relative abundance of DCs in A20fl/fl Cd11c-Cre mice (See Fig. 2). (c–d) Representative histograms (c) and relative expression (d) of CD80, CD86, and CD40 by cDCs (gated on upper box in b) from A20+/+ (black) and A20fl/fl Cd11c-Cre mice (red, shaded). Data in (d) is averaged from all mice tested and is normalized to expression on control A20+/+ Cd11c-Cre cDCs. (e) CD11clowDCs (gated on dashed box in b) were secondly gated on B220+Ly6C+ DCs to identify pDCs. Activated, MHC-IIhigh pDCs relative to total pDCs is shown. Representative histogram (f) and fold changes in expression (g) of CD80, CD86 and CD40 on pDCs in A20fl/fl Cd11c-Cre mice. Data in (b–g) is averaged from nine separate experiments including at least two mice of each genotype (*, P ≤ 0.001, **, P = 0.01)
Conventional DCs and plasmacytoid DCs, the two main DC populations in the spleen, were readily detectable in A20fl/fl Cd11c-Cre mice, indicating that A20 was not required for DC development (Fig. 1b). During steady state, DCs exhibit modest expression of MHC and T cell costimulatory molecules. Activation of DCs triggers the upregulation of these molecules at the cell surface. To determine whether A20 restricts DC activation, we analyzed cell surface expression of these activation markers on DCs from A20fl/fl Cd11c-Cre mice. cDC expression of the costimulatory molecules CD80 and CD40 (but not CD86) was increased when compared to control A20+/+ Cd11c-Cre mice, indicating that A20-deficient DCs were spontaneously activated in vivo (Fig. 1c,d). Similarly, while pDCs of control mice were uniformly low for MHC class II expression, approximately 50% of pDCs in A20fl/fl Cd11c-Cre mice expressed abundant MHC class II (Fig. 1e). Expression of costimulatory molecules CD80, CD86 and CD40 was increased more than 3-fold on A20-deficient pDCs (Fig. 1f,g). Thus, A20 expression in DCs prevents their spontaneous activation in vivo.
A20 prevents DC-triggered splenomegaly and lymphadenopathy
During overt immunization or infection, increased expression of costimulatory molecules and cytokines by DCs drives immune cell recruitment and activation. To determine whether A20 expression in DCs maintains immune homeostasis, we examined lymphoid organs of A20fl/fl Cd11c-Cre and control Cd11c-Cre mice. Despite appearing outwardly healthy, A20fl/fl Cd11c-Cre mice developed marked splenomegaly and lymphadenopathy within three weeks of birth (Supplementary Fig. 2), accumulating large numbers of CD11b+ inflammatory myeloid cells, including F4/80+ CD169− monocytes (Fig. 2a–d and data not shown). These cells comprised nearly 50% of all immune cells in the spleen and greater than 5% of lymph node cells (Fig. 2a,c). Compared to controls, lymph nodes from A20fl/fl Cd11c-Cre mice contained ten fold more inflammatory CD11b+ F4/80 cells and five fold more B cells and T cells, including CD4+ CD25+ FOXP3+ regulatory T cells (Fig. 2d). These findings indicate that A20 expression in DCs prevents expansion of both innate and adaptive immune cells at steady state.
Figure 2. A20-deficient DCs induce expansion of innate and adaptive immune cells.

(a) Relative abundance of splenic CD11b+ myeloid cells among non-erythroid immune cells of A20+/+, A20+/fl, and A20fl/fl Cd11c-Cre mice. (b) Absolute numbers of the indicated cell types in spleens of A20+/+ and A20fl/fl Cd11c-Cre mice; splenic populations of A20+/fl Cd11c-Cre mice were similar to control mice (data not shown). (c–d) Relative abundance of myeloid cells (c) and absolute numbers of CD11b+F4/80+ myeloid cells and lymphocytes (d) in lymph nodes of the indicated mice. (e–f) Sublethally irradiated WT mice (CD45.1) were reconstituted with congenic (CD45.2) A20+/+ Cd11c-Cre or A20fl/fl Cd11c-Cre bone marrow cells. Four weeks post reconstitution, the weight of the spleen (e) and splenic myeloid cell populations (f) was analyzed. Data is representative of three independent experiments each containing three recipient mice of each genotype. (g) BMDCs from A20+/+, A20 heterozygous (A20+/−), and A20-deficient mice (A20−/−) were stimulated with LPS. Seven hours post stimulation, cytokines in the cell supernatant were quanitified by ELISA. (h–i) Myeloid cells recruited to lymph nodes by LPS-stimulated control or A20−/− BMDCs (see methods) were quantified (*, P ≤ 0.01, **, P = 0.03). (h) The number of CD11b+F4/80+ monocytes (expressed as a percentage of total lymph node cells) recruited by BMDCs of the indicated genotypes. Data is representative of five independent experiments containing at least three recipient mice from each group. (i) Monocytes recruited by LPS-stimulated A20+/+ or A20−/− BMDCs in WT mice pre-treated with the indicated blocking antibodies or isotype control antibody. Data is pooled from three independent experiments each containing three recipients for each treatment. (j) ELISA quantification of IL-6 in sera from 6–10 week old mice of the indicated genotypes; each symbol represents one mouse.
Loss of DCs results in myeloid expansion28,29. We thus sought to determine whether the myeloid expansion observed in A20fl/fl Cd11c-Cre mice represents a loss or gain of DC functions. We generated radiation chimera wherein wild-type mice were reconstituted with roughly equal proportions of A20fl/fl Cd11c-Cre and wild-type hematopoietic cells. In such mice, the potential loss of immunosuppressive function(s) by A20-deficient DCs would be compensated for by the wild-type DCs. Four weeks after irradiation, spleens of hematopoietic chimeras were enlarged greater than four-fold and contained large numbers of activated monocytes compared to mice reconstituted with control A20+/+ Cd11c-Cre hematopoietic cells (Fig. 2e,f). These results suggest that A20-deficient DCs induce splenomegaly and myeloid cell expansion in a physiologically dominant fashion over wild-type DCs.
