

Abstract
Little is known about gene-environment interactions in Parkinson disease (PD). We examined potential interactions of smoking and caffeine intake with ten GWAS SNPs at or near the SNCA, MAPT, LRRK2 and HLA loci among 584 PD patients and 1,571 controls. The main effects of these SNPs and environmental exposures were consistent with previous reports. Family history of PD was associated with PD risk (OR=2.71, 95% CI: 1.97–3.74), which was little affected by further adjustment for these SNPs and environmental exposures. Overall, we did not find significant interactions of either smoking or caffeine intake with these SNPs. However, with a combined smoking and caffeine intake exposure, we found a significant interaction with rs2896905 at SLC2A13, near LRRK2 (p uncorrected= 0.0008). Each A allele was associated with a 35% higher PD risk among never smokers with low caffeine intake, but with a 32% lower risk among smokers with high caffeine intake. This study provides preliminary evidence of a potential gene-environment interaction for PD, which should be investigated in future studies.
Keywords: Parkinson disease (PD), gene-environment interactions, genome-wide association study
1. Introduction
Parkinson disease (PD) is the second most prevalent neurodegenerative disease and affects over one million Americans. It has been hypothesized that interactions between genetic and environmental factors may play important roles in the pathogeneses of late-onset sporadic PD. Recent genome-wide association studies (GWAS) (Hamza, et al., 2010,Satake, et al., 2009,Simon-Sanchez, et al., 2009) and later meta-analyses (Do, et al. 2011,Nalls, et al., 2011,IPDGC, 2011) have reported multiple genetic loci that contribute to risk for late onset PD. The data are most consistent for alpha-synuclein (SNCA) and microtubule-associated protein tau (MAPT), both of which are known PD susceptibility genes (Satake, et al., 2009,Simon-Sanchez, et al., 2009). In addition, several novel genetic loci have been identified in recent GWAS studies, including a single nucleotide polymorphism (SNP) in the human leucocyte antigen (HLA-DRA) region (Hamza, et al., 2010). However, little is known on whether these genetic associations with PD could be modified by environmental factors. Among environmental factors evaluated in epidemiological studies, cigarette smoking and coffee drinking have been consistently associated with lower PD risk (Ascherio, et al., 2001,Chen, et al., 2010,Ritz, et al., 2007,Ross, et al., 2000). Using data from a previous GWAS study replication genotyping, we examined the potential interactions between PD GWAS SNPs from the SNCA, MAPT, and HLA loci and smoking and caffeine intake on PD risk in a large case-control study with prospectively collected environmental data. In addition, we also included in the analysis 3 SNPs on chromosome 12 that are close to the leucine-rich repeat kinase 2 (LRRK2) locus.
2. Methods
2.1 Study population and PD case recruitment
The Parkinson’s Genes and Environmental study (PAGE) is a case-control study nested in the large prospective NIH-AARP Diet and Health Study. The cohort collected detailed information on cigarette smoking and the consumption of caffeine containing drinks as part of its baseline survey in 1995–1996, and asked life-time occurrence of PD at its follow-up survey in 2004–2006 (Chen, et al., 2010). Details of the PAGE study design have been published elsewhere (Gao, et al., 2011). Briefly, potential PD cases were first identified from self-reports from the cohort’s follow-up survey and then the PD diagnosis was either confirmed by their treating neurologists or via medical record review by a movement disorder specialist (Chen, et al., 2010). Only cases with a confirmed PD diagnosis were included in the analysis. For PD cases, we also asked for the dates of diagnosis and first symptoms and family history of PD defined as having at least one first-degree blood relative with physician diagnosed PD. Controls were randomly selected from cohort participants who did not report PD on the follow-up questionnaire, and were frequency matched to cases by gender, ethnicity, and year of birth in 5 year groups. A total of 838 confirmed PD cases and 1,703 controls had genotyping data. To avoid population stratification, we excluded 30 cases and 80 controls who were not Non-Hispanic Whites or who did not report ethnicity. As PD patients might have modified their smoking and coffee habits as a result of PD, to reduce potential impact of reverse causation on the analyses, we further excluded 208 patients whose PD diagnoses were before 1997. Finally, we excluded 16 cases and 52 controls for poor genotyping quality, leaving us a total of 584 cases and 1,571 controls for the final analyses.
