

Abstract
Overproduction of proinflammatory cytokines in the CNS has been implicated as a key contributor to pathophysiology progression in Alzheimer's disease (AD), and extensive studies with animal models have shown that selective suppression of excessive glial proinflammatory cytokines can improve neurologic outcomes. The prior art, therefore, raises the logical postulation that intervention with drugs targeting dysregulated glial proinflammatory cytokine production might be effective disease-modifying therapeutics if used in the appropriate biological time window. To test the hypothesis that early stage intervention with such drugs might be therapeutically beneficial, we examined the impact of intervention with MW01-2-151SRM (MW-151), an experimental therapeutic that selectively attenuates proinflammatory cytokine production at low doses. MW-151 was tested in an APP/PS1 knock-in mouse model that exhibits increases in AD-relevant pathology progression with age, including increases in proinflammatory cytokine levels. Drug was administered during two distinct but overlapping therapeutic time windows of early stage pathology development. MW-151 treatment attenuated the increase in microglial and astrocyte activation and proinflammatory cytokine production in the cortex and yielded improvement in neurologic outcomes, such as protection against synaptic protein loss and synaptic plasticity impairment. The results also demonstrate that the therapeutic time window is an important consideration in efficacy studies of drugs that modulate glia biological responses involved in pathology progression and suggest that such paradigms should be considered in the development of new therapeutic regimens that seek to delay the onset or slow the progression of AD.
Introduction
Alzheimer's disease (AD) and related neurodegenerative disorders have an unmet need for therapies that alter disease progression. Evidence suggests that disease progression involves uncontrolled glial activation and neuroinflammation that contribute to neuronal/synaptic dysfunction, which in turn drives a vicious cycle of further glial activation and neuronal damage (Mrak and Griffin, 2005). Dissecting this cycle to find points of potential therapeutic intervention is compounded by the complexity of this multifactorial disease, the multiple roles of glia in homeostasis and host defense, and the diversity of inflammatory and immune response mechanisms (Nelson et al., 2009, 2011; Cameron and Landreth, 2010; Huang and Mucke, 2012; Wyss-Coray and Rogers, 2012).
The diverse array of physiological functions that glia perform, which are context dependent, complicates the interpretation of experimental investigations and clinical observations related to AD pathology. For example, astrocytes and microglia interact with neurons at the synapse to modulate synaptic function and plasticity (Eroglu and Barres, 2010; Tremblay et al., 2011) and are also vital for host defense mechanisms and response to stress (Ransohoff and Perry, 2009). Therefore, health and homeostasis, disease progression and susceptibility, and therapeutic interventions involve a delicate balance in which glial responses must be modulated within a dynamic range.
Related to disease progression, extended overproduction of proinflammatory cytokines such as IL-1β can be particularly detrimental to neuronal function (Shaftel et al., 2008; Hein et al., 2010; Kitazawa et al., 2011). The potential for amelioration of cytokine-induced neuronal dysfunction through therapeutic intervention was shown by Hu et al. (2007) with the development of a small molecule experimental therapeutic, MW01-2-151SRM (here termed MW-151). MW-151 is a selective suppressor of β amyloid (Aβ)-induced glia proinflammatory cytokine production, with resultant attenuation of the cytokine-induced loss of synaptic marker proteins (e.g., synaptophysin and PSD-95) and cognitive deficits (Hu et al., 2007). A major unmet challenge in the field, however, is the question of a therapeutic time window in progressive neurological disorders like AD. The therapeutic time window is a component of drug dosing, a fundamental aspect of the pharmacological basis of drug action, and is linked to the clinical principle of pathology progression. For example, an optimal time window for therapeutic intervention is generally during a peak activity change in the targeted pathway involved in pathology progression.
To address the issue of whether glial activation and proinflammatory cytokine increases might be an early stage pathology progression feature that could be modulated by drugs such as MW-151 with resultant improvement in neurologic endpoints, we examined an APP/PS1 humanized knock-in mouse model that exhibits age-dependent AD pathology progression. We found that proinflammatory cytokines begin to increase early in the pathology progression and that repeat dose intervention with MW-151 starting during this period resulted in significantly improved outcomes. While improvements were also seen with later short-term intervention, the earlier and more prolonged intervention yielded more robust effects. Neither dosing paradigm had a detectable effect on amyloid plaque load or Aβ levels. The results indicate that selective inhibition of increasing proinflammatory cytokine production early in disease progression is beneficial at maintaining synaptic function.
Materials and Methods
Synthesis of MW-151.
MW01-2-151SRM [2-(4-(4-methyl-6-phenylpyridazin-3-yl)piperazin-1-yl)pyrimidine] was synthesized and characterized as reported previously (Hu et al., 2007). The active pharmaceutical used for biological studies was the dihydrochloride hydrate form of the water-soluble, acidic drug. Low doses per administration were employed as noted and are similar to those used previously for efficacy in diverse animal models of CNS diseases that involve excessive proinflammatory cytokine production as part of the pathology progression mechanism, including a model of early AD (Hu et al., 2007), TBI (Lloyd et al., 2008; Chrzaszcz et al., 2010), multiple sclerosis (Karpus et al., 2008), and seizures (Somera-Molina et al., 2007, 2009) at low doses similar to those used in this study.
APP/PS1 knock-in mice.
The AD mouse model used is the APPNLh/NLh × PS1P264L/P264L mutant mice originally developed at Cephalon (Flood et al., 2002). This double mutant mouse was generated by using gene targeting knock-in (KI) technology to introduce the Swedish FAD K670N/M671L mutations and humanize the mouse Aβ sequence (NLh) and to introduce the proline to leucine (P264L) mutation in the mouse PS-1 gene (Reaume et al., 1996; Siman et al., 2000). Expression of both genes is driven by the endogenous promoters, and thus this model shows AD pathology without APP or PS1 overproduction. The APP/PS1 KI mice were maintained on a CD-1/129 background, and wild-type (WT) mice were obtained from heterozygous APP/PS1 matings and maintained as a separate line for >20 generations of inbred mice used as controls. Mice were monitored for maintenance of the appropriate genotypes by PCR analysis of tail snip DNA (Anantharaman et al., 2006). Mice were maintained under 12 h light/dark cycles and provided food and water ad libitum. All animal protocols followed the principles of animal care and experimentation in the Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the University of Kentucky (Lexington, KY).
Treatment of mice.
