

Abstract
Varicella-zoster virus (VZV) is a neurotropic herpesvirus, which can cause a variety of complications during varicella infections. These range from meningoencephalitis to polyneuritis to retinitis. After primary VZV infection, VZV enters the dorsal root ganglia in a latent state. Reactivation from latency leads to zoster. Zoster ophthalmicus in particular can in turn lead to involvement of the cerebral arteries and subsequent stroke. The velocity of VZV is 13 cm per day, as virus travels from ganglion to skin. The live attenuated varicella vaccine virus is markedly less neurovirulent than the wild type virus. Nevertheless, a few cases of herpes zoster due to the vaccine virus have been documented. Usually, herpes zoster occurs in the same arm as the vaccination, often 3 or more years after vaccination. Thus, herpes zoster in a vaccinee often represents a reactivation of vaccine virus that was carried to the cervical dorsal root ganglia from a site of local replication in the arm. Finally, the role of autophagy during VZV is discussed. Autophagosome formation is a prominent feature in the skin vesicles during both varicella and herpes zoster. Therefore, autophagy is one of the innate immune mechanisms associated with VZV infection in humans.
Keywords: Varicella encephalitis, Herpes zoster, Stroke, Autophagy
INTRODUCTION
Varicella-zoster virus (VZV) is a neurotropic herpes virus. VZV infection causes a variety of manifestations in the central nervous system (CNS) and the peripheral nervous system. Primary VZV infection is called varicella (chickenpox)1. The incubation period of varicella is usually 14–15 days (Figure 1). The duration of the typical varicella exanthem is usually 5–7 days. Within a vesicle, the virus enters a sensory nerve and travels retrograde to the dorsal root ganglion, to establish a latent state. Decades later, the same virus often reactivates and travels anterograde in the sensory nerve to the skin, where local replication leads to zoster (shingles). Recent studies have established that the VZV velocity within a sensory neuron is around 13 cm per day2.
Figure 1.

Pathogenesis of central nervous disease caused by varicella. After a child contracts varicella, a series of events occur during the 14-day incubation period (left side). First, the virus replicates in the head and neck region, including the tonsils, for 4–6 days. Thereafter, a viremia occurs, which seeds the virus throughout the body. Finally the virus exits the capillaries and enters the epidermis, to cause the characteristic vesicular rash. The times when the virus can enter the CNS are listed on the right side.
In 1995, a live attenuated varicella vaccine was approved for administration to children in the United States3,4. Prior to 1995, there were between 100 and 150 deaths annually in the United States related to varicella. Because of universal varicella vaccination, the disease varicella has largely disappeared from the United States5. Canada has also instituted universal varicella immunization. However, in Europe, South America, Asia, and Africa, varicella vaccination is not widely recommended. Therefore, varicella and its neurologic complications will remain a common problem in many countries around the world. In fact, in Sweden, VZV infection is currently one of the most frequently diagnosed viral causes of meningoencephalitis6.
In this review, we will discuss the neurologic manifestations of varicella, congenital varicella, as well as the live attenuated varicella vaccine virus. We will also discuss the herpesvirus neurovirulence gene called ICP34.5 and its effect on autophagy.
NEUROVIRULENCE OF VARICELLA
The neurological complications of varicella have been well described. The most comprehensive review in the English language was written by the British physician Underwood in 19357. He described over 120 cases, including clinical vignettes on each case (Table 1). The most common diagnosis was meningoencephalitis. A major subgroup within the preceding diagnosis was acute cerebellitis. Other diagnoses included hemiparesis and hemiplegia, ascending and transverse myelitis, and peripheral neuritis, e.g., facial palsy. Eye findings included involvement of the optic nerve and anisocoria.
Table 1.
