

ABSTRACT
Results are presented supporting a regulatory role for the product of the MA3302 gene locus (designated MreA) previously annotated as a hypothetical protein in the methanogenic species Methanosarcina acetivorans of the domain Archaea. Sequence analysis of MreA revealed identity to the TrmB family of transcription factors, albeit the sequence is lacking the sensor domain analogous to TrmBL2, abundant in nonmethanogenic species of the domain Archaea. Transcription of mreA was highly upregulated during growth on acetate versus methylotrophic substrates, and an mreA deletion (ΔmreA) strain was impaired for growth with acetate in contrast to normal growth with methylotrophic substrates. Transcriptional profiling of acetate-grown cells identified 280 genes with altered expression in the ΔmreA strain versus the wild-type strain. Expression of genes unique to the acetate pathway decreased whereas expression of genes unique to methylotrophic metabolism increased in the ΔmreA strain relative to the wild type, results indicative of a dual role for MreA in either the direct or indirect activation of acetate-specific genes and repression of methylotrophic-specific genes. Gel shift experiments revealed specific binding of MreA to promoter regions of regulated genes. Homologs of MreA were identified in M. acetivorans and other Methanosarcina species for which expression patterns indicate roles in regulating methylotrophic pathways.
IMPORTANCE
Species in the domain Archaea utilize basal transcription machinery resembling that of the domain Eukarya, raising questions addressing the role of numerous putative transcription factors identified in sequenced archaeal genomes. Species in the genus Methanosarcina are ideally suited for investigating principles of archaeal transcription through analysis of the capacity to utilize a diversity of substrates for growth and methanogenesis. Methanosarcina species switch pathways in response to the most energetically favorable substrate, metabolizing methylotrophic substrates in preference to acetate marked by substantial regulation of gene expression. Although conversion of the methyl group of acetate accounts for most of the methane produced in Earth’s biosphere, no proteins involved in the regulation of genes in the acetate pathway have been reported. The results presented here establish that MreA participates in the global regulation of diverse methanogenic pathways in the genus Methanosarcina. Finally, the results contribute to a broader understanding of transcriptional regulation in the domain Archaea.
Introduction
Microbes in the domain Archaea utilize basal transcription machinery resembling that of members of the domain Eukarya which includes a TATA box promoter sequence, a TATA box–binding protein (TBP), a homologue of the transcription factor TFIIB (TFB), and a polymerase (Pol) II-like RNA polymerase containing between 8 and 13 subunits (1). In contrast, the majority of characterized and putative transcription factors in the domain Archaea are homologous to activators and repressors in the domain Bacteria (1–8). A recent analysis of 52 sequenced archaeal genomes revealed 3,918 putative TFs, among which the HTH_3, AsnC, TrmB, and ArsR families were universally abundant in all 52 genomes (9). Interestingly, the proportion of small TF’s (100 to 200 residues) was considerably larger than the proportion encoded in bacterial genomes. This understanding raises basic questions regarding the physiological roles of putative TF’s and their transcriptional regulatory networks. Although methane-producing species (methanogens) are the largest and most broadly studied group in the domain Archaea, relatively few are archaean specific (8, 10, 11, 12), and bacterium-like (13, 14) TF’s have been identified which regulate genes involved in electron transport (13, 14), nitrogen fixation and assimilation (8, 11), oxidative stress (10), and biosynthesis of tryptophan (12).
Methanogens have ancient origins and are terminal organisms of anaerobic microbial food chains which function in diverse anaerobic environments of Earth’s biosphere to convert complex organic matter to methane, an integral component of the global carbon cycle. The nearly 1 billion metric tons produced annually contributes to the accumulation of atmospheric methane, a greenhouse gas severalfold more effective than CO2 in radiating light energy back to Earth (15). Implementation of this natural process for the large-scale conversion of renewable biomass and organic wastes to methane is a viable alternative to the use of fossil fuels. Microbes, primarily of the domain Bacteria, initiate the food chain by decomposing complex organic matter to acetate, methylamines, methanol, formate, H2, and CO2, which are growth substrates for methanogens. Species of the genus Methanosarcina are the only methanogens known to grow and produce methane from acetate and the methylotrophic substrates methanol, methylamines, and methylsulfide. These versatile methanogens switch pathways in response to availability of the most energetically favorable substrate, metabolizing methylotrophic substrates in preference to acetate, as marked by substantial regulation of gene expression (16, 17). Thus, Methanosarcina species are ideally suited for investigating complex regulation in the domain Archaea. Furthermore, robust genetic systems are available for several Methanosarcina species (18–20).
Although much is known of enzymes essential to the diverse methanogenic pathways of Methanosarcina species, relatively little is known regarding regulation of gene expression. A recent genetic investigation identified a class of regulatory proteins in Methanosarcina acetivorans which coordinate expression of methanol-grown and methylsulfide-specific methyltransferase genes (21, 22). However, although acetate is the major substrate for biological methane formation in Earth’s biosphere, no proteins involved in the regulation of genes essential to the pathway for methanogenesis from acetate have been reported.