MW-151 was dissolved in 0.9% sterile NaCl (saline; Hospira NDC 0409-4888-10). Two experimental paradigms were used to test MW-151 efficacy in the APP/PS1 mice. In the extended administration prevention study (chronic intervention paradigm), MW-151 was administered at a low dose (2.5 mg/kg) by intraperitoneal (i.p.) injection three times per week (Monday, Wednesday, Friday), beginning when mice were 6 months old and ending at 11 months. In the short-term treatment study (acute intervention paradigm), MW-151 (2.5 mg/kg) was administered i.p. to 11-month-old mice once daily for 1 week. Controls included APP/PS1 KI or WT mice administered vehicle control (saline). To conserve on animal numbers, WT mice were not administered MW-151. Each experimental group (WT plus vehicle, APP/PS1 KI plus vehicle, APP/PS1 KI plus MW-151) started with 12 mice of either sex, and the numbers of males and females in each group were matched as closely as possible.
Euthanasia and brain tissue harvesting.
Mice were euthanized with an overdose of sodium pentobarbital and transcardially perfused with ice-cold PBS for 5 min. The brains were rapidly removed and bisected along the sagittal plane. The right hemisphere was further dissected and flash frozen in liquid nitrogen and stored at −80°C for subsequent biochemical evaluation. The left hemisphere was immersion fixed in 4% paraformaldehyde overnight before cryoprotection in a 30% sucrose/PBS solution.
Biochemical endpoint analyses.
Neocortical tissue was homogenized using an Omni TH homogenizer in a lysis buffer (1:10 w/v) containing PBS, 1 mm EDTA, and protease inhibitors (1 mm PMSF, 1 μg/ml leupeptin). Extracts were centrifuged at 12,000 × g for 20 min at 4°C in a Beckman Microfuge 18, and supernatants were used as the PBS soluble fraction. Pellets were suspended and rehomogenized in a detergent-containing lysis buffer (tissue protein extraction reagent with Halt Protease and Phosphatase Inhibitor Cocktail; Thermo Scientific) at the same volume used for the first homogenization. Extracts were centrifuged at 12,000 × g for 20 min at 4°C, and supernatants were used for measurement of synaptic proteins in the detergent-soluble fraction. The pellet was then resuspended and rehomogenized in 70% formic acid (FA). The FA homogenate was centrifuged at 1,000,000 × g at 4°C for 60 min. The supernatant was neutralized 1:20 with 1 m Tris (pH11) and used as the FA soluble fraction. Levels of Aβ40 and Aβ42 were measured by ELISA assay using kits from Meso Scale Discovery (MSD) according to the manufacturer's instructions. Levels of cytokines (IL-1β and IL-10) were measured by ELISA assay using kits from MSD according to the manufacturer's instructions with minor modifications. Briefly, 50 μl of the PBS soluble fraction was loaded per well of the MSD plate (100–200 μg of protein). The sample was incubated in the plate overnight at 4°C. All incubation steps were done using an Eppendorf MixMate at 1000 rpm. Cytokine levels were normalized to the total amount of protein in the sample loaded as determined by BCA Protein Assay (Thermo Scientific). The detection limits of the MSD assays are 1.5 pg/ml for IL-1β and 42 pg/ml for IL-10 in serum/plasma.
Levels of synaptic proteins were measured by Western blots as described previously (Xing et al., 2011). Briefly, 1 μg of the detergent-solubilized fraction was prepared in SDS buffer (LI-COR Biosciences). The sample was separated on a 10% NuPAGE Novex Bis-Tris Midi Gel (Invitrogen). Proteins were transferred to nitrocellulose membrane using a dry blotting system (iBlot, Invitrogen). Blots were probed using reagents and manufacturer recommendations for Odyssey Infrared Imaging System (LI-COR Biosciences). Blots were probed for the following primary antibodies: rabbit anti-PSD95 (1:2000, Cell Signaling Technology, catalog no. 3450); mouse anti-synaptophysin (clone SY38) (1:30,000, Millipore, catalog no. MAB5258); rabbit anti-syntaxin 1 (1:100,000, Millipore, catalog no. ab5820); mouse anti-SNAP 25 (clone 20) (1:10,000, BD Biosciences, catalog no. 610366). Rabbit anti-GAPDH (clone 14C10) (1:10,000, Cell Signaling Technology, catalog no. 2118) was used as a loading control. Blots were visualized and analyzed on the Odyssey Infrared Imaging System (LI-COR Biosciences), and integrated intensity values were used in statistics.
For gene expression, dissected neocortical tissues stored at −80°C were used for RNA isolation using RNeasy mini-columns (Qiagen, catalog no. 74104) with on-column DNase treatment (Qiagen, catalog no. 79254) according to the manufacturer's protocol. RNA quantity and quality were determined using A260/A280 readings by NanoDrop (Thermo Scientific). Reverse transcription (RT) was done following the manufacturer's protocol using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, catalog no. 4368814). A no template and a no RT control were conducted to control for contamination. Real-time PCR was performed using the TaqMan Gene Expression Assay Kit (Applied Biosystems, catalog no. 4444964) according to the manufacturer's instructions on a ViiA 7 Real-Time PCR System (Applied Biosystems). The following TaqMan probes (Applied Biosystems) were used: GFAP (Mm00546086_m1), S100B (Mm00485897_m1), vimentin (Mm01333430_m1), Actb (Mm00607939_s1), and IL-1β (Mm00434228_m1) (see Table 1). Relative gene expression was calculated by the 2−ΔΔCT method.
Table 1.
Selective effects of MW-151 on inflammation-related gene expression markers
| Δ > 20% | p < 0.05 (*) | Vehicle | MW-151 | Gene/TaqMan assay ID | |
|---|---|---|---|---|---|
| IBA1 | ↓ | * | 100 ± 9 | 75 ± 4 | Aif1/Mm00479862_g1 |
| CD45 | 100 ± 8 | 88 ± 10 | Ptprc/Mm01293575_m1 | ||
| CD68 | ↓ | * | 100 ± 9 | 74 ± 2 | Cd68/Mm00839636_g1 |
| TLR2 | 100 ± 13 | 118 ± 17 | Tlr2/Mm00442346_m1 | ||
| TLR4 | ↑ | 100 ± 15 | 150 ± 34 | Tlr4/Mm00445273_m1 | |
| NOS2 | 100 ± 15 | 113 ± 18 | Nos2/Mm00440502_m1 | ||
| TNFα | ↓ | 100 ± 12 | 62 ± 18 | Tnf/Mm00443258_m1 | |
| TGFβ | ↓ | * | 100 ± 7 | 75 ± 3 | Tgfb1/Mm00441726_m1 |
| IGF1 | ↓ | 100 ± 28 | 76 ± 17 | Igf1/Mm00439561_m1 | |
| CD200 | ↑ | * | 100 ± 9 | 218 ± 57 | Cd200/Mm00487740_m1 |
| Fractalkine | ↑ | * | 100 ± 9 | 134 ± 6 | Cx3cl1/Mm00436454_m1 |
| HMGB1 | ↓ | * | 100 ± 9 | 70 ± 3 | Hmgb1/Mm00849805_gH |
Immunohistochemical staining.