2.2 DNA extraction and genotyping
These PD cases and controls were included as the primary replication samples in one of the previous GWAS studies (Simon-Sanchez, et al., 2009). A total of 384 SNPs were genotyped in the replication stage of the GWAS study based on their main effect p-values at the initial genome-wide screening. Our analyses included 6 SNPs that showed genome-wide association in the previous GWAS analysis (Simon-Sanchez, et al., 2009), 3 from SNCA and 3 from MAPT. In addition, we also included 3 SNPs at the SLC2A13 locus on chromosome 12, which is close to a known PD gene LRRK2; we included these 3 SNPs also because they surpassed stage-1 screening in the previous GWAS study and showed nominal associations with PD (P ~10−3-10−5) in combined samples (Simon-Sanchez, et al., 2009). Further, a companion GWAS analysis among Japanese population also mapped to this region (Satake, et al. 2009). Finally, we genotyped rs3129882 at the HLA-DRA locus that was linked to PD risk in a later GWAS (Hamza, et al., 2010). The GWAS replication genotyping was done by collaborators at the National Institute on Aging with customized GoldenGate assays (Illumina, San Diego, CA) (Scholz, et al., 2009,Simon-Sanchez, et al., 2009), with an overall call rate of 97%. The other SNP (HLA- rs3129882) was separately genotyped by BioServe Biotechnologies, Ltd. (Beltsville, MD) using MassARRAY iPLEXTM platform. Call rate for this SNP was also 97%.
2.3 Assessments of Environmental Exposures
Information on smoking and caffeine intake was obtained from the cohort’s baseline survey in 1995–1996 (Schatzkin, et al., 2001). Participants were asked whether they had ever smoked more than 100 cigarettes during their lifetime; and for ever smokers, the typical amount of smoking, current smoking status, and years since last smoking (Chen, et al., 2010). Caffeine intake was derived from a food frequency survey based on consumptions of coffee and other caffeine containing drinks and foods in the year before the baseline survey. The baseline survey also collected information on date of birth, sex, and ethnicity.
2.4 Statistical analyses
For each SNP, we first examined the main effect based on log-additive model, and then its interactions with environmental exposures. We first examined interactions with smoking (never v.s. ever) or caffeine intake (≤ median (low) v.s. > median (high)) separately. We then combined the exposure of smoking with caffeine intake as both nicotine and caffeine are brain stimulants and both exposures are linked to lower PD risk. We defined the combined exposure as 1) never smokers with low caffeine intake, 2) ever smokers with low caffeine intake or never smokers with high caffeine intake, and 3) ever smokers with high caffeine intake. Odds ratios (ORs) and 95% confidence intervals (CIs) were derived from logistic regression models, adjusting for year of birth, gender, smoking status, and daily caffeine intake when appropriate. The statistical significance for interaction was examined by including a multiplicative term between environmental exposure and each SNP with the Wald test. The main effects of SNPs were saturated in the model to improve its ability to find a significant gene-environment interaction. Statistical analyses were performed using SAS version 9.2 (SAS Institute Inc, Cary, NC) and Plink v1.07 (Purcell, et al., 2007). Power calculation was performed with Quanto V1.2.4.
2.5 Standard protocol approvals, registrations and patient consent
The study protocol was approved by the Institutional Review Board of the National Institute of Environmental Health Sciences and all study participants provided informed written consent.