To begin to determine how A20-deficient DCs cause myeloid expansion, we first stimulated A20−/− or A20+/+ bone marrow-derived dendritic cells (BMDCs) with lipopolysaccharide (LPS). After LPS stimulation, A20−/− DCs produced three-fold more IL-6 and ten-fold more TNF and IL-12 than control DCs (Fig. 2g). Thus, A20 directly restricts DC responses to LPS. Production of IL-10 and type I interferons (IFNs) were also enhanced in A20−/− DCs, indicating that loss of A20 did not preclude expression of these cytokines (Fig. 2g, right, Supplementary Fig. 3). To test the ability of A20−/− DCs to acutely recruit myeloid cells in vivo, we labeled LPS-activated DCs with CFSE, injected them into the footpads of wild-type mice and then quantified both the number of DCs (CFSE+) that had migrated to the draining lymph nodes and the numbers of monocytes (CFSE−CD11b + F4/80+) recruited to these lymph node after 24 hours. While the number of A20−/− and control DCs obtained from draining lymph nodes was similar (< 0.025% of all cells, data not shown), greater numbers of monocytes were recruited to lymph nodes in recipients of A20−/− DCs than mice receiving control DCs (Fig. 2h). Therefore, activated A20−/− DCs induce more myeloid inflammation than wild-type DCs.
To determine whether A20−/− DCs drive myeloid inflammation by producing pro-inflammatory cytokines, we tested the involvement of TNF and IL-6 in this acute process. After adoptive transfer of LPS-activated A20−/− DCs into footpads of wild-type mice, antibody-mediated blockade of either IL-6 or TNF partially inhibited myeloid cell recruitment into draining lymph nodes (Fig. 2i). Combined with the observation that A20fl/fl Cd11c-Cre mice express twenty five fold elevated serum concentrations of IL-6 (Fig. 2j), these experiments suggest that A20 suppresses DC production of inflammatory cytokines, thereby preserving myeloid cell homeostasis.
A20 expression in DCs preserves T cell homeostasis
Development, homeostasis and activation of T lymphocytes are dependent on DC functions. Intrathymic T cell development, including that of regulatory CD4+CD25+FOXP3+ T cells was normal in A20fl/fl Cd11c-Cre mice (Fig. 3a,b). In the periphery, regulatory T cell populations (percentage of total CD4+ T cells) and their relative expression of FOXP3 were normal in A20fl/fl Cd11c-Cre mice and were numerically increased, similar to the expansion we observed for conventional CD4+ and CD8+ T cells (Fig. 3b, 2d and data not shown). Thus, DCs do not require A20 to support development and maintenance of regulatory T cells. In contrast, peripheral homeostasis of conventional T cells was grossly disrupted in A20fl/fl Cd11c-Cre mice. Nearly 50% of splenic T cells expressed the early activation marker CD69, compared to only 10% in control A20++Cd11c-Cre mice (Fig. 3c,d). Additionally, the percentages and numbers of activated CD44hiCD62Llo CD4+ and CD8+ T cells were increased in A20fl/fl Cd11c-Cre mice (Fig. 3e,f). These results indicate that A20 expression in DCs prevents the spontaneous activation of T cells. In addition, although heterozygous A20+/fl Cd11c-Cre mice did not exhibit splenomegaly or expansion of myeloid cells, the numbers of activated T cells in these mice were consistently increased more than two-fold when compared to A20++Cd11c-Cre control mice (Figs. 2a,c, Supplementary Fig. 2, 3e,f). Thus, haplo-insufficient expression of A20 in DCs compromises their ability to preserve T cell quiescence.
Figure 3. A20 expression in DCs is required to prevent aberrant T cell activation.

(a) Representative thymocyte populations from four analyses of control and A20fl/fl Cd11c-Cre mice. (b) Regulatory CD4+ CD25+ FOXP3+ T cells among total CD4 single positive thymocytes and lymph node CD4+ T cells. Data is averaged from four mice from each genotype. (c–f) Splenic T cells (TCRβ gated) from mice of the indicated genotype were analyzed for expression of CD69 (c,d), or CD44 and CD62L (e,f). The percentage of CD44hiCD62Llo activated CD4+(top) or CD8+ (bottom) T cells is shown. Data in (d,f) is averaged from ten independent experiments with at least one mouse of each genotype (*, P ≤ 0.02). (g–h) Polyclonal, CFSE-labeled, WT CD8+ T cells were adoptively transferred into congenic A20fl/fl Cd11c-Cre mice or control mice. (g) On the indicated day, the percentage of adoptively transferred T cells that had been converted into activated CD44hiCD62Llo T cells was quantified. (h) CFSE dilution was analyzed in tandem to assess T cell proliferation; shown are representative histograms of CFSE expression in CD8+ T cells ten days post transfer. Data in (g,h) is representative of three experiments containing two mice of each genotype per timepoint. (i) Ovalbumin specific OT-II T cells were adoptively transferred into A20fl/fl Cd11c-Cre mice or control mice, followed by injection of PBS (−) or OVAp (+). Ten days later, the percentage of OT-II T cells among total CD4+ T cells (left) and the absolute number of OT-II cells (right) in peripheral lymph nodes was quantified. Shown is one of two experiments containing one PBS recipient and two OVAp recipients per genotype.
Spontaneous DC activation, T cell activation and T cell expansion occurred by three weeks of age in A20fl/fl Cd11c-Cre mice. These findings suggest that A20-deficient DCs potently induce naive T cell activation and proliferation. Consistent with this hypothesis, A20−/− DCs drove aberrant T cell activation in a physiologically dominant fashion over wild-type DCs in mixed hematopoietic chimeras (Supplementary Fig. 4). Additionally, antigen-pulsed A20−/− BMDCs induced robust T cell responses as did A20+/fl Cd11c-Cre mice immunized with ovalbumin (Supplementary Fig. 5). To further determine how A20-deficient DCs disrupt T cell homeostasis, we adoptively transferred CFSE-labeled, naive, wild-type CD8+ T cells into A20fl/fl Cd11c-Cre and control A20++ Cd11c-Cre mice. These polyclonal T cells developed normally in wild-type mice and hence their homeostasis in recipient A20fl/fl Cd11c-Cre mice depended entirely on regulatory controls in peripheral lymphoid organs. Ten days after adoptive transfer, greater than 95% of transferred T cells remained naive in control Cd11c-Cre mice (Fig. 3g). In contrast, within three days, a significant proportion of transferred CD8+ T cells were converted into CD44hiCD62Llo activated T cells in A20fl/fl Cd11c-Cre mice, and nearly all transferred cells were activated ten days post transfer. In addition, within these ten days, transferred CD8+ T cells underwent multiple cellular divisions in A20fl/fl Cd11c-Cre mice, while no significant proliferation occurred in control Cd11c-Cre mice (Fig. 3h). Similar results were observed with TCR transgenic OT-I CD8+ T cells (Supplementary Fig. 6). These observations indicate that A20-deficient DCs deliver potent signals that induce aberrant activation and proliferation of naive CD8+ T cells even in the absence of overt immunization.