Coronal sections (30 μm) of the left hemisphere were made using a sliding microtome and a freezing stage and stored in cryoprotectant at −20°C. Staining procedures were conducted on free-floating sections using every twelfth section of the entire left cerebral cortex. Primary and secondary antibodies were diluted in 3% normal goat serum (NGS) (LAMPIRE Biological Laboratories, catalog no. 7332500) with 0.2% Triton X-100. Amyloid plaque burden was detected by rabbit polyclonal anti-Aβ N-terminal antibody (1:10,000, Invitrogen, catalog no. 44338100). Sections were pretreated with 70% formic acid and endogenous peroxidase activity was quenched with 3% H2O2 in methanol, before the tissue blocking in 10% NGS with 0.2% Triton X-100. The biotinylated secondary antibody (1:500, Jackson ImmunoResearch Laboratories, catalog no. 111-065-144) was amplified using avidin-biotin substrate (ABC solution, Vector Laboratories catalog no. PK-6100) followed by color development in 3,3′-diaminobenzidine tetra-hydrochloride (DAB; Sigma, catalog no. D5637). Glial activation was detected with the following antibodies: rabbit anti-GFAP (1:10,000 Dako, catalog no. Z0334) for astrocytes and rabbit anti-IBA1 (1:10,000, Wako, catalog no. 019-19741) for microglia. Endogenous peroxidase activity was quenched with 3% H2O2 in methanol, followed by blocking in 10% NGS with 0.2% Triton X-100. The horseradish peroxidase secondary antibodies (1:500, Jackson ImmunoResearch Laboratories, catalog no. 111-035-003) were visualized with DAB.
Quantitative image analysis of immunohistochemical stains.
The Aperio ScanScope XT digital slide scanner was used to image the entire stained slide at 20× magnification to create a single high-resolution digital image. The neocortex and hippocampus were outlined using the Aperio ImageScope software. The Aperio-positive pixel count algorithm (version 9) was used to quantify the amount of specific staining in the region. The number of positive pixels was normalized to the number of total pixels (positive and negative) to account for variations in the size of the region sampled. The Aperio nuclear algorithm (version 9) was used to quantify the number of cells or the number and size of Aβ plaques in the outlined region. To quantify the number of plaques as a function of size, a threshold was set in the nuclear algorithm so only plaques that were contained in the size bin would be quantified. Color and intensity thresholds were established to detect the immunostaining as positive pixels and background staining as negative pixels. Once conditions were established for an immunohistochemical stain, the entire batch of slides was analyzed with the same parameters. The resulting color markup of the analysis was confirmed for each slide. All quantifications were done by a blinded observer.
Slice electrophysiology.
Methods for preparing acute brain slices for assessment of CA1 synaptic function and plasticity were similar to our previous methods (Norris and Scheff, 2009; Mathis et al., 2011). Briefly, 12-month-old WT and APP/PS1 KI mice were decapitated under CO2 anesthesia, and brains were rapidly removed and stored in ice-cold artificial cerebrospinal fluid (ACSF) containing: 124 mm NaCl, 2 mm KCl, 1.25 mm KH2PO4, 2 mm MgSO4, 26 mm NaHCO3, 10 mm dextrose, saturated with 95% O2 and 5% CO2. Cerebral hemispheres were blocked, mounted, and sectioned (400 μm) in ice-cold Ca2+-free ACSF with a vibrating microtome (Leica Biosystems). Slices were then stored on nets in a humidified interface holding chamber and bathed in CaCl2 (2 mm)-containing ACSF (32°C) with saline vehicle or 30 μm MW-151. After a 1.5–2 h incubation period, slices were transferred to a modified RC-22 chamber (Warner Instruments) affixed to the stage of a Nikon E600 microscope (Nikon Instruments) and perfused (1–2 ml/min) with oxygenated CaCl2 (2 mm)-containing ACSF (32°C). Synaptic responses were evoked by electrical stimulation of CA3 Schaffer collaterals and recorded in CA1 stratum radiatum as described previously (Norris and Scheff, 2009; Mathis et al. 2011). To generate synaptic strength curves, EPSP amplitudes were measured at nine stimulus intensity levels (30, 50, 100, 150, 200, 250, 300, 400, and 500 mA) and plotted against corresponding fiber volley (FV) amplitudes. For some slices, stimulus intensity was reset after the synaptic strength curve to evoke a 1 mV EPSP every 30 s for at least 20 min before the delivery of two 1 s duration trains of 100 Hz stimulation (10 s intertrain interval) for induction of long term potentiation (LTP). Synaptic responses were then collected for an additional 60 min. All electrophysiological recordings were conducted and analyzed by personnel who were blind to genotype and drug treatment conditions.
Statistics.
Statistical analysis was conducted using GraphPad prism software version 5 (GraphPad Software). Values are expressed as mean ± SEM. Groups of two were compared by unpaired t test. Groups of three or more were compared by one-way ANOVA, followed by the Bonferroni Multiple Comparison Test. Significance was defined as p < 0.05. Synaptic strength curves were fit with a three parameter (i.e., max EPSP amplitude, half-maximal FV amplitude, and curve slope) sigmoidal function and compared across treatment groups using Z tests (Z values ≥ |2| were considered statistically significant). For each slice, LTP amplitudes were averaged 50–60 min after delivery of 100 Hz stimulation and normalized to the pre-100 Hz baseline. Significant effects of drug treatment on LTP were determined within genotype using paired t tests. Differences in LTP amplitude between WT and APP/PS1 KI mice were compared using an unpaired t test.