3. Results
Population characteristics by incident PD cases diagnosed after 1997 and controls are listed in Table 1. Cases and controls were matched by year of birth and gender. The average age of PD diagnosis was 69.3 ± 5.4 years and the average age of PD onset was 68.2 ± 5.7 years. Compared with controls, PD cases were less likely to smoke and had lower caffeine intake. After adjusting for matching factors, the OR comparing ever with never smokers was 0.66 (95% CI: 0.54 – 0.80) and OR comparing high v.s. low caffeine intake was 0.81 (95% CI: 0.67–0.99). Ever smokers who also reported high caffeine intake had a 45% lower PD risk as compared with never smokers with low caffeine intake (OR= 0.55, 95% CI: 0.42 – 0.73). Further, PD cases were more likely to report a PD diagnosis among first degree blood relatives (OR=2.71, 95% CI: 1.97 – 3.74).
Table 1.
Population characteristics according to case-control status*
| PD cases | Controls | OR (95% CI) | |
|---|---|---|---|
| N | 584 | 1,571 | |
| Gender, (%) | Matching factor | ||
| Men | 449 (76.9) | 1,237 (78.7) | |
| Women | 135 (23.1) | 334 (21.3) | |
| Year of Birth | 1932 ± 4.9 | 1932 ± 4.8 | Matching factor |
| Smoking, (%) | |||
| Never | 263 (45.7) | 553 (35.6) | Referent |
| Ever | 313 (54.3) | 1,002 (64.4) | 0.66 (0.54 0.80) |
| Caffeine intake, (%) | |||
| ≤Median (low) | 325 (55.7) | 793 (50.5) | Referent |
| > Median (high) | 259(44.4) | 778 (49.5) | 0.81 (0.67 0.99) |
| Combined exposure, (%) | |||
| Never smokers with low caffeine intake | 153 (26.6) | 295 (19.0) | Referent |
| Never smokers with high intake or ever smokers with low intake |
277 (48.1) | 748 (48.1) | 0.72 (0.57 0.91) |
| Ever smokers with high intake | 146 (25.4) | 512 (32.9) | 0.55 (0.42 0.73) |
| Family history of PD, (%) | |||
| No | 494 (85.5) | 1,351 (94.1) | Referent |
| Yes | 84 (14.5) | 84 (5.9) | 2.71 (1.97 3.74) |
Odds ratios (OR) and 95% confidence intervals (CI) were adjusted for gender and year of birth; the number of cases and controls may not add up to total due to missing.
As expected, multiple SNPs from the SNCA and MAPT loci showed marginal main effects with PD (Table 2). SNPs near the LRRK2 and HLA-DRA loci did not show statistically significance with PD risk, but the strength and direction of the associations were in line with previous GWAS studies.
Table 2.
Main effects of SNPs on the risk of Parkinson disease and potential interactions with environmental factors
| F_A* | F_U† | Main effect | P for interaction | |||||
|---|---|---|---|---|---|---|---|---|
| Locus | Mapped Gene | |||||||
| (%) | (%) | OR (95%CI) ‡ | P | Smoking | Caffeine intake | Combined | ||
| CHR4 | ||||||||
| rs11931074 G>T | GPRIN3||SNCA | 10.1 | 7.5 | 1.45 (1.14–1.84) | 0.002 | 0.23 | 0.36 | 0.14 |
| rs3857059A>G | SNCA | 10.1 | 7.4 | 1.46 (1.15–1.86) | 0.002 | 0.33 | 0.33 | 0.17 |
| rs2736990T>C | SNCA | 49.1 | 45.5 | 1.17 (1.02–1.34) | 0.02 | 0.69 | 0.44 | 0.44 |
| CHR17 | ||||||||
| rs393152A>G | C17orf69 | 19.6 | 23.2 | 0.81 (0.68–0.96) | 0.02 | 0.95 | 0.91 | 0.99 |
| rs17563986A>G | MAPT | 18.9 | 22.1 | 0.83 (0.70–0.99) | 0.04 | 0.75 | 0.91 | 0.69 |
| rs199533C>T | NSF | 17.9 | 20.5 | 0.85 (0.71–1.01) | 0.07 | 0.86 | 0.68 | 0.76 |
| CHR12 | ||||||||
| rs11564162 A>G | SLC2A13 | 18.9 | 20.8 | 0.90 (0.76–1.07) | 0.23 | 0.76 | 0.21 | 0.25 |
| rs2896905G>A | SLC2A13 | 38.2 | 38.9 | 0.98 (0.86–1.13) | 0.82 | 0.02 | 0.01 | 0.0008¥ |
| rs1491923T>C | SLC2A13||LRRK2 | 32.8 | 30.1 | 1.12 (0.97–1.30) | 0.11 | 0.14 | 0.048 | 0.02 |
| CHR6 | ||||||||
| rs3129882A>G | HLA||DRA | 43.7 | 42.1 | 1.06 (0.93–1.21) | 0.41 | 0.42 | 0.42 | 0.89 |
F_A: minor allele frequency in cases;
F_U: minor allele frequency in controls
Odds ratios (OR) and 95% confidence intervals (CI) were adjusted for year of birth, gender, smoking status (never vs. ever), and daily caffeine intake (≤ vs. > median)
Statistically significant after Bonferroni correction (p≤0.0017).