At steady state, DCs can support antigen-specific T cell tolerance in peripheral lymphoid organs11,30–32. To determine whether aberrant T cell activation driven by A20-deficient DCs compromised induction of peripheral tolerance, we adoptively transferred OT-II CD4+ T cells into five week old A20fl/fl Cd11c-Cre and control Cd11c-Cre mice, and then administered a tolerizing dose of cognate ovalbumin-derived peptide (OVAp)12,33. Ten days after peptide injection, A20+ Cd11c-Cre recipients of OVAp contained ten fold fewer OT-II T cells than did mock treated Cd11c-Cre mice (Fig. 3i), consistent with tolerization via T cell deletion. In marked contrast, injection of OVAp into A20fl/fl Cd11c-Cre mice induced three-fold expansion of OT-II T cells. Taken together, peptide-induced proliferation resulted in thirty-fold more OT-II T cells in A20fl/fl Cd11c-Cre mice than in tolerized control Cd11c-Cre mice (Fig. 3i, right). Thus, A20fl/fl Cd11c-Cre mice not only failed to induce peripheral deletion of OT-II CD4+ T cells, but also induced remarkable antigen-specific expansion of these T cells under tolerizing conditions. These results indicate that A20 expression in DCs prevents promiscuous T cell activation, aberrant proliferation and disruption of peripheral T cell tolerance.
A20 inhibits MyD88-independent signals in DCs to preserve DC and T cell quiescence
DC activation in A20fl/fl Cd11c-Cre mice in the absence of overt stimulation suggests that A20 restricts intracellular signaling cascades in DCs that are triggered by commensal and/or endogenous ligands during steady state conditions. The nature of such signals and how they regulate steady state DC functions are unknown. As A20-deficient DCs were hyper-responsive to TLR ligands, we interrogated the role of MyD88 signals in DCs by interbreeding A20fl/fl Cd11c-Cre mice with Myd88fl/fl mice34. The resulting compound A20fl/flMyd88fl/fl Cd11c-Cre mice lack both A20 and MyD88 specifically in DCs. We then used these mice to determine which A20-restricted signals in DCs were MyD88-dependent and which were MyD88-independent.
To determine the nature of steady state signals that are regulated by A20, we first analyzed expression of activation markers on DCs from A20fl/flMyd88fl/fl Cd11c-Cre and control mice. Comparable to A20fl/fl Cd11c-Cre mice, DCs from A20fl/flMyd88fl/fl Cd11c-Cre mice spontaneously expressed high levels of CD80 and CD40 (Fig. 4a). Thus, MyD88 signals were not required to trigger spontaneous activation of A20-deficient DCs in vivo. Similar results were obtained from A20fl/fl Cd11c-Cre mice that were globally MyD88-deficient (Myd88−/− mice) (Fig. 4b), confirming that steady-state stimuli that trigger DC activation in A20fl/fl Cd11c-Cre mice do not require MyD88 signaling in DCs or any other cell type. Thus, in resting animals, A20 restricts MyD88-independent signals in DCs to prevent spontaneous DC activation.
Figure 4. MyD88-independent signals trigger DC activation and drive aberrant T cell activation in A20FL/FL CD11c-Cre mice.

(a) Splenic cDCs (top) and pDCs (bottom) from A20+/+ Myd88fl/fl Cd11c-Cre mice (solid, black) and A20fl/fl Myd88fl/fl Cd11c-Cre mice (red, shaded) were analyzed for expression of CD80 and CD40. For comparison, DCs from age matched A20fl/fl Cd11c-Cre mice (dashed, black) were analyzed in parallel. (b) cDCs and pDCs from A20+/+ Cd11c-Cre Myd88−/− (solid, black), A20fl/fl Cd11c-Cre Myd88−/− (red, shaded) and A20fl/fl Cd11c-Cre Myd88+/− mice (dashed, black) were analyzed as in (a). (c) The percentage of CD44hiCD62Llo activated CD4+ (top) or CD8+ (bottom) T cells in lymph nodes from mice of the indicated genotype is shown. Data in (a–c) is representative of five independent experiments containing at least one mouse per genotype. (d–i) Polyclonal, CFSE-labeled, WT CD8+ T cells were adoptively transferred into A20fl/fl Cd11c-Cre mice or control mice. Recipient mice were treated with isotype control antibodies (d,g) or a combination of anti-CD80 and anti-CD86 (e,h). The percentage of adoptively transferred T cells that had been converted into activated CD44hiCD62Llo T cells (d–f) or induced to proliferate (g–i) was quantified nine days post transfer. Data in (f,i) is averaged from two independent experiments each including two mice per group.
To determine the consequences of these disregulated MyD88-independent signals on T cell homeostasis, we analyzed lymph node T cells from A20fl/flMyd88fl/fl Cd11c-Cre and control mice. The percentages of activated CD44hiCD62Llo CD4+ and CD8+ T cells were increased four to five fold when compared to control Myd88fl/fl Cd11c-Cre mice (Fig. 4c). Moreover, the percentages of activated T cells in A20fl/flMyd88fl/fl Cd11c-Cre mice were comparable to that of A20fl/fl Cd11c-Cre mice, indicating that MyD88-independent signals that trigger DC activation also drive aberrant activation of T cells.
The findings above suggest that high expression of costimulatory molecules on A20-deficient DCs inappropriately stimulates T cells and disrupts T cell homeostasis. To test this notion, we adoptively transferred polyclonal, naive wild-type CD8+ T cells into A20fl/fl Cd11c-Cre mice and administered non-depleting blocking antibodies to CD80 and CD86 over the course of nine days. While the majority (69%) of transferred T cells became CD44hiCD62Llo in A20fl/fl Cd11c-Cre recipients of isotype control antibodies, blockade of CD80 and CD86 reduced the percentage of activated T cells in A20fl/fl Cd11c-Cre mice to just 23% (Fig. 4d,e). On average, blockade of costimulatory molecules in A20fl/fl Cd11c-Cre mice inhibited the aberrant activation of adoptively transferred CD8+ T cells by more than 60% (Fig. 4f). Aberrant proliferation of adoptively transferred CD8+ T cells in A20fl/fl Cd11c-Cre mice was also antagonized by CD80 CD86 blockade (Fig. 4g,i). Taken together, these findings indicate that A20 restricts MyD88-independent signals in DCs that can trigger spontaneous DC activation, upregulation of costimulatory molecules and drive aberrant T cell activation.