Results
Validation of mechanism of action endpoint in APPxPS1 mice
We reported previously (Hu et al., 2007) that MW-151 is efficacious in a mouse model of AD pathology that involves infusion of human oligomeric Aβ1–42 and represents disease in its early stages (Craft et al., 2004a,b). However, the effect of the compound in an AD model that exhibits more severe amyloid plaque pathology and age-dependent disease progression had not been examined. The first necessary step was to choose an AD mouse model that exhibits appropriate changes in the process that is being targeted by our small molecule compound. Since the mechanism of action of MW-151 is to attenuate cytokine overproduction from activated glia, it was important to select an animal model that exhibits increases in glial activation and cytokine production during the pathology progression. We selected the APPNLh/NLh × PS1P264L/P264L mutant mouse model (APP/PS1) originally developed at Cephalon (Flood et al., 2002), where gene expression in this humanized APP/PS1 double knock-in model is driven by endogenous promoters of the APP and PS-1 genes. This model preserves physiologically relevant APP expression levels and demonstrates progressive AD pathology without APP overproduction. Most importantly, we verified that this model shows an age-dependent increase in levels of proinflammatory cytokines above that of WT mice. As shown in Figure 1A, the levels of IL-1β mRNA and protein in the cortex increase with age in both the WT and APP/PS1 KI mice, with the KI mice showing a significantly higher level of IL-1β at 11 months compared to the WT mice. Thus, this is a relevant animal model in which to test in vivo the efficacy of a cytokine response modulator.
Figure 1.

Increase in IL-1β levels in the APP/PS1 KI mouse as a function of animal age, and selective attenuation of IL-1β by extended administration of MW-151 in a chronic intervention paradigm. A, Neocortical tissue from WT (open circles) or APP/PS1 mice (closed circles) was dissected from the brains of mice at different ages, and levels of IL-1β mRNA and protein were determined by qPCR or MSD ELISA, respectively. B, APP/PS1 mice were treated in a chronic intervention paradigm with either saline vehicle (veh; open bars) or MW-151 (closed bars; 2.5 mg/kg i.p. three times weekly) beginning at early age (6 months old). At the end of the treatment period, when mice were 11 months old, neocortical tissue was analyzed by MSD ELISA for the levels of IL-1β and IL-10. MW-151 selectively attenuated the proinflammatory cytokine IL-1β, but not the anti-inflammatory cytokine IL-10. *p < 0.05 compared to WT mice at 11 months old.
Extended administration of MW-151 in chronic intervention paradigm reduces proinflammatory cytokine levels
Based on the time course of the cytokine rise, we designed a chronic dosing, prevention study paradigm where MW-151 (2.5 mg/kg i.p.) or saline vehicle was administered three times per week (Monday, Wednesday, Friday) beginning at 6 months (when cytokine levels are just beginning to increase) and ending at 11 months (when cytokine overproduction is easily apparent). The mice tolerated well the extended administration of MW-151, exhibiting no significant differences in survival or weight. As shown in Figure 1B, APP/PS1 KI mice administered MW-151 showed lower levels of brain IL-1β compared to vehicle-treated mice, but no decrease in the anti-inflammatory cytokine IL-10. These data confirm that MW-151 exhibits its mechanism of action, i.e., selective attenuation of brain proinflammatory cytokine levels, in this animal model.
Chronic MW-151 administration attenuates microglial and astrocyte activation
The effect of MW-151 treatment on glial activation was examined by immunohistochemistry (IHC) using anti-IBA1 antibody to label microglia and anti-GFAP antibody to label astrocytes. Immunostained sections were scanned and converted into a high-resolution digital image using the Aperio ScanScope digital slide scanner and the positive pixels algorithm or the nuclear algorithm to quantify the immunohistochemical staining. The Aperio ScanScope enables high-resolution image recognition and rigorous digital pathological quantitation. Figure 2A shows a representative field of IBA1-stained microglia in the cortex and digital images of the algorithms used to calculate microglial activation characteristics. MW-151 treatment of APP/PS1 KI mice reduced the microglial activation when data were expressed as either the number of positive pixels (Fig. 2B) or the number of stained cells/mm2 (Fig. 2C).
Figure 2.

Chronic MW-151 administration attenuates microglial activation. A, Representative images of IBA1-stained microglia in cortex of 11-month old APP/PS1 mouse, demonstrating the positive pixel and nuclear algorithms used for quantification of the staining. A computer-generated markup of the algorithm is created by the ScanScope software, which validates the specificity of the method. In the positive pixel algorithm, the microglia shown in shades of gray (indicating intensity of positive staining) is segregated from the background shown as black. The markup in the nuclear algorithm shows the reliable detection and quantification of individual cells (shown by the black markup). B, C, The APP/PS1 mice treated with vehicle (veh; gray bars) show increased microglial staining compared to WT mice treated with vehicle (white bars). MW-151 treatment of the APP/PS1 mice (black bars) reduced the microglial activation as assessed by either the stained area (B) or the number of stained microglia (C). *p < 0.05 compared to vehicle-treated APP/PS1 mice. **p < 0.01 compared to vehicle-treated APP/PS1 mice.
In addition to effects on microglia activation, MW-151 administration also attenuated astrocyte activation. Figure 3A illustrates the GFAP-labeled hemi-brain sections. MW-151 treatment reduced the astrocyte activation in cortex (Fig. 3B) and hippocampus (data not shown), as quantified from the IHC. Interestingly, MW-151 had no effect on GFAP mRNA levels (Fig. 3C). MW-151 treatment did, however, reduce the mRNA levels of two other astrocyte markers. There was a significant decrease in S100B mRNA and a trend toward a decrease in vimentin mRNA (Fig. 3C).
Figure 3.

Chronic MW-151 administration attenuates astrocyte activation. A, Representative images of GFAP-stained sections. B, The APP/PS1 mice treated with vehicle (veh; gray bars) show increased astrocyte staining in cortex compared to WT mice treated with vehicle (white bars). MW-151 treatment of the APP/PS1 mice (black bars) reduced the astrocyte activation. C, MW-151 treatment had no effect on GFAP mRNA levels but did reduce mRNA levels of two other astrocyte markers, S100B and vimentin. **p < 0.01 compared to vehicle-treated APP/PS1 mice.
Chronic MW-151 administration prevents synaptic protein loss
The ability of chronic administration of MW-151 to lead to beneficial neuronal changes was tested by measuring the levels of a panel of synaptic protein markers by Western blot analysis. The APP/PS1 KI model exhibits synaptic dysfunction as seen by a reduction in the levels of synaptic proteins compared to WT mice (Fig. 4). MW-151 treatment of APP/PS1 KI mice prevented the loss of the synaptic proteins PSD95 (Fig. 4A), synaptophysin (Fig. 4B), syntaxin (Fig. 4C), and SNAP25 (Fig. 4D).
Figure 4.