Neurological complications of varicella described by Underwood
| Condition | Number of Patients |
|---|---|
| Meningo-encephalitis/Encephalomyelitis | 52 |
| Cerebral encephalitis – 12 | |
| Meningo-encephalitis – 11 | |
| Acute cerebral tremor – 6 | |
| Convulsions – 5 | |
| Meningitis – 4 | |
| Encephalomyelitis – 3 | |
| Meningomyelitis – 2 | |
| Non-characterized encephalitis – 2 | |
| Ascending myelitis – 1 | |
| Coma and convulsions – 1 | |
| Delirium and convulsions – 1 | |
| Disseminated sclerosis – 1 | |
| Insanity – 1 | |
| Meningeal syndrome – 1 | |
| Violent agitation – 1 | |
| Cerebellar ataxia | 30 |
| Cerebellar ataxia – 29 | |
| Cerebellar syndrome – 1 | |
| Poliomyelitis | 16 |
| Anterior poliomyelitis – 6 | |
| Myelitis – 4 | |
| Poliomyelitis – 2 | |
| Hemiparesis – 1 | |
| Hemiplegia – 1 | |
| Polio-encephalitis – 1 | |
| Transverse myelitis – 1 | |
| Choreo-athetosis | 8 |
| Neuro-retinitis/Eye | 6 |
| Choked disc – 1 | |
| Neuro-retinitis – 1 | |
| Ophthalmoplegia – 1 | |
| Optic neuritis – 1 | |
| Paralysis of iris – 1 | |
| Transitory blindness – 1 | |
| Polyneuritis | 5 |
| Polyneuritis – 4 | |
| Peripheral neuritis – 1 | |
| Cranial neuritis | 2 |
| Bilateral ptosis and divergent squint – 1 | |
| Facial paralysis – 1 |
Most of the neurological manifestations of varicella arise after the characteristic vesicular rash has appeared (Figure 1). However, on rare occasions, the first neurological sign can appear in the late incubation period before any vesicles have appeared on the skin. One report described a 5 year old boy who developed headache, nausea, and malaise, followed on 2 successive days by vomiting and ataxia8. Upon neurologic examination 3 days later he was found to have slow and slurred speech, impaired ability to perform coordinating and alternating movements, and ataxic gait. Examination of the cerebrospinal fluid revealed a total cell count of 52 per milliliter (elevated) and a total protein of 17 mg per 100 ml (normal). A macular rash appeared on the face and neck 11 days after the onset of symptoms, followed in 1 day by a generalized vesicular rash. This case is unusual because of the long interval between onset of neurologic symptoms and vesicular rash. If one reexamines the schema for pathogenesis (Fig. 1), it would appear that the most likely explanation is invasion of the central nervous system (CNS) by virus during or immediately after the initial period of replication in the regional lymph nodes of the head and neck, before the viremia9.
NEUROVIRULENCE PRESENTING AS STROKE
Stroke can also be associated with recent herpes zoster and to a lesser extent recent varicella10. Stroke after VZV infection in children was the subject of a recent review and commentary11,12. Altogether 70 cases of stroke in children with VZV infection were found in a literature search. The median age of the children was 5 years, with a range of 6 months to 11 years. One child developed signs of stroke during the incubation period of varicella. Most children, however, manifested a stroke 4–5 months after their bout of varicella.
The pathogenesis of stroke after varicella involves the cerebral arteries. Imaging of the stroke cases showed inflammation of the middle cerebral artery, anterior cerebral artery and internal carotid artery, in decreasing order. These arteries are innervated by the ophthalmic branch of the trigeminal nerve (Figure 2). Therefore, the most likely scenario involves retrograde trafficking of virus from vesicles on the face to the trigeminal ganglion at time of varicella (Figure 2). Some virions presumably then traffic from the trigeminal ganglion to the cerebral artery, where they incite inflammation of the endothelial lining, thrombosis and subsequent stroke. These events often occur concomitantly with zoster ophthalmicus.
Figure 2.