A20 inhibits MyD88-dependent signals in DCs to prevent aberrant T cell expansion
Comparisons of A20fl/flMyd88fl/fl Cd11c-Cre mice and A20fl/fl Cd11c-Cre mice also allowed us to determine the role of MyD88-dependent signals in DCs that control immune homeostasis. Total cellularity and lymphocyte numbers were largely normal in A20fl/flMyd88fl/fl Cd11c-Cre mice, in marked contrast with gross lymphadenopathy in A20fl/fl Cd11c-Cre mice (Fig. 5a). This finding indicates that hyper-responsiveness of A20-deficient DCs to steady-state MyD88 signals drives aberrant lymphocyte expansion in A20fl/fl Cd11c-Cre mice.
Figure 5. MyD88-dependent signals in DCs drive T cell expansion in A20FL/FL CD11c-Cre mice.

(a) Lymph nodes from the indicated mice were analyzed for total cell number, CD4+ T cells and CD8+ T cells. Data is averaged from five independent experiments including one mouse of each genotype. (b) IL-6 and TNF produced by LPS-stimulated A20+/+, A20−/−, A20+/+ Myd88−/− and A20−/− Myd88−/− BMDCs was quantified by ELISA. (c) Proliferation (left) and activation (right) of polyclonal, WT CD8+ T cells adoptively transferred into A20fl/fl Cd11c-Cre mice or control mice. Recipients were treated with anti-IL-6, anti-TNF, or isotype control antibody over the period of nine days. Data is averaged from two independent experiments each including two mice per group.
To determine how unrestricted MyD88 signaling in DCs perturb T cell homeostasis, we measured the cytokines produced by A20−/−, A20−/− Myd88−/− DCs, and other control DCs in response to LPS. While A20−/− DCs produced markedly more TNF and IL-6 than wild-type DCs, A20−/− Myd8−/− DCs produced only 10% that of A20−/− DCs. Hence, A20 inhibits the production of these MyD88-dependent cytokines in DCs (Fig. 5b). To test whether these DC products may support T cell expansion in A20fl/fl Cd11c-Cre mice, we adoptively transferred wild-type CD8+ T cells into A20fl/fl Cd11c-Cre mice and administered blocking antibodies to either IL-6 or TNF. Blockade of either of these cytokines resulted in approximately 50% reduction in CD8+ T cell proliferation, indicating that overexpression of these cytokines in A20fl/fl Cd11c-Cre mice induce T cell expansion (Fig. 5c). Antibody-mediated blockade also inhibited T cell activation, suggesting IL-6 and TNF additionally contribute to aberrant DC-driven T cell activation. Thus, A20 restricts MyD88-dependent signals in DCs to limit production of cytokines that drive aberrant T cell expansion.
A20-dependent DC functions prevent colitis
Given the genetic association of TNFAIP3 SNPs with human autoimmune diseases, we analyzed A20fl/fl Cd11c-Cre mice for the development of these conditions. We did not detect an increase in serum immunoglobulin, DNA-specific antibodies or immunoglobulin deposits in kidneys of A20fl/fl Cd11c-Cre mice (data not shown). However, by five months of age, 100% of A20fl/fl Cd11c-Cre mice developed an inflammatory bowel disease in which colons were enlarged in diameter, contained foci of expanded immune cells in the lamina propria, and were depleted of goblet cells (Fig. 6a,b). These mice also had increased numbers of splenic IFN-γ producing CD4+ T cells (Fig. 6c). Although two to three month old A20fl/fl Cd11c-Cre mice did not exhibit overt colitis, these younger mice had markedly elevated titers of serum immunoglobulin A, a pathology consistent with perturbed bowel homeostasis (Fig. 6d)35,36. Moreover, these pre-colitic A20fl/fl Cd11c-Cre mice exhibited increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. DSS treated A20fl/fl Cd11c-Cre mice suffered exaggerated weight loss, with several mice requiring euthanization, while control Cd11c-Cre mice showed only moderate weight loss and diarrhea (Fig. 6e). Taken together, these results indicate that DCs require A20 to maintain intestinal immune homeostasis and to restrict epithelial damage-triggered colitis.
Figure 6. A20 function in DCs preserves intestinal homeostasis.

(a–c) A20fl/fl Cd11c-Cre mice of at least five months of age were analyzed for spontaneous development of inflammatory bowel disease. (a) Colons of littermate A20+/+ Cd11c-Cre control mice (top) and A20fl/fl Cd11c-Cre mice (bottom) are shown. (b) Representative H+E histology of colons from control and A20fl/fl Cd11c-Cre mice (original magnification 100x). (c) Representative plot of IL-17 and IFN-γ production by splenic CD4+ T cells from five analyses of six month old A20fl/fl Cd11c-Cre and littermate control mice. (d) ELISA of IgA in sera of pre-colitic, 2–3 month old mice. Data is averaged from ten mice. (e) DSS was added to the drinking water of twelve week old A20+/+ (●), A20+/fl(○), and A20fl/fl Cd11c-Cre mice (□) for five days (see methods). Experiment included three mice of each genotype; shown is the average body weight on the indicated day post DSS-treatment, expressed as a percentage of initial body weight. Asterisks (*) adjacent to symbols indicates death or obligatory euthanasia of mice. No A20fl/fl Cd11c-Cre mice survived beyond seven days. (f–g) A20+/+ Cd11c-Cre Rag1−/− mice and A20fl/fl Cd11c-Cre Rag1−/− mice were analyzed for inflammatory CD11b+ myeloid cells in the spleen (f) and development of spontaneous colitis (g). Representative H+E colon histology from six month old control and A20fl/fl Cd11c-Cre Rag1−/− mice is shown (original magnification 100x). Data in (f–g) is representative of five mice of each genotype.