We also examined whether selected GWAS SNPs and/or environmental factors could explain the association of family history with PD. The OR was barely affected by the adjustment: with the adjustment of both environmental and genetic variables, the OR for family history changed to 2.65 (95% CI: 1.92 – 3.67).
The results for the interactions between the 10 examined SNPs and smoking and/or caffeine intake are presented in Table 2. We found one SNP (rs2896905) at the SLC2A13 locus near LRRK2 suggested potential interactions with smoking or caffeine intake. The P value without Bonferroni correction was 0.02 for interaction with smoking and 0.01 for interaction with caffeine intake. The interaction term became statistically significant (p uncorrected = 0.0008) when smoking and caffeine intake were combined, even after Bonferroni correction (with 30 independent tests, P =0.024). We did not identify statistically significant interactions between the other SNPs examined and smoking and/or caffeine intake.
We further conducted stratified analyses to examine rs2896905 in relation to PD among subgroups of smoking and caffeine intake (Figure 1). Among never smokers with low caffeine intake, each A allele was associated with 35% higher PD risk (OR=1.35, 95% CI: 1.01 – 1.82, p = 0.04); the corresponding OR was 1.01 (95% CI: 0.83 – 1.23, p = 0.91) among smokers with low caffeine intake or never smokers with high caffeine intake, and was 0.68 (95% CI: 0.51 – 0.90, p = 0.007) for smokers with high caffeine intake. Similar observations were made when analyses were stratified by smoking or caffeine intake separately. The OR for each A allele was 1.18 (95% CI: 0.95 – 1.47, p = 0.13) among never smokers, 0.85 (95% CI: 0.70 – 1.05, p = 0.08) among ever smokers, 1.15 (95% CI: 0.96 – 1.39, p = 0.14) among participants with low caffeine intake, and 0.81 (95% CI: 0.66 – 1.00, p = 0.049) among participants with high caffeine intake.
Figure 1. Subgroup analyses of rs2896905 in relation to Parkinson disease according to combined exposure to smoking and caffeine intake.