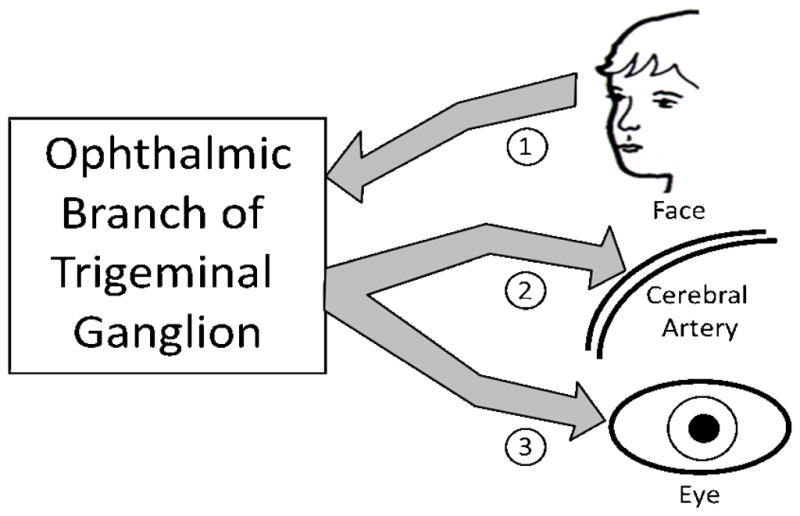
Stroke after varicella infection. Stroke after varicella infection requires viral transit to the cerebral arteries. Since the varicella exanthem commonly involves the face, the most likely route involves retrograde spread of virus from the face to the trigeminal ganglion via the ophthalmic branch (arrow 1). From the trigeminal ganglion, virus can spread via anterograde circuits to a cerebral artery (arrow 2) or the eye (arrow 3). Viral replication in the endothelial lining of an artery leads to inflammation and subsequent stroke. These events are most obvious when zoster ophthalmicus is present, but varicella related stroke can occur without an overt zosteriform rash on the face.
NEUROVIRULENCE OF VARICELLA IN THE UNITED STATES
Universal varicella vaccination was instituted in the United States shortly after the live attenuated varicella vaccine was approved in 1995. During the first decade of varicella vaccination, the number of hospitalizations for varicella-related illnesses fell dramatically13. Furthermore, the number of varicella-related deaths dropped from 100–150 per year to less than 20. Obviously, therefore, varicella vaccination has greatly reduced both the morbidity and the mortality surrounding this childhood illness.
In a recent review of the neurological complications of varicella in California, the authors analyzed data from 26 patients with CNS disease between 1998–200914. The most important finding was that only one of the 29 cases was related to a side-effect of varicella vaccination. All the other cases were caused by community-acquired (wild-type) varicella. The complications included meningitis (50%), encephalitis (42%), and acute disseminated encephalomyelitis (ADEM) (8%). Seven patients were less than 18 years of age.
The one child with vaccine virus related disease was 12 years old. She had had a single varicella vaccination at age 1 year. Then 11 years later she presented with zoster of the C4/C5 dermatomes. She recovered completely after receiving oral acyclovir. Of note, at least 4 children with wild-type CNS disease had had one varicella vaccination. We now know that one dose is insufficient to assure immunity throughout childhood. Therefore these children undoubtedly had breakthrough varicella. Two doses of varicella vaccine during early childhood are now recommended to avoid the problem of breakthrough infection with wild-type varicella15.
In summary, the above data from the most populous state in the United States demonstrate a dramatic reduction in the CNS complications of wild-type varicella in children. Implementation of the two-dose varicella vaccination policy should eliminate the few cases of breakthrough disease. The detection of only one case of vaccine virus related zoster demonstrates a diminished neurovirulence of the live attenuated vaccine.
NEUROVIRULENCE OF VARICELLA IN SWEDEN
In the previous section, we analyzed the decreasing complications of varicella in the United States, a country that initiated universal varicella vaccination in 1995. In this section, we analyze the continuing neurological consequences of varicella in Sweden, a country that has decided not to institute universal varicella vaccination of young children.