Misregulated activation of either innate or adaptive immune cells can compromise bowel homeostasis37,38. To determine whether spontaneous colitis in A20fl/fl Cd11c-Cre mice required lymphocytes or was mediated solely by activated myeloid cells we interbred A20fl/fl Cd11c-Cre mice with lymphocyte-deficient Rag1−/− mice. Although high numbers of myeloid cells accumulated in the spleen, histological analysis of colons from compound A20fl/fl Cd11c-Cre Rag1−/− mice indicated that bowel inflammation did not occur (Fig. 6f,g). Thus, spontaneous colitis in A20fl/fl Cd11c-Cre mice requires DC-induced disruption of adaptive immune cell homeostasis.
The absence of colitis in A20fl/fl Cd11c-Cre Rag1−/− suggested that A20-deficient DCs stimulated excessive T cell activation in intestines of A20fl/fl Cd11c-Cre mice to cause inflammatory bowel disease. To directly test this pathophysiological mechanism, we adoptively transferred naive wild-type CD4+ T cells into A20fl/fl Cd11c-Cre Rag1−/− and control Cd11c-Cre Rag1−/− mice, and monitored these animals for the development of colitis. T cell complemented A20fl/fl Cd11c-Cre Rag1−/− mice rapidly lost weight and developed diarrhea within weeks after T cell transfer, frequently necessitating euthanasia due to excessive weight loss (Fig. 7a). During the same time period, T cell complemented A20+/fl Cd11c-Cre Rag1−/− and A20+ Cd11c-Cre Rag1−/− mice lost only modest amounts of weight. Grossly, spleens of A20fl/fl Cd11c-Cre Rag1−/− mice were more enlarged, and colons were shorter and thicker than those of control mice (Fig. 7b, Supplementary Fig. 7a). Histological analysis confirmed that the colonic lamina propria of A20fl/fl Cd11c-Cre Rag1−/− mice were infiltrated with large numbers of inflammatory cells (Supplementary Fig. 7b,c). Taken together, these results indicate that A20-deficient DCs induce rapid activation and proliferation of T cells that leads to T cell-mediated colitis in A20fl/fl Cd11c-Cre mice.
Figure 7. A20-deficient DCs activate naive T cells and drive T cell mediated colitis.

Naive, WT CD4+CD62L+CD25− T cells were transferred into mice of the indicated genotypes. All mice were Cd11c-Cre Rag1−/−. (a) Body weight of mice on the indicated day post T cell transfer, expressed as a percentage of initial weights. (b) Weights of spleens from A20+/+, A20+/fl, and A20fl/fl Cd11c-Cre Rag1−/− mice 24 days post T cell transfer. Data is representative of two independent experiments including at least two mice per genotype.
A20 SNPs are associated with human Crohn’s disease
Single nucleotide polymorphisms (SNPs) in the human TNFAIP3 locus have been associated with several autoimmune diseases, including systemic lupus erythematosus (SLE)20,21, and rheumatoid arthritis22,23. Because A20fl/fl Cd11c-Cre mice did not manifest overt autoimmunity but had perturbed intestinal homeostasis, we investigated whether TNFAIP3 SNPs may be associated with human Crohn’s disease (CD), an inflammatory bowel disease (IBD). We analyzed a whole genome scan of CD patients and healthy controls from the Welcome Trust Case-Control Consortium (WTCCC)39. Out of 443 genotyped or imputed SNPs from the TNFAIP3 locus, we identified 28 SNPs (P < 0.01), 11 of which were highly significant (P < 10−5) for association with CD (Supplementary Fig. 8). We confirmed an association with SNP rs7753394 39 (P = 3.9 × 10−6), and identified 3 new SNPs of even higher significance (P > 2 × 10−6) among CD patients. Our top SNP was imputed SNP rs2683064, with a P -value of 1.8 × 10−6, and odds ratio of 1.25 (95% CI 1.14 – 1.36). Furthermore, conditional analysis of each locus to the top SNP indicated that the multiple signals in this region were due to linkage disequilibrium, with a single CD-associated locus. These associations suggest that in addition to other autoimmune diseases, A20 regulates susceptibility to human inflammatory bowel disease. Taken together with the susceptibility of A20fl/fl Cd11c-Cre mice to colitis, these results reinforce A20 functions in DCs as critical to prevent inflammatory responses and preserve intestinal homeostasis.
A20fl/fl Cd11c-Cre mice develop IBD-associated arthritis
Colitis in human patients is frequently associated with a subset of sero-negative arthritic diseases. We thus examined A20fl/fl Cd11c-Cre mice for the development of arthritis. Intriguingly, approximately 10% of A20fl/fl Cd11c-Cre mice spontaneously developed gross limb joint inflammation within 4–6 months of age (Supplementary Fig. 9). Histopathological analyses revealed marked synovitis and inflammatory infiltrates surrounding the tendon and entheses in arthritic A20fl/fl Cd11c-Cre mice (Fig. 8a–f). Acute arthritis and macroscopic joint swelling resolved within weeks after disease onset (data not shown), but histopathology of joints from one year old mice revealed ankylosis, bone cysts and abnormal formation of bone and cartilage at joint surfaces (Fig. 8g–j). Thus, upon the onset of arthritis, chronic inflammation of the joint and bone likely persisted throughout the lifetime of the afflicted animals. During the acute and chronic phase of arthritic disease, we consistently observed enthesitis at tendon insertion sites of the tibial-talar joints (Fig. 8d, g–j). This finding was particularly intriguing since enthesitis is a hallmark of spondyloarthropathies, a heterogeneous group of sero-negative rheumatic diseases that occur in a subset of human IBD patients. To immunophenotype the spontaneous arthritis observed in A20fl/fl Cd11c-Cre mice, we tested sera from arthritic animals (acute and chronic) for the presence of autoantibodies related to arthritic disease. All animals had negligible serum titers of rheumatoid factor, anti-nuclear antibodies, and anti-cyclic citrullinated peptide (CCP), the best characterized immunoglobulin determinants of rheumatoid arthritis (Fig. 8k). Thus, A20fl/fl Cd11c-Cre mice spontaneously develop chronic, sero-negative, ankylosing arthritis with severe enthesitis.
Figure 8. A20FL/FL Cd11c-Cre mice spontaneously develop arthritic disease with pathologies similar to human IBD-associated arthritis.