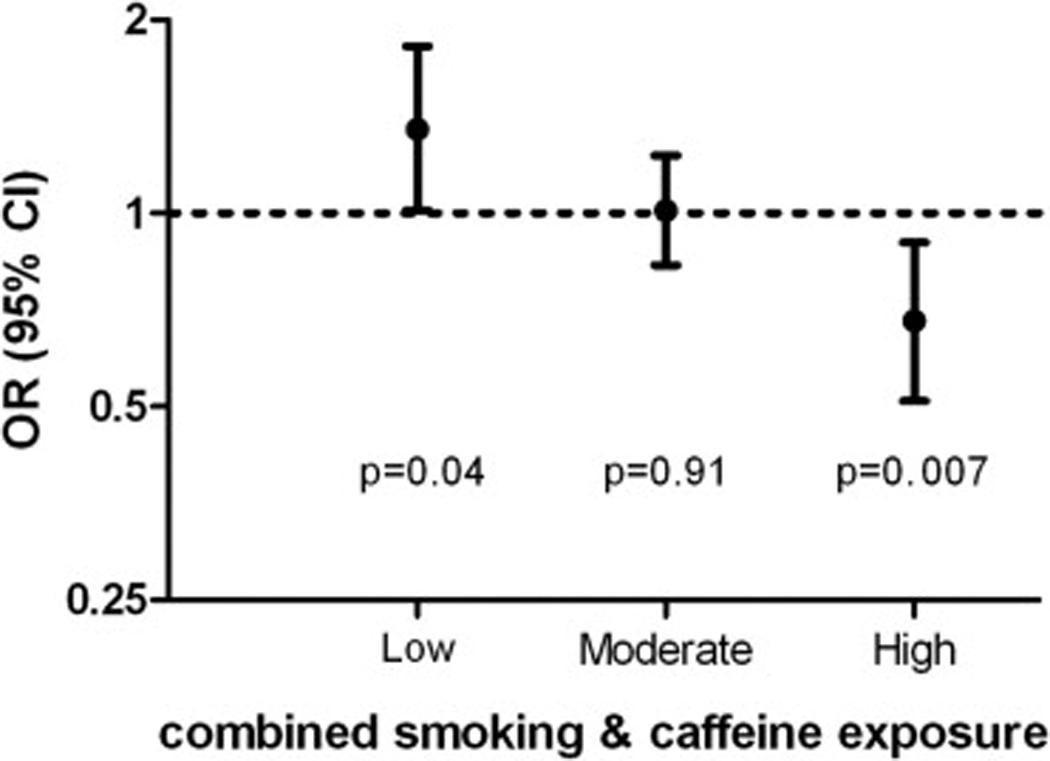
Low: never smokers with lower than median caffeine intake; Moderate: never smokers with higher than median caffeine intake or ever smokers with lower than median caffeine intake; High: ever smokers with higher than median caffeine intake.
4. Discussion
Both genetic and environmental factors have been implicated in the development of PD. Recent PD GWAS studies confirmed associations of over a dozen common polymorphisms (Simon-Sanchez, et al., 2009). The three loci (SNCA, MAPT, and HLA-DRA) included in the current analyses were among the most consistent findings. The other three SNPs are on chromosome 12 near LRRK2, a known PD gene (Satake, et al., 2009,Simon-Sanchez, et al., 2009). On the environment side, both cigarette smoking and higher caffeine intake have been consistently associated with lower PD risk across epidemiological studies (Ascherio, et al., 2001,Chen, et al., 2010,Ritz, et al., 2007,Ross, et al., 2000). This study simultaneously considered these genetic and environmental factors; we not only confirmed known associations, but also evaluated potential interactions between these key genetic and environment risk factors for PD.
It has long been hypothesized that genetic factors may interact with environmental factors in causing PD. Nicotine stabilizes soluble alpha-synuclein oligomers and inhibits alpha-synuclein fibrillation in a concentration-dependent manner (Hong, et al., 2009). Interactions of these known PD genes and environmental factors, however, have rarely been examined in epidemiological studies. In a study of 932 prevalent PD cases and 664 controls (McCulloch, et al., 2008), an interaction was suggested between smoking and the REP1 repeat of alpha-synuclein (P=0.02), while no interaction was suggested for the H1 haplotype of MAPT with smoking (P =0.97) nor with coffee consumption (P =0.73). A later study further examined SNCA REP1 microsatellites and smoking in relation to PD (333 PD cases and 336 controls) (Gatto, et al., 2010), the authors found that the 263-bp genotype was associated with higher PD risk only among never smokers (P for interaction=0.06). A third study identified no significant interactions between the LRRK2 Gly2385Arg variant and smoking on PD risk among 229 prevalent cases and 358 controls in Japan (Miyake, et al., 2010). These studies raised the possibility of a potential interaction between α-synuclein genetic variations and smoking on PD risk; however, alternative explanations such as chance could not be excluded. Further, none of the previous studies accounted for multiple comparisons.