At the University Hospital in Gothenburg, Sweden, a registry was kept of all VZV-related neurological complications between 1995 and 20016. The investigators had records of 97 patients who had a variety of neurological manifestations. These included: (i) encephalitis or encephalopathy, (ii) viral meningitis, (iii) stroke, and (iv) cranial nerve involvement, presenting as facial palsy or Ramsay Hunt Syndrome. Of the 97 patients, 7 were 12 years old or younger. Although the mortality rate was low (2 deaths), nearly one-third of the patients had long lived neurological deficits after the acute VZV-related disease. The authors also state the CNS disease related to varicella is now more common in their population than the neurological complications of herpes simplex virus.
NEUROVIRULENCE OF CONGENITAL VARICELLA
When a pregnant woman is exposed to varicella, she can contract the disease if she never had chickenpox as a child or if she has never received varicella vaccination as a child. Varicella in pregnant women is a serious illness because of the high risk of varicella pneumonitis. The fetus is also at risk of contracting an intrauterine varicella infection16. However, the risk of a symptomatic infection is low, certainly less than 2%.
Chickenpox in the pregnant woman occasionally is followed by in utero infection of the fetus with VZ virus. The consequences of such an infection to the fetus may be minimal to inapparent; alternately the newborn may present multiple congenital abnormalities. Laforet and Lynch published the first report of a congenital defects syndrome in an infant who was born to a mother who had contracted chickenpox during week 8 of pregnancy17. At birth the infant was noted to have a malformed right leg. Clinically, the right leg was shorter than the left and had obvious muscle atrophy. The toes of the right foot were described only as “small horny pegs.” Roentgenographic examination revealed underdeveloped long bones and absent phalanges. Knee and ankle jerks were also absent in the same leg. The left leg was normal in size, but reddish pigmented areas (3–5 mm in diameter) were observed on the medial aspect. The infant was also noted to have insufficiency of vesical and anal sphincters. Within the first 3 months of life, the baby manifested generalized cortical atrophy as well as chorioretinitis with bilateral optic atrophy.
Savage and colleagues reported another infant with congenital defects born to a mother with a history of chickenpox during week 11 of pregnancy18. This full-term baby had a hypoplastic left arm with rudimentary digits. Cutaneous cicatrices (“zig-zag” scars) were prominent along the entire length of the left arm and were also present to a lesser degree on the scalp and the otherwise normal-appearing right leg. Ptosis and meiosis of the left eye indicated Horner’s syndrome, but there was no involvement of the retina. Electromyographic and nerve conduction studies of the affected arm gave evidence of denervation. A third case, reported by McKendry and Bailey during the same year, was remarkably similar to the previous two cases19. Multiple congenital defects were noted in an infant born to a mother who also had chickenpox during week 11 of gestation. This baby girl had hypoplasia of the right arm and shoulder; radiographs revealed corresponding hypoplasia of the right scapula, clavicle, and humerus. The skin over the right shoulder contained four deep scars. Superficial scars were present on the knees, scalp, and left hip. Chorioretinitis and nystagmoid eye movements developed within a few months.
The congenital defects syndrome may occur following VZV infection during any stage of embryogenesis or fetal growth. However, the period of greatest risk is the first trimester. In order to understand the pathogenesis of the fetal malformations it is essential to be cognizant of certain aspects of intrauterine development of the nervous system, especially the spinal cord. In utero the cervical and lumbosacral cord comprise greater than 70% of the total weight, since these two areas innervate the developing upper and lower extremities, respectively, whereas the thoracic cord serves mainly as a conduit. Destruction of any of the cervical or lumbosacral plexi during embryogenesis would lead to denervation and subsequent hypoplasia of the corresponding limb bud.