Chronic MW-151 administration prevents synaptic loss. Neocortex tissue was analyzed for the levels of synaptic proteins by Western blots. Compared to WT mice treated with vehicle (veh; white bars), the APP/PS1 mice treated with vehicle (gray bars) show reduced levels of the synaptic proteins PSD95 (A), synaptophysin (B), syntaxin (C), and SNAP25 (D). MW-151 treatment of the APP/PS1 mice protected against this loss of synaptic protein levels, restoring the levels back toward WT values. *p < 0.05 compared to vehicle-treated APP/PS1 mice.
Selectivity of MW-151
In the AD brain, microglia exhibit a complex and heterogeneous set of responses in attempts to restore homeostasis. Although our understanding of the consequences of particular microglia responses to the neurodegenerative process in AD is limited, the classical inflammatory activation state (termed M1) can lead to overproduction of proinflammatory cytokines and subsequent neuronal damage, while an alternative activation state (termed M2) may be involved in attempts to prevent further damage or promote tissue repair. To test the selectivity of MW-151 action in terms of its effects on selected microglia and neuron responses, we measured a panel of markers by quantitative PCR (qPCR) in the cortex from the vehicle-treated or MW-151-treated APP/PS1 KI mice. The endpoints tested represent microglia markers, toll-like receptors, M1 and M2 endpoints, neuroimmune regulatory molecules, and a damage-associated molecular pattern (DAMP) molecule. Table 1 shows that MW-151 modulates some endpoints but not others. For example, TNFα is reduced but NOS2 is not, even though both are considered M1 inflammatory molecules. Similarly, one of the M2 markers is changed (TGFβ), but not the other (IGF1). Other M2 markers in the qPCR array (fizz, Arg1, BDNF) could not be measured accurately because they were at very low to undetected levels in the assay. These results with M2 markers are consistent with our previous observations (data not shown) that in the absence of external stressor stimuli, this APP/PS1 KI mouse model does not express many of the standard M2 markers to an appreciable degree. Other microglia markers also showed selective responses in that IBA1 and CD68 are reduced but CD45 is not. The neuroimmune regulatory molecules CD200 and fractalkine are increased, and the DAMP HMGB1 is decreased by MW-151 treatment. Although the functional significance of changes in these specific responses was not explored as part of this study, these data confirm that MW-151 is selective in its action and not a pan-suppressor of microglia and neuron responses.
Short-term MW-151 treatment in acute intervention paradigm
To test whether MW-151 is efficacious in older mice where disease pathology is already present, we administered MW-151 (2.5 mg/kg i.p.) or saline vehicle to 11-month-old mice once daily for 1 week. As shown in Figure 5A, short-term administration of MW-151 in the acute intervention treatment paradigm attenuated IL-1β production without significant reductions in IL-10, and had no effect on microglial (IBA1) or astrocyte (GFAP) activation. Analysis of synaptic protein levels (Fig. 5B) showed that MW-151 significantly protected the brain against loss of PSD95 and synaptophysin but was less effective against loss of syntaxin and SNAP25.
Figure 5.

Effects of short-term MW-151 treatment in acute intervention paradigm. Eleven-month-old mice were treated with either saline vehicle (veh; white bars, WT; gray bars, APP/PS1) or with 2.5 mg/kg/d MW-151 i.p. (black bars, APP/PS) once daily for 1 week. Neocortex tissue was harvested and endpoints were measured. A, Short-term MW-151 treatment reduced the levels of IL-1β, but not IL-10, and had no effect on microglial (IBA1) or astrocyte (GFAP) activation. B, MW-151 treatment prevented the loss of PSD95 and synaptophysin, but the protection against syntaxin and SNAP25 loss did not reach significance. *p < 0.05 compared to vehicle-treated APP/PS1 mice.
No effects of MW-151 on amyloid plaque load or Aβ40 or Aβ42
We examined the effect of MW-151 treatment on amyloid plaque burden in both the chronic and acute dosing paradigms by staining brain sections with a polyclonal anti-Aβ N-terminal antibody. This antibody reacts with both Aβ40 and Aβ42 and is specific for human Aβ. As shown in Figure 6A, the 11-month-old APP/PS1 mice exhibit abundant numbers of amyloid plaques at this age, similar to previous reports using this model (Flood et al., 2002; Anantharaman et al., 2006; Murphy et al., 2007). Analysis of amyloid plaque staining in the cortex demonstrates that MW-151 treatment in either the chronic intervention paradigm (Fig. 6B) or the acute intervention paradigm (Fig. 6C) had no significant effect on amyloid plaque burden. There was also no effect of MW-151 treatment on amyloid plaque load in the hippocampus (data not shown). The lack of effect was seen when the data were expressed either as percentage of the area stained with anti-Aβ antibody or as number of plaques per mm2. In addition, MW-151 treatment had no effect on the size of the amyloid plaques detected. To determine whether there was any effect of MW-151 treatment on the levels of Aβ, we prepared PBS soluble, detergent soluble, and FA soluble fractions of cortex and measured the levels of Aβ40 and Aβ42 by quantitative Aβ ELISA. No appreciable Aβ was detected in the detergent soluble fraction (data not shown). The amount of Aβ40 or Aβ42 in either the PBS soluble or FA soluble fractions was not significantly different in the APP/PS1 KI mice treated with vehicle or MW-151 in either the chronic (Fig. 6B) or acute (Fig. 6C) intervention paradigm. These data are consistent with the lack of effect on amyloid plaque burden and show that Aβ pathology overall was not significantly changed by MW-151 treatment.
Figure 6.

No effect of MW-151 treatment on Aβ pathology. A, Representative images of Aβ-stained hemi-brain sections. B, C, In either the chronic intervention paradigm (B) or the acute intervention paradigm (C), MW-151 treatment of APP/PS1 mice did not reduce the amyloid plaque load when measured as a percentage of the area stained or as the number of plaques per unit area. There were also no differences in the number of small (20–300), medium (301–1000), or large (1001-∞) amyloid plaques detected. MW-151 treatment of APP/PS1 mice also had no significant effect on the levels of soluble or aggregated forms of Aβ, as measured by levels of Aβ40 or Aβ42 in PBS-soluble and FA-soluble fractions. veh, Vehicle.