Our study took a different approach to this question and utilized genetic findings from recent GWAS studies. The GWAS findings on tag-SNPs at the SNCA and MAPT loci have been confirmed in multiple studies. Further, tag SNPs in the 3′ region of the SNCA gene are in moderate linkage disequilibrium with the REP1 microsatellites (e.g. R2 = 0.37 with rs3857059), while the MAPT tag SNPs form a large block that is in strong LD with the H1 haplotype (e.g. R2 = 0.76 for rs393152) (Simon-Sanchez, et al., 2009). Our analyses only included incident PD cases and thus were less likely to be prone to reverse causality. In this study, we also observed similar ORs for main effects at three SNPs on chromosome 12 near LRRK2 locus in comparison with meta-analysis results (pdgene.org).
Overall, we did not see evidence for interaction between smoking and/or caffeine intake and GWAS SNPs at the SNCA, MAPT, or HLA-RDA locus. However, our analysis suggests a possible interaction of these environmental exposures with rs2896905. This SNP maps to the SLC2A13 gene and is 0.27Mb upstream to LRRK2 (Simon-Sanchez, et al., 2009). A previous GWAS study among Japanese PD patients and controls also mapped to the SLC2A13 locus (Satake, et al., 2009); this gene is close to LRRK2; given our existing knowledge on LRRK2 and PD (Ross, et al., 2011), it has been hypothesized that the association may be due to LRRK2 (Satake, et al., 2009,Simon-Sanchez, et al., 2009). As our analysis was secondary to a recent GWAS study, we did not directly genotype the rare LRRK2 mutations that are known to be related to PD. Therefore our preliminary finding needs to be confirmed, and the possibility that SLC2A13 or LRRK2 interact with environmental factors with PD should be investigated in future studies.
As expected, a positive family history was strongly associated with PD risk. Family members often share common genetic background and environmental exposures. Interestingly, our analysis suggests that the association of family history with PD was unlikely to be explained by the GWAS SNPs and lifestyle factors that were examined in the current study. However, our data on genetic and environmental factors were limited and other genetic and environmental factors may contribute to the association of family history with PD.
Although the current study is among the largest population-based case-control studies with both genetic and prospectively collected environmental data, the sample size may not be adequate to detect moderate gene-environment interactions. For example, assuming 50% of the participants were environmentally exposed, a minor allele frequency of 0.2 in controls, an OR of 1.5 for gene, an OR of 0.6 for the environmental factor, and α of 0.05, we had about 80% power to detect an OR for gene-environmental interaction of ≤ 0.61 or > 1.55. Further, as one of the first analysis on interactions of known PD genes with known non-genetic factors, the analyses are exploratory in nature. In addition, our analyses were primarily based on genotyping results from a previous GWAS replication sample, therefore we did not directly assess the known PD related variants in these susceptibility genes nor have a complete coverage of genetic variations in these genes.
In summary, this large population based study did not find significant interactions of smoking or caffeine intake with several SNPs at or near the SNCA, MAPT, and HLA-DRA loci that are significantly associated with PD risk in previous GWAS studies. The possibility of an interaction of SLC2A13 or LRRK2 with environmental exposures should be further investigated.
Acknowledgements
The authors are grateful to the continuous contribution of the NIH-AARP Diet and Health Study participants. This study was supported by the intramural research program of the National Institute of Environmental Health Sciences, National Cancer Institute, National Institute on Aging, National Institute of Neurological Disorders and Stroke, the National Human Genome Research Institute of the National Institutes of Health (Z01-ES-101986, Z01 CP010196-02, Z01-AG000949-02, NIA human subjects protocol 2003-077). This work was also supported by the US Department of Defense (award number W81XWH-09-2-0128); National Institutes of Health (grants NS057105 and RR024992) and a NIH extramural grant (R01 NS060722) to Dr Huang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests: The authors have declared that no competing interests exist.
Disclosure statement
The authors disclose no conflicts of interest. All authors contributed to this study and approved its submission. The study protocol was approved by NIHES IRB with written consent from study participants.
None of the authors have financial conflict of interest for this work.