(a–f) Representative H+E of hindpaws of a healthy control (a,c,e) and littermate A20fl/fl
Cd11c-Cre mouse (b,d,f) suffering acute arthritis at five months of age. Ti=tibia, Ta=talus, N=navicular. Boxes with asterisks are entheses shown in high magnification in (c,d). Synovitis and inflammatory infiltrates surrounding the tendon and entheses of arthritic A20fl/fl
Cd11c-Cre mice (b,d). Marked synovitis and joint destruction is seen in (f) compared with control (e). (g–j) Joint pathology from two different one year old A20fl/fl
Cd11c-Cre mice. (g,h) Bone cysts (○), pannus (*), joint ankylosis and new bone formation (
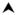 ). (i) Sites of cartilage erosion are indicated by arrows. (j) New bone formation and chondrogenesis are indicated by arrows. Bars indicate 1mm (a,b,e–h), 100 μm (c,d) or 500 μM (i,j). (k) Serum from five month old and one year old mice of the indicated genotypes was analyzed for the indicated auto-antibodies: rheumatoid factor, anti-nuclear antigens, and anti-cyclic citrullinated peptide (CCP). Data is representative of two experiments containing at least seven mice per genotype.
). (i) Sites of cartilage erosion are indicated by arrows. (j) New bone formation and chondrogenesis are indicated by arrows. Bars indicate 1mm (a,b,e–h), 100 μm (c,d) or 500 μM (i,j). (k) Serum from five month old and one year old mice of the indicated genotypes was analyzed for the indicated auto-antibodies: rheumatoid factor, anti-nuclear antigens, and anti-cyclic citrullinated peptide (CCP). Data is representative of two experiments containing at least seven mice per genotype.
In human IBD patients, these pathologies often include spondyloarthritis involving the axial skeleton40. To determine whether arthritis in A20fl/fl Cd11c-Cre mice extended beyond the peripheral joints, we analyzed micro-computed tomography (micro-CT) scans of A20fl/fl Cd11c-Cre and control mice. Micro-CT analyses confirmed the presence of bone erosions throughout the limb joints and vertebrae of A20fl/fl Cd11c-Cre mice (Supplementary Fig. 10). Thus, A20fl/fl Cd11c-Cre mice spontaneously develop sero-negative arthritis, enthesitis, and spondyloarthritis resembling stereotypic IBD-associated arthritis.
Discussion
DCs, well recognized for their abilities to activate immune responses, are emerging as critical regulators of immune homeostasis. Loss of DCs has been shown to lead to expansion of myeloid cells, a phenotype ascribed to increased Flt3 ligand levels28,29. Prolonged survival of DCs leads to gradual autoimmunity, perhaps due to DC mediated stimulation of T cells for inappropriate periods of time41,42. The inability of DCs to expand regulatory T cells or respond to tolerizing IL-10 or β-catenin signals also perturbs intestinal immune homeostasis35,43,44. Distinct from these mechanisms, we now find that multiple signaling pathways in DCs must be tightly restricted by A20 to maintain homeostasis of both innate and adaptive immune cells. The profound splenomegaly and lymphadenopathy that occurs within 2–3 weeks of birth in A20fl/fl Cd11c-Cre mice contrasts with the more subtle phenotypes of many of the strains above. Thus, restriction of intracellular signaling cascades triggered by steady state stimuli may be particularly critical to DC maintenance of immune homeostasis.
A20 restricts the duration and/or magnitude of overt TLR, CD40, and NOD signals by limiting NF-κ B signaling16–19. These functions likely contribute to A20’s capacity to restrict the immunogenicity of DC based vaccines, which make use of TLR-stimulated DCs activated in vitro45,46. By interrogating A20 function in DCs during steady state, we have revealed that A20 is also required to restrict the more subtle signals sensed by DCs under resting conditions. As A20 may set a signaling threshold for TNF induced NF-κ B to prevent unwanted inflammatory responses47, A20 may perform similar function for TLR and other signals to prevent DC activation in response to naturally occurring stimuli in vivo.
The nature and origin of steady state signals sensed by DCs and how DCs use these signals to regulate immune homeostasis is key to understanding how DC functions may prevent or contribute to disease. To dissect the physiological signals sensed by DCs, we have performed DC-specific epistasis experiments. By combining floxed A20 and Myd88 alleles with the Cd11c-Cre transgene, we have discovered that A20 restricts both MyD88-dependent and MyD88-independent signals in DCs under resting conditions. MyD88-dependent signals might include TLR, IL-1, IL-18, or IL-33 triggered signals. MyD88-independent signals may include TRIF (also known as Ticam1)-dependent TLR signals, other microbial sensors (e.g., Nod like receptors, RIG-1 like receptors), nucleic acid sensors (e.g., RNA or DNA sensors), or other pro-inflammatory proteins (e.g., TNFR family receptors)14,16–19. As our results indicate that multiple intracellular signaling pathways are triggered in DCs, DCs may engage multiple ligands during steady state conditions.
Our studies also unveil immunological consequences of absent A20 regulation of MyD88-dependent and MyD88-independent DC signals. A20 restricts MyD88-independent DC signals that upregulate T cell costimulatory molecules, and blockade of CD80 and CD86 significantly antagonized the activation of adoptively transferred naive T cells. In addition to limiting MyD88-independent signals in DCs, A20 restricts MyD88-dependent signals that drive IL-6 and TNF secretion, T cell expansion, and lymphadenopathy. Hence, a model of DC-regulated T cell homeostasis emerges in which steady state stimuli such as commensal TLR ligands trigger both MyD88-independent and MyD88-dependent intracellular signaling cascades in DCs that are normally restricted by A20. In the absence of A20, unrestrained MyD88-independent signals increase co-stimulatory molecule expression and cause spontaneous T cell activation, and unrestrained MyD88-dependent signals increase DC secretion of cytokines such as IL-6 that drive T cell and myeloid expansion. Future studies with other floxed alleles of molecules that regulate DC signaling, should further clarify the nature of steady state signals that regulate DC functions in resting mice.
In contrast to several other strains of mice bearing perturbed DCs, A20fl/fl Cd11c-Cre mice did not develop autoantibodies41,42,48. This result was somewhat surprising, as A20-deficient DCs were hyperresponsive to MyD88 signals and several reports have attributed TLRs and MyD88 signaling to the development of spontaneous autoimmunity9,10,49. Moreover, lymphadenopathy in A20fl/fl Cd11c-Cre mice involved the expansion of both T cells and B cells, yet with the exception of IgA, serum Ig levels were unperturbed. One potential explanation for the absence of autoantibodies in A20fl/fl Cd11c-Cre mice might be myeloid cell mediated inhibition or cytolysis of activated B cells. By contrast, mice lacking A20 specifically in B cells (A20fl/fl Cd19-Cre mice) had expanded numbers of germinal center B cells and spontaneously developed autoantibodies and lupus-like disease18. Thus, A20 preserves immune homeostasis and prevents autoimmune disease via several cell-type specific functions.