MW-151 treatment rescues impairments in LTP
LTP is a widely studied experimental model of synaptic plasticity implicated in learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). Because of the well established link between elevated proinflammatory cytokine levels and impairments in LTP, along with observations that anti-cytokine strategies rescue LTP deficits (Rowan et al., 2007; Lynch, 2010; Yirmiya and Goshen, 2011), we investigated the effect of MW-151 on synaptic function and plasticity by LTP measurements. Coronal brain slices from 12-month-old APP/PS1 KI mice were prepared and incubated for ≥1.5 h in oxygenated ACSF-containing saline vehicle or 30 μm MW-151. Electrically evoked EPSPs were obtained in hippocampal CA1 stratum radiatum, and LTP was induced (Fig. 7A) using 100 Hz stimulus trains as described previously (Mathis et al., 2011; Norris and Scheff, 2009). Similar to previous findings with other APP/PS1 mouse models (e.g., Trinchese et al., 2004), slices from vehicle-treated APP/PS1 mice exhibited significantly reduced LTP compared to slices from age-matched WT controls (n = 5) (Fig. 7B,C). In contrast, LTP in MW-151-treated slices from the APP/PS1 group was similar in magnitude to the WT group and significantly greater than that shown by vehicle-treated APP/PS1 mice (p < 0.05). No genotype or drug treatment effects were observed for other synaptic function markers, including basal synaptic strength (Fig. 7D) and paired-pulse facilitation (data not shown). The results suggest that MW-151 treatment selectively enhances synaptic plasticity mechanisms in APP/PS1 KI mice.
Figure 7.
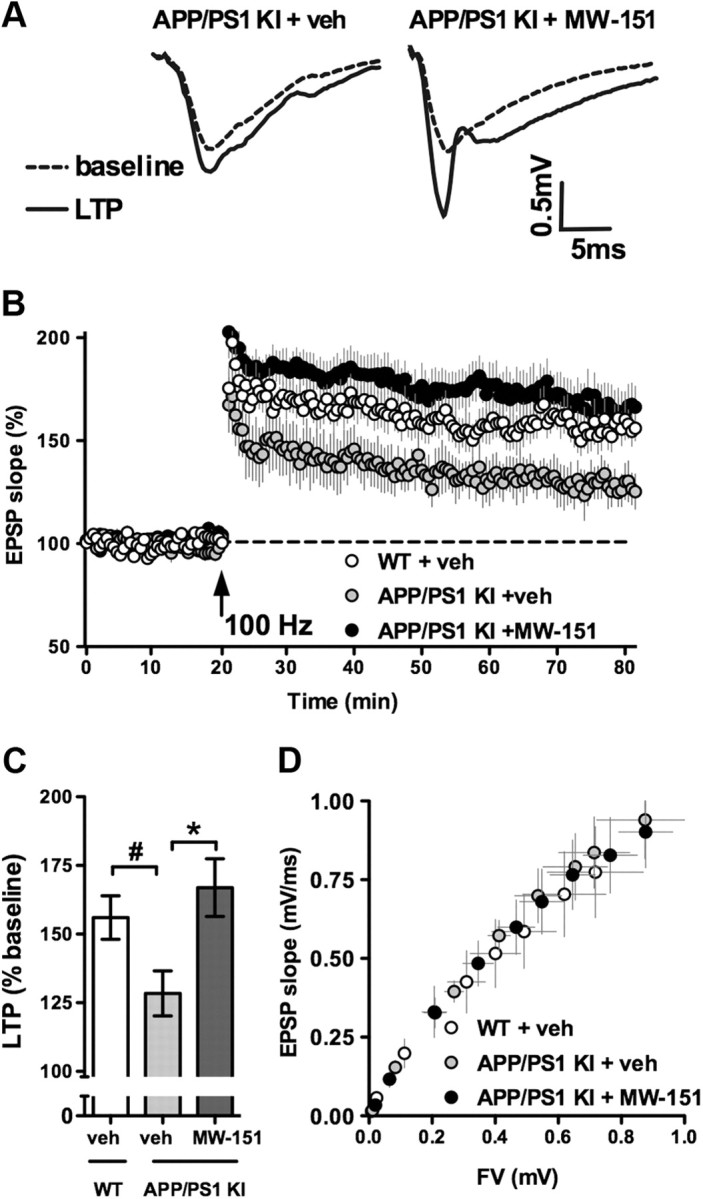
MW-151 increases LTP in the hippocampus of 12-month-old APP/PS1 mice. A, Representative CA3-CA1 synaptic responses from vehicle (veh)-treated and 30 μm MW-151-treated hippocampal slices from APP/PS1 mice, collected immediately before (dashed line) and 60 min after (solid line) LTP induction. B, Time plot of EPSP slope measures taken before and after 100 Hz stimulation in slices from WT mice (white circles) and APP/PS1 mice treated with (black circles) or without (gray circles) MW-151. C, Bar graph shows LTP levels across treatment conditions. Number symbol (#) indicates a significant decrease (p < 0.05) in LTP in vehicle-treated slices from APP/PS1 mice compared to vehicle-treated slices from WT mice. Asterisk indicates a significant increase (p < 0.05) in LTP in MW-151-treated slices from APP/PS1 mice relative to the APP/PS1 vehicle group. D, EPSP slope measures plotted against FV measures in each treatment condition. No effects of genotype or MW-151 treatment on synaptic strength were observed.
Discussion
We report here several findings with important implications for future glia-targeted AD therapeutic strategies. First, we show in an APP/PS1 KI mouse model of AD pathology that pharmacological intervention with a selective, small molecule inhibitor of glial proinflammatory cytokine overproduction is efficacious at attenuating microglial and astrocyte activation and proinflammatory cytokine levels, which prevents loss of synaptic proteins and impairments in synaptic plasticity (LTP). Second, there were no adverse effects observed with extended treatment of aging mice with a therapy that targets excessive proinflammatory cytokine production. Third, the beneficial effects of MW-151 occurred in the absence of a detectable change in amyloid plaque load or in levels of soluble or aggregated forms of Aβ40 or Aβ42 measured in PBS soluble and FA soluble neocortical fractions. Fourth, although MW-151 is effective when administration is started at either early or later stages of disease pathology progression, the compound is more effective in a preventative mode where administration is initiated before full later stage pathology is emerging. Overall, our results show that selective inhibition of the increasing glia proinflammatory cytokine response in early time windows of disease progression is beneficial at maintaining synaptic function, suggesting the importance of therapeutic time window considerations in future design of efficacy studies of neuroinflammation-targeted drugs.
The observation that MW-151 was more efficacious when administered in a preventative paradigm before onset of overt pathology implies that drugs that target neuroinflammation may be most beneficial when used early in the course of disease pathogenesis. This is consistent with many studies demonstrating that glial activation and neuroinflammation are early events in AD pathogenesis (e.g., see Cagnin et al., 2001; Mrak and Griffin, 2005; Griffin and Barger, 2010; Carter et al., 2012), recent genome-wide association study data showing that many of the genes associated with increased AD risk are inflammation-related (Sleegers et al., 2010; Guerreiro et al., 2012), and epidemiological and population-based clinical studies that suggest an association between early use of nonsteroidal anti-inflammatory drugs (NSAIDs) and reduced risk of developing AD (McGeer et al., 1996; in't Veld et al., 2001; Zandi and Breitner, 2001).