References
- Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE, Willett WC. Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol. 2001;50(1):56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Chen H, Huang X, Guo X, Mailman RB, Park Y, Kamel F, Umbach DM, Xu Q, Hollenbeck A, Schatzkin A, Blair A. Smoking duration, intensity, and risk of Parkinson disease. Neurology. 2010;74(11):878–884. doi: 10.1212/WNL.0b013e3181d55f38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, Wojcicki A, Eriksson N. Web-based genome-wide association study identifies two novel Loci and a substantial genetic component for Parkinson's disease. PLoS Genet. 2011;7(6):e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Xu H, Weinberg C, Huang X, Park Y, Hollenbeck A, Blair A, Schatzkin A, Burch L, Chen H. An Exploratory Study on the CHRNA3-CHRNA5-CHRNB4 Cluster, Smoking, and Parkinson's Disease. Neurodegener Dis. 2011 doi: 10.1159/000323190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto NM, Rhodes SL, Manthripragada AD, Bronstein J, Cockburn M, Farrer M, Ritz B. alpha-Synuclein gene may interact with environmental factors in increasing risk of Parkinson's disease. Neuroepidemiology. 2010;35(3):191–195. doi: 10.1159/000315157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. 2010;42(9):781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DP, Fink AL, Uversky VN. Smoking and Parkinson's disease: does nicotine affect alpha-synuclein fibrillation? Biochim Biophys Acta. 2009;1794(2):282–290. doi: 10.1016/j.bbapap.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Parkinson's Disease Genomics Consortium (IPDGC), Wellcome Trust Case Control Consortium 2 (WTCCC2) A two-stage meta-analysis identifies several new Loci for Parkinson's disease. PLoS Genet. 2011;7(6):e1002142. doi: 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch CC, Kay DM, Factor SA, Samii A, Nutt JG, Higgins DS, Griffith A, Roberts JW, Leis BC, Montimurro JS, Zabetian CP, Payami H. Exploring gene-environment interactions in Parkinson's disease. Hum Genet. 2008;123(3):257–265. doi: 10.1007/s00439-008-0466-z. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Tsuboi Y, Koyanagi M, Fujimoto T, Shirasawa S, Kiyohara C, Tanaka K, Fukushima W, Sasaki S, Yamada T, Oeda T, Miki T, Kawamura N, Sakae N, Fukuyama H, Hirota Y, Nagai M. LRRK2 Gly2385Arg polymorphism, cigarette smoking, and risk of sporadic Parkinson's disease: a case-control study in Japan. J Neurol Sci. 2010;297(1–2):15–18. doi: 10.1016/j.jns.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377(9766):641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for 17 whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA, Ross GW, Strickland D, Van Den Eeden SK, Gorell J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol. 2007;64(7):990–997. doi: 10.1001/archneur.64.7.990. [DOI] [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283(20):2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin MC, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JP, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lesage S, Lin CH, Lynch T, Maraganore DM, Mellick GD, Mutez E, Nilsson C, Opala G, Park SS, Puschmann A, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Theuns J, Tomiyama H, Uitti RJ, Valente EM, van de Loo S, Vassilatis DK, Vilarino-Guell C, White LR, Wirdefeldt K, Wszolek ZK, Wu RM, Farrer MJ. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case-control study. Lancet Neurol. 2011;10(10):898–908. doi: 10.1016/S1474-4422(11)70175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41(12):1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Schatzkin A, Subar AF, Thompson FE, Harlan LC, Tangrea J, Hollenbeck AR, Hurwitz PE, Coyle L, Schussler N, Michaud DS, Freedman LS, Brown CC, Midthune D, Kipnis V. Design and serendipity in establishing a large cohort with wide dietary intake distributions : the National Institutes of Health-American Association of Retired Persons Diet and Health Study. Am J Epidemiol. 2001;154(12):1119–1125. doi: 10.1093/aje/154.12.1119. [DOI] [PubMed] [Google Scholar]
- Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Gasser T, Del Sorbo F, Schneider S, Bhatia KP. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65(5):610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41(12):1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]