Our studies reinforce the notion that A20 is an enzyme whose expression levels are closely tied to the ability to maintain immune quiescence. Hypomorphic expression of A20 in B cells in A20+fl Cd19-Cre mice perturbs B cell homeostasis and causes autoantibody production18. Similarly, A20+fl Cd11c-Cre mice contained increased numbers of spontaneously activated T cells, and proliferation of naive CD4+ T cells in A20+fl Cd11c-Cre Rag1−/− mice was intermediate to that of A20+ Cd11c-Cre Rag1−/− and A20fl/fl Cd11c-Cre Rag1−/− mice. As our genetic studies strengthen the association of non-coding A20/TNFAIP3 SNPs with susceptibility to human Crohn’s disease and as non-coding A20/TNFAIP3 SNPs (and one coding SNP) have also been linked to susceptibility to rheumatoid arthritis, SLE, psoriasis, and celiac sprue, and as at least one SNP conveys decreased A20 mRNA expression, decreased expression of A20 may well explain how non-coding A20 SNPs predispose to human inflammatory and autoimmune diseases20–26,50.
Our findings that A20-dependent DC functions preserve intestinal homeostasis in A20fl/fl Cd11c-Cre mice provide mechanistic insights into how A20 deficiency may cause susceptibility to IBD and IBD-associated arthritides. Even in a tolerogenic setting, A20-deficient DCs potently induced T cell expansion and drove T cell mediated colitis despite elaborating high levels of IL-10. Hence, in addition to secreting tolerogenic proteins, DCs must restrict their expression of inflammatory proteins to maintain intestinal homeostasis. The spontaneous development of sero-negative arthritis, spondyloarthritis and enthesitis in A20fl/fl Cd11c-Cre mice provides further insights into the pathophysiology of these conditions, particularly as these conditions develop in combination with colitis. IBD-associated arthritis is a family member of the spondylarthopathies in humans, a heterogenous group of arthritic disease strongly associated with HLA-B27 (~90% prevalence)51. Like A20fl/fl Cd11c-Cre mice, HLA-B27 transgenic rats and mice that overexpress TNF (TNFΔARE) mice also exhibit enthesopathy accompanied by intestinal inflammation36,52. A20-deficient DCs produce higher levels of TNF upon activation and it is plausible that A20fl/fl Cd11c-Cre mice and TNFARE mice have partially overlapping pathophysiologies. The responsiveness of human spondyloarthritis patients to anti-TNF therapy further suggests that TNF-driven pathology is intimately linked to human disease51,53. Increased levels of serum IgA and IL-6 observed in A20fl/fl Cd11c-Cre mice are also associated with human spondyloarthopathies53,54 and gene expression profiling of peripheral blood mononuclear cells suggests that A20 may be deficiently expressed in spondyloarthritis patients55,56. The rare combination of spontaneously occurring colitis, sero-negative arthritis, and enthesitis in mice lacking A20 in DCs suggests that disruption of A20-dependent DC functions may underlie colitis and colitis-associated arthritides in human patients. Therapies directed at DCs and A20 may be particularly useful for these diseases.
Methods
Mice
Mice globally deficient in A20 (A20−/−), mice with loxP sites flanking exon 2 of A20 (also known as TNFAIP3) (A20fl/fl mice), Myd88fl/fl mice and Cd11c-Cre transgenic mice have been described14,18,27,34. Unless otherwise indicated, mice were analyzed between 6–12 weeks of age. WT C57BL/6J mice purchased from Jackson Laboratories were used as recipients for adoptive transfer of DCs and as a source of WT polyclonal T cells. To generate bone marrow chimera containing mixed populations of hematopoietic cells, CD45.1 mice (purchased from National Cancer Institute; NCI-Frederick) were sub-lethally irradiated and reconstituted with 2 × 106 bone marrow cells from A20fl/fl Cd11c-Cre mice or A20+/+ Cd11c-Cre control mice. Chimera were analyzed 4–5 weeks post reconstitution; the ratio of CD45.2 (donor) and CD45.1 (WT host) hematopoietic cells was routinely between 0.6 – 1.1. All animals were cared for in accordance with University of California San Francisco (UCSF) institutional guidelines.
Cell preparations and flow cytometry
Splenic dendritic cells were isolated by digesting spleens in 0.2 μg/ml Liberase Blendzyme II or Liberase Blendzyme III, followed by Dynabead depletion of TER119 (TER-119) erythrocyte-lineage cells. For flow cytometry and cell sorting, CD3− (145-2C11) CD19− (1D3) NK1.1− (PK136) cells were gated and cDCs and pDCs distinguished by CD11c (HL3), MHC-II (AF6-120.1), B220 (RA3-6B2), CD11b (M1/70) and Ly6C (AL-21) expression. Samples to be sorted were first enriched by EasySep Stemcell Technologies CD11c positive selection. All antibodies were purchased from BD biosciences or Invitrogen, cells analyzed on an LSR II, or sorted using a MoFlo XDP.
Preparation of bone marrow derived DCs and adoptive transfer of DCs
BMDCs were generated by culturing bone marrow cells in media containing GMCSF for 7 days, after which CD11c+ BMDCs were enriched by autoMACs. To analyze DC responses to LPS, BMDCs were stimulated for 7 h with 1 μg/ml LPS. Cytokines secreted into the supernatant were quantified by ELISA (BD Bioscience). To analyze DC-mediated myeloid recruitment in vivo, BMDCs were stimulated with 0.1 μg/ml LPS for 2.5 h, labeled with CFSE (10 μM), and then 0.5 × 106 DCs were injected into the footpads of wild-type recipients. 24 h post-transfer, the number of CFSE+ DCs in the draining lymph node and the number of myeloid cells recruited to the draining lymph node was analyzed. For some experiments, recipient animals were first injected with 20 μg of rat IgG1 or blocking antibody to IL-6 (MP5-20F3) or TNF (XT3.11), all purchased from the UCSF hybridoma core.