In addition, analysis of data (Breitner et al., 2011; Leoutsakos et al., 2012) from the randomized clinical trial ADAPT (Alzheimer's Disease Anti-inflammatory Prevention Trial) suggest that the treatment effects of NSAIDs differ depending on the stage of disease, with potential beneficial effects in asymptomatic individuals with little or no cognitive decline at the time of drug use, but harmful effects in later stages of AD pathogenesis. Although MW-151 is not an NSAID, the results reported here are consistent with the idea that suppression of CNS inflammation is likely to be effective as a prevention strategy before the self-propagating, proinflammatory cytokine cycle is too far advanced during disease progression. However, MW-151 was also able to block cytokine production and prevent the decrease in some synaptic proteins even when administered at a later stage of pathology progression (to 11-month old mice for only a week). These initial observations will need to be followed up with more detailed studies on the therapeutic window, but they suggest that future anti-inflammatory strategies aimed at selective fine tuning of neuroinflammation might be useful not only to prevent or delay onset of AD, but potentially as part of therapeutic strategies to treat existing disease, which would involve co-therapy with other drugs.
MW-151 was discovered using a functional in vivo screen (Ralay Ranaivo et al., 2006; Wing et al., 2006; Hu et al., 2007; Van Eldik et al., 2007) that employed an animal model of injury induced by toxic aggregates of human Aβ1–42. That model lacked an age-dependent progressive pathology and did not have an amyloid plaque-rich environment. The APP/PS1 KI model used here exhibits progressive amyloid plaque deposition, beginning at around 9 months of age and increasing with age (Flood et al., 2002; Anantharaman et al., 2006; Murphy et al., 2007). This model also exhibits progressive pathological alterations in bidirectional synaptic plasticity, cognitive measures, synaptosomal membrane lipids, and oxidative stress markers as a function of animal development and maturation (Anantharaman et al., 2006; Chang et al., 2006; Bruce-Keller et al., 2011). We have shown here that the APP/PS1 KI model also demonstrates an early and progressive increase in brain proinflammatory cytokine levels, and that dampening of the proinflammatory cytokine production and the associated glial activation is beneficial to synaptic function. In this regard, it is interesting to note that these beneficial effects of MW-151 occurred in the absence of a detectable effect on amyloid plaque load or a change in PBS soluble or FA soluble Aβ40/42 levels, demonstrating that MW-151 treatment does not shift the levels of soluble toxic Aβ oligomers or insoluble fibrils.
There is inconsistency in the literature with respect to the effect of blocking cytokine signaling on Aβ pathology. Some studies have reported that interference with cytokine signaling by anti-inflammatory drug treatment or genetic deletion strategies results in reduced amyloid plaque load (Lim et al., 2000; Jantzen et al., 2002; Yan et al., 2003; Morihara et al., 2005; He et al., 2007; Yamamoto et al., 2007). However, other studies have shown that cytokine-suppressing strategies can reduce brain inflammatory responses and attenuate markers of neuronal pathology with little or no effect on amyloid plaque pathology (Ralay Ranaivo et al., 2006; Kitazawa et al., 2011). Therefore, it is possible that an impact on amyloid plaques might be due to the model being used, the pharmacodynamic profile of the particular drug, the therapeutic time window of treatment and pathology development, or any combination of these variables. Our results with MW-151 pharmacodynamics in this particular model clearly demonstrate that synaptic and glia endpoints, related respectively to efficacy and in vivo mechanism of drug action, can be modulated independently of effects on Aβ. The linkage to tau pathology or its interface with Aβ pathology (Zilka et al., 2012) cannot be addressed in this model because the APP/PS1 KI mouse lacks AD-relevant tau pathology. Thus, the interplay between glia proinflammatory cytokine-linked neuropathologies and AD-relevant tau neurofibrillary pathology remains to be addressed in future studies.
The linkage between changes in proinflammatory cytokine levels and neurologic outcomes, revealed by the improvement in synaptic endpoints (synaptic protein levels and LTP) after attenuation of proinflammatory cytokine levels by MW-151 treatment, is consistent with previous reports of MW-151 efficacy in other rodent CNS injury models, including oligomeric Aβ-induced injury (Hu et al., 2007), kainic acid-induced seizures (Somera-Molina et al., 2007, 2009), Traumatic Brain Injury models (Lloyd et al., 2008; Chrzaszcz et al., 2010), and the Experimental Autoimmune Encephalomyelitis model of multiple sclerosis (Karpus et al., 2008). These previous studies documented that MW-151 efficacy at reducing injury-induced upregulation of proinflammatory cytokine levels was associated with a reduction in long-term neuronal injury and attenuation of hippocampal-dependent cognitive deficits. Our findings are also consistent with reports that have linked other anti-neuroinflammatory interventions with changes in synaptic plasticity. For example, pharmacological interventions and/or genetic manipulations that suppress proinflammatory cytokine signaling normalize synaptic function in primary neural cultures (Sama et al., 2008) and ameliorate LTP deficits associated with aging (Griffin et al., 2006; Cowley et al., 2012) and Aβ (Rowan et al., 2007; Kotilinek et al., 2008; Yirmiya et al., 2011).
Overall, the data reported here add to the growing body of evidence that has identified dysregulated glial activation and proinflammatory cytokine overproduction as a common early pathophysiologic mechanism and potential therapeutic target in diverse neurodegenerative disorders. In addition, the results extend this state of knowledge to therapeutic intervention possibilities, especially the importance of therapeutic time window considerations in the design of intervention strategies.
Footnotes
This research was supported in part by funds from the American Health Assistance Foundation (L.J.V.E.), an Alzheimer's Association Zenith award (L.J.V.E.), a gift from the Kleberg Foundation (C.M.N.), and NIH Grants P01 AG005119 (L.J.V.E., D.S.C.), R01 AG027297 (C.M.N.), R01 NS056051 (D.M. Watterson), R01 AG031311 (D.M. Watterson), and S10 RR026489. A.D.B. is a postdoctoral fellow supported by NIH F32 AG037280. We thank Edgardo Dimayuga, Saktimayee M. Roy, James Schavocky, and Bin Xing for their assistance with various aspects of this work.
The authors declare no competing financial interests.