In this regard, it is of interest to review the timetable of musculoskeletal development which has been constructed after examination of aborted fetuses. The major differentiation and innervation of the limb primordia occur between 6 and 12 weeks of gestation. This critical time interval correlates precisely with the chickenpox histories of mothers who delivered severely affected children. Thus the agenesis or hypoplasia of the extremities is most likely the result of viral localization and replication in areas of the cervical or lumbosacral cord. In a similar manner, optic atrophy and chorioretinitis would follow VZV infection of the developing optic tracts, while involvement of the sympathetic fibers in the cervical and lumbosacral cord would lead to such divergent effects as Horner’s syndrome and bladder or anal sphincter dysfunction. The commonly seen zig-zag cicatrices represent the cutaneous residua of viral infection of the sensory nerves. In summary it appears that after infection of the fetus, VZV often manifests its known neurotropic potential and preferentially localizes in the nervous system. In other words, the malformations associated with the congenital defects syndrome are the sequelae of neurologic damage following this viral infection (Table 2).
Table 2.
Sequelae of fetal Infection with varicella-zoster virus
| Damage to sensory nerves |
| Cutaneous manifestations |
| Zig-zag (cicatricial) skin lesions |
| Hypopigmentation |
| Damage to optic stalk, optic cup, and lens vesicles |
| Microphthalmia |
| Cataracts |
| Chorioretinitis |
| Optic atrophy |
| Damage to cervical and lumbosacral cord |
| Hypoplasia of upper/lower extremities |
| Motor/sensory deficits |
| Absent deep tendon reflexes |
| Anisocoria/Horner’s syndrome |
| Anal/vesical sphincter dysfunction |
| Damage to brain |
| Encephalitis |
| Microcephaly |
NEUROVIRULENCE OF THE LIVE ATTENUATED VARICELLA VACCINATION
Varicella vaccination is usually administered by subcutaneous injection into the arm. Occasionally a small number of vesicles will arise around the site of injection, a sign of viral replication. However, the infection usually remains localized in the arm and adjacent lymph nodes. In around 5% of immunized children, a viremia has been documented by PCR detection of VZV DNA in peripheral blood cells.
The following neurological complications of varicella vaccine have been reported (Table 3). Case 1 was vaccinated at 12 months of age in his right thigh20. At 15 months the patient developed zoster lesions in the right thigh. The zoster occurred at the same site as vaccination. The child had an underlying neuroblastoma. Biopsy from a zoster lesion and subsequent DNA analysis confirmed the presence of Oka VZV. Case 2 was vaccinated at 22 months of age21. At 3.5 years the girl developed zoster ophthalmicus and encephalitis. The article does not state where the patient was vaccinated. Polymerase chain reaction (PCR) of the cerebral spinal fluid (CSF) revealed the patient was positive for Oka VZV. Case 3 was vaccinated at 16 months of age22. At 4 years a zoster rash developed on the right arm, followed by meningitis. The article does not state where the patient was vaccinated. CSF DNA was sent to the CDC for strain typing, and the presence of Oka VZV was confirmed. PCR of zoster lesions also identified VZV, but genotyping was not performed.
Table 3.