T cell activation and tolerance
Peripheral T cells were stained with BD bioscience antibodies CD44 (IM7) and CD62L (MEL-14) to quantify activated T cell populations. In some experiments, splenic cells were stimulated with PMA and ionomycin to detect cytokine production by CD4 T cells. For adoptive transfer experiments, polyclonal T cells from WT, CD45.1 female mice were labeled with 3 μM CFSE and 2 × 106 T cells were injected into A20fl/fl Cd11c-Cre mice or A20 Cd11c-Cre control mice (CD45.2). Activation and proliferation of CD45.1 CD4 and CD8 T cells in the spleen was analyzed over the period of 10 days. In some experiments, recipient mice were injected every other day with 100 μg of rat IgG1, hamster IgG1 (G235–2356; BD bioscience) or blocking antibodies to CD80 (16-10A1), CD86 (GL-1), IL-6 (MP5-20F3) or TNF (XT3.11) (all purchased from the UCSF hybridoma core).
To assess antigen-specific tolerance, 5 × 106 OT-II T cells (CD45.1) were adoptively transferred into A20fl/fl Cd11c-Cre mice or A20 Cd11c-Cre control mice. On the following day, recipient mice were injected intravenously with 300 μg of OVAp or Hanks buffered saline solution alone. The number of OT-II T cells in each recipient was analyzed 10 days later.
Dextran Sodium Sulfate and T cell induced colitis
DSS (3%, MP Biomedicals) was added to the drinking water of co-housed, 10–12 week old A20fl/fl Cd11c-Cre mice and control Cd11c-Cre mice for 5 days. Mice were assessed daily for diarrhea, bloody stool and body weight. To analyze T cell induced colitis, naive, CD4CD62L CD25 T cells from wild-type mice were sorted by flow cytometry and 1 × 106 cells were injected intravenously into A20fl/fl Cd11c-Cre Rag1 and control Cd11c-Cre Rag1 mice. Body weights were recorded daily.
Histology and micro-computed tomography
Tissues were fixed in 10% paraformaldehyde prior to sectioning and staining with hematoxylin and eosin. Hindpaws were additionally decalcified with Cal-Ex II prior to sectioning. For micro-CT scans, bones were serially fixed in 4% formaldehyde and 70% ethanol and scanned by high resolution micro-CT (vivaCT 40, Scanco Medical) with a slice increment of 25.00 μm. The segmentation values were set at 0.5/2/350. Three-dimensional reconstruction and structural parameters quantification were calculated using Scanco Medical software.
Statistical analysis
Statistical analysis was performed using student’s T test analysis with GraphPad Prism software.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH and the CCFA (to A.M.), a Damon Runyon Postdoctoral Fellowship (to G.E.H.), a CCFA Career Development Award (to S.O.), the Kenneth Rainin Foundation and the UCSF Liver Center. This study makes use of data generated by the Wellcome Trust Case-Control Consortium that was funded by the Wellcome Trust under Awards 076113 and 08475. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
Footnotes
Author contributions
GEH designed and performed cellular and molecular analyses of immune homeostasis, tolerance, arthritis, and colitis experiments in A20fl/fl Cd11c-Cre and related mice; EET generated A20fl/fl Cd11c-Cre mice and initiated analyses of these mice; BR generated Cd11c-Cre mice; BH and AF generated Myd88fl/fl mice; SO assisted with colitis experiments; KET and LAC performed genetic analyses of A20 SNPs using Wellcome Trust data; CJF, EJH and MCN performed micro-CT scans and histological analyses of arthritic joints; RA and JB assisted with breeding, genotyping, and radiation chimera experiments; AM directed the study; AM and GEH wrote the manuscript, with input from BAM.
The authors declare no competing financial interests.
References
- 1.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3.Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- 4.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–338. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 5.Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234–247. doi: 10.1111/j.1600-065X.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 7.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 8.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 9.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 12.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opipari AW, Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- 14.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 16.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 17.Hitotsumatsu O, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavares RM, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–191. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med. 2008;205:451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musone SL, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008 doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton PR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair RP, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elder JT. Genome-wide association scan yields new insights into the immunopathogenesis of psoriasis. Genes Immun. 2009;10:201–209. doi: 10.1038/gene.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trynka G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–1083. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 27.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birnberg T, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. 2008;29:986–997. doi: 10.1016/j.immuni.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Ohnmacht C, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. 2009;206:549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawiger D, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275–284. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Luckashenak N, et al. Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity. 2008;28:521–532. doi: 10.1016/j.immuni.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 33.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 34.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Travis MA, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taurog JD, et al. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–223. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 37.Uhlig HH, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 38.Garrett WS, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D’Agostino MA, Olivieri I. Enthesitis. Best Pract Res Clin Rheumatol. 2006;20:473–486. doi: 10.1016/j.berh.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Chen M, et al. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 42.Stranges PB, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melillo JA, et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184:2638–2645. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manicassamy S, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329:849–853. doi: 10.1126/science.1188510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song XT, et al. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14:258–265. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breckpot K, et al. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol. 2009;182:860–870. doi: 10.4049/jimmunol.182.2.860. [DOI] [PubMed] [Google Scholar]
- 47.Werner SL, et al. Encoding NF-kappaB temporal control in response to TNF: distinct roles for the negative regulators IkappaBalpha and A20. Genes Dev. 2008;22:2093–2101. doi: 10.1101/gad.1680708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanada T, et al. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19:437–450. doi: 10.1016/s1074-7613(03)00240-1. [DOI] [PubMed] [Google Scholar]
- 49.Deane JA, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adrianto I, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43:253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melis L, Elewaut D. Progress in spondylarthritis. Immunopathogenesis of spondyloarthritis: which cells drive disease? Arthritis Res Ther. 2009;11:233. doi: 10.1186/ar2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armaka M, et al. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205:331–337. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keller C, Webb A, Davis J. Cytokines in the seronegative spondyloarthropathies and their modification by TNF blockade: a brief report and literature review. Ann Rheum Dis. 2003;62:1128–1132. doi: 10.1136/ard.2003.011023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Archer JR. Ankylosing spondylitis, IgA, and transforming growth factors. Ann Rheum Dis. 1995;54:544–546. doi: 10.1136/ard.54.7.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith JA, et al. Gene expression analysis of macrophages derived from ankylosing spondylitis patients reveals interferon-gamma dysregulation. Arthritis Rheum. 2008;58:1640–1649. doi: 10.1002/art.23512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duan R, Leo P, Bradbury L, Brown MA, Thomas G. Gene expression profiling reveals a downregulation in immune-associated genes in patients with AS. Ann Rheum Dis. 2010;69:1724–1729. doi: 10.1136/ard.2009.111690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.