References
- Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. β-Amyloid mediated nitration of manganese superoxide dismutase: Implications for oxidative stress in a APPNLh/NLh × PS-1P264L/P264L double knock-in mouse model of Alzheimer's disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- ADAP Research Group. Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7:402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMulle JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, Keller JN. Cognitive impairment in humanized APP×PS1 mice is linked to Aβ1–42 and NOX activation. Neurobiol Dis. 2011;44:317–326. doi: 10.1016/j.nbd.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cameron B, Landreth GE. Inflammation, microglia and Alzheimer's disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, Nordberg A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-l-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53:37–46. doi: 10.2967/jnumed.110.087031. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzaszcz M, Venkatesan C, Dragisic T, Watterson DM, Wainwright MS. Minozac treatment prevents increased seizure susceptibility in a mouse “two-hit” model of closed skull traumatic brain injury and electroconvulsive shock-induced seizures. J Neurotrauma. 2010;27:1283–1295. doi: 10.1089/neu.2009.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley TR, O'Sullivan J, Blau C, Deighan BF, Jones R, Kerskens C, Richardson JC, Virley D, Upton N, Lynch MA. Rosiglitazone attenuates the age-related changes in astrocytosis and the deficit in LTP. Neurobiol Aging. 2012;33:162–175. doi: 10.1016/j.neurobiolaging.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ. Aminopyridazines inhibit β-amyloid induced glial activation and neuronal damage in vivo. Neurobiol Aging. 2004a;25:1283–1292. doi: 10.1016/j.neurobiolaging.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM. Aminopyridazines attenuate hippocampus dependent behavioral deficits induced by human Aβ in a murine model of neuroinflammation. J Mol Neurosci. 2004b;24:115–122. doi: 10.1385/JMN:24:1:115. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased Aβ42 and Aβ deposition without APP overproduction. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99:1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- Griffin WST, Barger SW. Neuroinflammatory cytokines – the common thread in Alzheimer's pathogenesis. US Neurol. 2010;6:19–27. [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Gustafson DR, Hardy J. The genetic architecture of Alzheimer's disease: beyond APP, PSENs and APOE. Neurobiol Aging. 2012;33:437–456. doi: 10.1016/j.neurobiolaging.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa ACS, O'Banion MK. Sustained hippocampal IL-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Ralay Ranaivo H, Roy SM, Behanna HA, Wing LK, Munoz L, Guo L, Van Eldik LJ, Watterson DM. Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. Bioorg Med Chem Lett. 2007;17:414–418. doi: 10.1016/j.bmcl.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in't Veld BA, Ruitenberg A, Hofman A, Launer LJ, Van Duijn CM, Stijnen T, Breteler MMB, Stricker BHC. Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and β-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus WJ, Reynolds N, Behanna HA, Van Eldik LJ, Watterson DM. Inhibition of experimental autoimmune encephalomyelitis by a novel small molecular weight pro-inflammatory cytokine suppressing drug. J Neuroimmunol. 2008;203:73–78. doi: 10.1016/j.jneuroim.2008.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer's disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the ADAP Research Team. Leoutsakos J-MS, Muthen BO, Breitner JCS, Lyketsos CG. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer's disease anti-inflammatory prevention trial. Int J Geriatr Psychiatry. 2012;27:364–374. doi: 10.1002/gps.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina KC, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurology outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Age-related neuroinflammatory changes negatively impact on neuronal function. Front Aging Neurosci. 2010;1:6. doi: 10.3389/neuro.24.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation – a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mathis DM, Furman JL, Norris CM. Preparation of acute hippocampal slices from rats and transgenic mice for the study of synaptic alterations during aging and amyloid pathology. J Vis Exp. 2011;pii:2330. doi: 10.3791/2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM. Ibuprofen suppresses interleukin-1β induction of pro-amyloidogenic α2-antichymotrypsin to ameliorate β-amyloid (Aβ) pathology in Alzheimer's models. Neuropsychopharmacology. 2005;30:1111–1120. doi: 10.1038/sj.npp.1300668. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WST. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Aβ solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, Abner EL, Smith CD, Van Eldik LJ, Kryscio RJ, Scheff SW. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121:571–587. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Scheff SW. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma. 2009;26:2269–2278. doi: 10.1089/neu.2009.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, Watterson DM. Glia as a therapeutic target: selective suppression of human amyloid-β-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–670. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer's disease mutations and a “humanized” Aβ sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer's disease amyloid β-peptide-induced dysfunction. Biochem Soc Trans. 2007;35:1219–1223. doi: 10.1042/BST0351219. [DOI] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1β-dependent signaling between glia and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WST, O'Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on Aβ production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleegers K, Lambert JC, Bertram L, Cruts M, Amouyel P, Van Broeckhoven C. The pursuit of susceptibility genes for Alzheimer's disease: progress and prospects. Trends Genet. 2010;26:84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Somera-Molina KC, Robin B, Somera CA, Anderson C, Koh S, Behanna HA, Van Eldik LJ, Watterson DM, Wainwright MS. Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of pro-inflammatory cytokine upregulation. Epilepsia. 2007;48:1785–1800. doi: 10.1111/j.1528-1167.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- Somera-Molina KC, Nair S, Van Eldik LJ, Watterson DM, Wainwright MS. Enhanced microglial activation and proinflammatory cytokine upregulation are linked to increased susceptibility to seizures and neurologic injury in a ‘two-hit’ seizure model. Brain Res. 2009;1282:162–172. doi: 10.1016/j.brainres.2009.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MÈ, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol. 2004;55:801–814. doi: 10.1002/ana.20101. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Watterson DM. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–296. doi: 10.1016/S0074-7742(07)82015-0. [DOI] [PubMed] [Google Scholar]
- Wing LK, Behanna HA, Van Eldik LJ, Watterson DM, Ralay Ranaivo H. De novo and molecular target-independent discovery of orally bioavailable lead compounds for neurological disorders. Curr Alzheimer Res. 2006;3:205–214. doi: 10.2174/156720506777632844. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease – a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B, Bachstetter AD, Van Eldik LJ. Microglia p38α MAPK is critical for LPS-induced neuron degeneration through a mechanism involving TNFα. Mol Neurodegener. 2011;6:84. doi: 10.1186/1750-1326-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Breitner JCS. Do NSAIDs prevent Alzheimer's disease? And, if so, why? The epidemiological evidence. Neurobiol Aging. 2001;22:811–817. doi: 10.1016/s0197-4580(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M. Who fans the flames of Alzheimer's disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi: 10.1186/1742-2094-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]