Children with neurological complications caused by vaccine virus
| Case Number
|
Age of Vaccination
|
Age of Complication
|
Underlying Condition
|
Complication
|
Reference
|
|---|---|---|---|---|---|
| 1 | 12 months | 15 months | Neuroblastoma | Zoster and meningitis | 20 |
| 2 | 22 months | 3.5 years | Healthy | Zoster and encephalitis | 21 |
| 3 | 16 months | 4 years | Healthy | Zoster and meningitis | 22 |
| 4 | 29 months | 4 years | Leukemia | Zoster and meningitis | 22 |
| 5 | 1 year | 8 years | Healthy | Zoster and meningitis | 23 |
| 6 | 1 year | 9 years | Healthy | Zoster and meningitis | 24 |
| 7 | 1 year | 12 years | Healthy | Zoster and meningitis | 14 |
Case 4 was vaccinated at 29 months of age22. At 4 years the child developed zoster in the C6-C7 dermatome, followed by meningitis. The article does not state where the patient was vaccinated. The patient was undergoing chemotherapy for leukemia when they developed zoster. Both zoster lesions and CSF confirmed the presence of Oka VZV. Case 5 was vaccinated at 1 year of age in the left deltoid region23. At 8 years the boy developed zoster lesions, followed by meningitis. The zoster occurred at the same site as vaccination. PCR of the patient’s CSF identified Oka VZV. Case 6 was vaccinated at 1 year of age24. At 9 years the child developed zoster lesions on his proximal left arm which progressed to the forearm, followed by meningitis. The article does not state where the patient was vaccinated. The CDC confirmed the presence of Oka VZV in both zoster lesions and CSF from the patient. Case 7 was vaccinated at 1 year of age14. At 12 years the girl developed zoster lesions and meningitis. The article does not state where the patient was vaccinated. PCR of CSF confirmed the presence of Oka VZV. With an 11 year interval, this case represents the longest period between Oka VZV vaccination and presentation of CNS disease found in the literature to date. Of note, in several cases the zoster event occurred in the same extremity as had received the varicella vaccination or was the likely site of vaccination (Figure 3).
Figure 3.
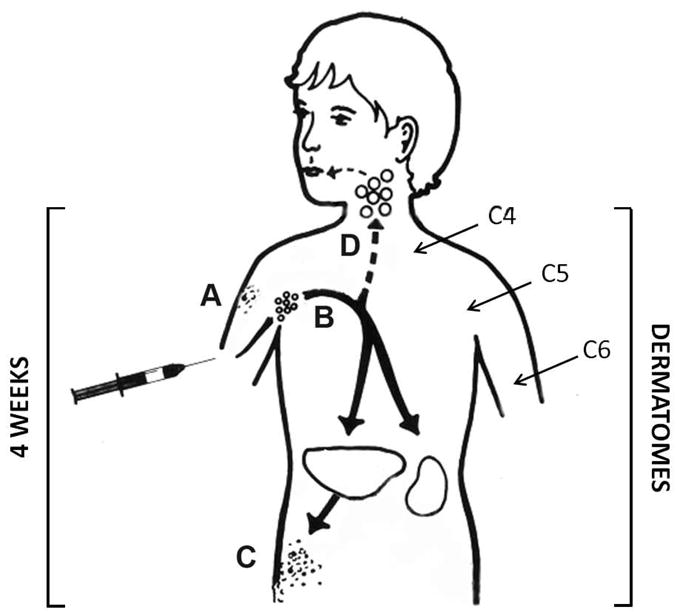
Zoster caused by varicella vaccination. After vaccination in the arm, the virus usually replicates locally (A), after which the virus travels retrograde to the dorsal root ganglia in the cervical spinal cord. The majority of cases of zoster that have been documented to be caused by vaccine virus have occurred at or near the site of the initial vaccination. In most cases, this site was the upper arm. The dermatomes which innervate the upper arm are shown in the figure. In about 5% of vaccinated children, the virus replicates to a greater extent and enters the blood stream (B). During the viremia, a rash can appear at a distant site (C); virus even can enter the head and neck region and transit into the brain (D).
Since 4 million children are immunized yearly in the United States, the neurovirulence of the live attenuated vaccine is dramatically less than that of the wild-type infection. Only a few cases of zoster and meningitis secondary to vaccine virus have been reported since 1995 (Table 3). Nevertheless, these cases illustrate that vaccine virus can occasionally establish latency in the dorsal root ganglia, especially those providing sensory fibers to the arm.
NEUROVIRULENCE GENES AND AUTOPHAGY
The HSV neurovirulence gene is called ICP34.525. The VZV genome lacks this gene. Nevertheless, a brief discussion of the neurovirulence gene will provide insight into one of the important processes involved in virulence. The ICP34.5 gene facilitates HSV replication in the CNS. Mutant HSV viruses lacking ICP34.5 replicate poorly when inoculated into the brain tissues of mice. The ICP34.5 protein contains 263 amino acids; these include a 159-amino terminal domain, ten repeats of the tripeptide AlaThrPro, followed by a 74-amino acid carboxy-terminal domain. The carboxy terminus binds to protein phosphatase 1 alpha (PP1); in turn, this complex dephosphorylates eukaryotic translation factor 2 alpha. Of interest, the HSV ICP34.5 carboxy terminus is homologous to that of GADD34.5 (growth arrest and DNA damage protein). GADD34 functions as a sensor of ER stress stimuli, after which it interacts with PP1 to dephosphorylate the transcription factor. The final result is a negative feedback loop to recover protein synthesis within the cell.
Autophagy is a newly recognized innate defense mechanism during several childhood infections. The neurovirulence marker of the HSV ICP34.5 gene is directly related to autophagy. In short, ICP34.5 acts to inhibit autophagy and thereby enhance neurovirulence. The mechanism involves the binding of ICP34.5 to the Beclin protein. In turn, this binding diminishes the interaction of Beclin with the class III phosphatidyl-inositol 3-kinase, a critical early step in the formation of the autophagosome. Without autophagy, HSV achieves its maximal neurovirulence.
In sharp contrast, the VZV genome lacks an ICP34.5 homolog. Although we initially postulated that VZV would down regulate autophagy by an alternative mechanism, that hypothesis was incorrect. Autophagosome formation is abundant within the skin vesicles during both varicella and zoster26. Since the skin vesicle is the final site of virion assembly and envelopment, autophagy does not appear to suppress VZV replication and decrease virus later, as is the case within the murine brain infected with HSV.
At the present time, we do not know which VZV genes regulate neurovirulence. However, the live attenuated varicella vaccine is markedly less virulent overall and certainly less neurovirulent. Therefore, some of the markers of attenuation, e.g., single nucleotide polymorphisms (SNPs), within the major regulatory protein called IE62, may provide insight into which wild-type sequences are determinants of VZV neurovirulence.
Acknowledgments
Research on varicella virus in the C. Grose laboratory is supported by grants from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Corey Horien, Email: charles-grose@uiowa.edu.
Charles Grose, Email: charles-grose@uiowa.edu.
References
- 1.Weller TH. Varicella and herpes zoster. Changing concepts of the natural history, control, and importance of a not-so-benign virus. N Engl J Med. 1983;309:1434–40. doi: 10.1056/NEJM198312083092306. [DOI] [PubMed] [Google Scholar]
- 2.Tannous R, Grose C. Calculation of the anterograde velocity of varicella-zoster virions in a human sciatic nerve during shingles. J Infect Dis. 2011;203:324–6. doi: 10.1093/infdis/jiq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi M. Effectiveness of live varicella vaccine. Expert Opin Biol Ther. 2004;4:199–216. doi: 10.1517/14712598.4.2.199. [DOI] [PubMed] [Google Scholar]
- 4.Gershon AA, LaRussa P, Hardy I, Steinberg S, Silverstein S. Varicella vaccine: the American experience. J Infect Dis. 1992;166 (Suppl 1):S63–8. doi: 10.1093/infdis/166.supplement_1.s63. [DOI] [PubMed] [Google Scholar]
- 5.Grose C. Varicella vaccination of children in the United States: assessment after the first decade 1995–2005. J Clin Virol. 2005;33:89–95. doi: 10.1016/j.jcv.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Persson A, Bergstrom T, Lindh M, Namvar L, Studahl M. Varicella-zoster virus CNS disease--viral load, clinical manifestations and sequels. J Clin Virol. 2009;46:249–53. doi: 10.1016/j.jcv.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Underwood E. The neurological complications of varicella: a clinical and epidemiological study. Br J Child Dis. 1935;32:83–127. 77–96, 241–63. [Google Scholar]
- 8.Goldston AS, Millichap JG, Miller RH. Cerebellar ataxia with preeruptive Varicella. Am J Dis Child. 1963;106:197–200. doi: 10.1001/archpedi.1963.02080050199013. [DOI] [PubMed] [Google Scholar]
- 9.Grose C. Variation on a theme by Fenner: the pathogenesis of chickenpox. Pediatrics. 1981;68:735–7. [PubMed] [Google Scholar]
- 10.Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 2009;8:731–40. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciccone S, Faggioli R, Calzolari F, Sartori S, Calderone M, Borgna-Pignatti C. Stroke after varicella-zoster infection: Report of a case and review of literature. Pediatr Infect Dis J. 2010;29:864–7. doi: 10.1097/inf.0b013e3181ddefb6. [DOI] [PubMed] [Google Scholar]
- 12.Grose C. Stroke after varicella and zoster ophthalmicus: another indication for treatment and immunization. Pediatr Infect Dis J. 2010;29:868–9. doi: 10.1097/INF.0b013e3181ddef9e. [DOI] [PubMed] [Google Scholar]
- 13.Lopez AS, Zhang J, Brown C, Bialek S. Varicella-related hospitalizations in the United States, 2000–2006: the 1-dose varicella vaccination era. Pediatrics. 2011;127:238–45. doi: 10.1542/peds.2010-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pahud BA, Glaser CA, Dekker CL, Arvin AM, Schmid DS. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316–23. doi: 10.1093/infdis/jiq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro ED, Vazquez M, Esposito D, et al. Effectiveness of 2 doses of varicella vaccine in children. J Infect Dis. 2011;203:312–5. doi: 10.1093/infdis/jiq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grose C. Congenital infections caused by varicella zoster virus and herpes simplex virus. Semin Pediatr Neurol. 1994;1:43–9. [PubMed] [Google Scholar]
- 17.Laforet EG, Lynch CL., Jr Multiple congenital defects following maternal varicella; report of a case. N Engl J Med. 1947;236:534–7. doi: 10.1056/NEJM194704102361504. [DOI] [PubMed] [Google Scholar]
- 18.Savage MO, Moosa A, Gordon RR. Maternal varicella infection as a cause of fetal malformations. Lancet. 1973;1:352–4. doi: 10.1016/s0140-6736(73)90134-7. [DOI] [PubMed] [Google Scholar]
- 19.McKendry JB, Bailey JD. Congenital varicella associated with multiple defects. Can Med Assoc J. 1973;108:66–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Levin MJ, Dahl KM, Weinberg A, Giller R, Patel A, Krause PR. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. J Infect Dis. 2003;188:954–9. doi: 10.1086/378502. [DOI] [PubMed] [Google Scholar]
- 21.Chouliaras G, Spoulou V, Quinlivan M, Breuer J, Theodoridou M. Vaccine-associated herpes zoster ophthalmicus [correction of opthalmicus] and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969–72. doi: 10.1542/peds.2009-2633. [DOI] [PubMed] [Google Scholar]
- 22.Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995–2005. J Infect Dis. 2008;197 (Suppl 2):S170–7. doi: 10.1086/522161. [DOI] [PubMed] [Google Scholar]
- 23.Levin MJ, DeBiasi RL, Bostik V, Schmid DS. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella-zoster virus vaccine. J Infect Dis. 2008;198:1444–7. doi: 10.1086/592452. [DOI] [PubMed] [Google Scholar]
- 24.Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792–5. doi: 10.1016/j.annemergmed.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34. 5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Carpenter JE, Jackson W, Benetti L, Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J Virol. 2011 doi: 10.1128/JVI.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]