A previous proteomics investigation of M. acetivorans C2A identified numerous proteins with elevated levels during growth on acetate versus methanol, indicating potential roles during growth on acetate (17). Bioinformatics analysis of one of the proteins (encoded by the MA3302 gene locus), previously annotated as hypothetical and upregulated 250-fold, revealed sequence identity to the TrmB family of archaeal regulatory proteins. Thus, MA3302 was investigated with a combination of genetics, comparative bioinformatics, and transcriptomics to ascertain the potential for regulation of genes unique to acetate metabolism and to broaden the understanding of regulation in the domain Archaea. The results reveal that the product of MA3302 is a previously unrecognized transcriptional regulatory protein that we designate MreA (Methanosarcina regulator of energy-converting metabolism). MreA was shown to be involved in the activation of genes encoding enzymes unique to the pathway of methanogenesis from acetate and the repression of genes encoding enzymes unique to pathways of methanogenesis from methylotrophic substrates. The results also indicate that MreA is involved in repression of genes encoding the previously described Msr family of transcriptional regulators and MreA homologs that may also regulate genes encoding enzymes unique to methylotrophic pathways. Overall, the results indicate that MreA plays a key role in the global regulation of diverse methanogenic pathways.
RESULTS AND DISCUSSION
Characterization of MA3302.
Previous proteomics analyses revealed elevated levels in acetate-grown versus methanol-grown M. acetivorans (17) of the protein encoded by MA3302 that we designate MreA. The expression pattern of mreA was reassessed by two additional quantitative approaches, quantitative reverse-transcription PCR (qRT-PCR) and a β-glucuronidase-based reporter gene system wherein the 1,099-bp region upstream of the mreA translational start site (TSS) was fused to uidA. This region contains putative TATA and purine-rich B recognition element (BRE) sequences consistent with a promoter region driving transcription of mreA (see Fig. S1 in the supplemental material). The two independent approaches were in good agreement (Table 1), revealing expression of mreA elevated approximately 18- to 38-fold during growth on acetate versus methanol or trimethylamine (TMA). The results indicate a role for MreA specific for genes essential for growth with acetate. Further, the successful expression of β-glucuronidase validated the identification of the putative promoter region driving transcription of mreA.
TABLE 1 .
Expression of mreA during growth of wild-type M. acetivorans strain WWM75 on acetate, methanol, or trimethylamine
| Substrate | Expression level |
|
|---|---|---|
| qRT-PCR (ΔΔCT)a | Gus (nmol·min−1 ⋅ mg protein−1)b | |
| Acetate | 38.0 ± 12.1 | 9.70 ± 2.26 |
| TMA | 2.22 ± 0.68 | 0.325 ± 0.075 |
| Methanol | 1.00 ± 0.17 | 0.276 ± 0.024 |
Calculated using the threshold cycle (ΔΔCT) method with the 16s rRNA gene used as an invariant control. The ΔCT values for methanol cells were used as the calibrator. Values represent the averages and standard deviations of the results of two biological replicate experiments assayed in triplicate. Primer and probe sequences are listed in Table S4 in the supplemental material.
β-Glucuronidase activity was measured in cell lysate collected at mid-log growth. Values represent the averages and standard deviations of the results of three biological replicate experiments.
Although MA3302 is annotated as encoding a hypothetical protein (24), a BLAST search revealed identity (E value, 1.9 10−40) of MreA to the conserved domain of the COG4738 family of predicted transcriptional regulators (25), of which none have been investigated. The search also revealed identity of MreA to several global transcriptional regulators of the archaeal TrmB (Thermococcus regulator of maltose binding) family which regulates sugar metabolism (2, 26) in Thermococcus kodakaraensis and Pyrococcus furiosus. The full-length MreA aligns with the first 111 N-terminal residues of the 346-residue TrmB regulator from T. kodakaraensis with 68% similarity, including conservation of a helix-turn-helix motif (see Fig. S2 in the supplemental material). Thus, MreA lacks the extended TrmB 235-residue C-terminal domain involved in sugar binding, suggesting that MreA lacks a small-molecule sensor domain. MreA is best described as a homolog of TrmBL2 which lacks the TrmB family sugar binding domain and is evolutionarily the most conserved regulator found in all sequenced genomes in the taxonomic order Thermococcales (3, 4). Although TrmBL2 of Pyrococcus furiosus recognizes the promoter of the maltodextrin-specific ABC transporter system, no pattern with respect to other promoter sequences can be deduced (4). Thus, it is unknown what DNA motifs or substrates, if any, are recognized by TrmBL2 homologs. Remarkably, it was recently shown that the TrmBL2 homolog of T. kodakaraensis is an abundant chromosomal protein which binds to both coding and intergenic regions, conferring a thick fibrous structure contributing to the overall DNA architecture (27). Analysis of a deletion mutant showed that TrmBL2 is involved in global regulation, although the mechanism is unknown.
Binding of MreA to promoter DNA.
MreA was overexpressed in Escherichia coli, purified to homogeneity, and used in electrophoretic mobility shift assays (Fig. 1) to determine if MreA binds to promoter DNA. Pronounced band shifts were observed in the presence of increasing amounts of MreA for DNA fragments covering the promoter regions of mreA and for the pta and fpo operons which represent genes from the acetotrophic and methylotrophic pathways previously shown to be regulated in response to the respective substrates (17, 28). Significant smearing, potentially attributable to binding of MreA at multiple sites analogous to abundant binding of TrmBL2 to the chromosome of T. kodakaraensis (27), was observed with the M. acetivorans DNA probes. No shift of poly(dI-dC)/poly(dI-dC) was observed (Fig.1D), confirming specificity of binding to the promoter regions of M. acetivorans DNA. Addition of acetate, methanol, or TMA (10 to 100 mM) had no significant effect on DNA binding, and manual inspection failed to identify a motif common to any of the promoter regions investigated, results consistent with properties reported for TrmBL2. Features of MreA which parallel TrmBL2 call for more-extensive investigations to determine other proteins required to interact with MreA to sense effector molecules and determine if MreA and MreA homologs bind DNA contributing to chromosome architecture similarly to TrmBL2.
FIG 1 .
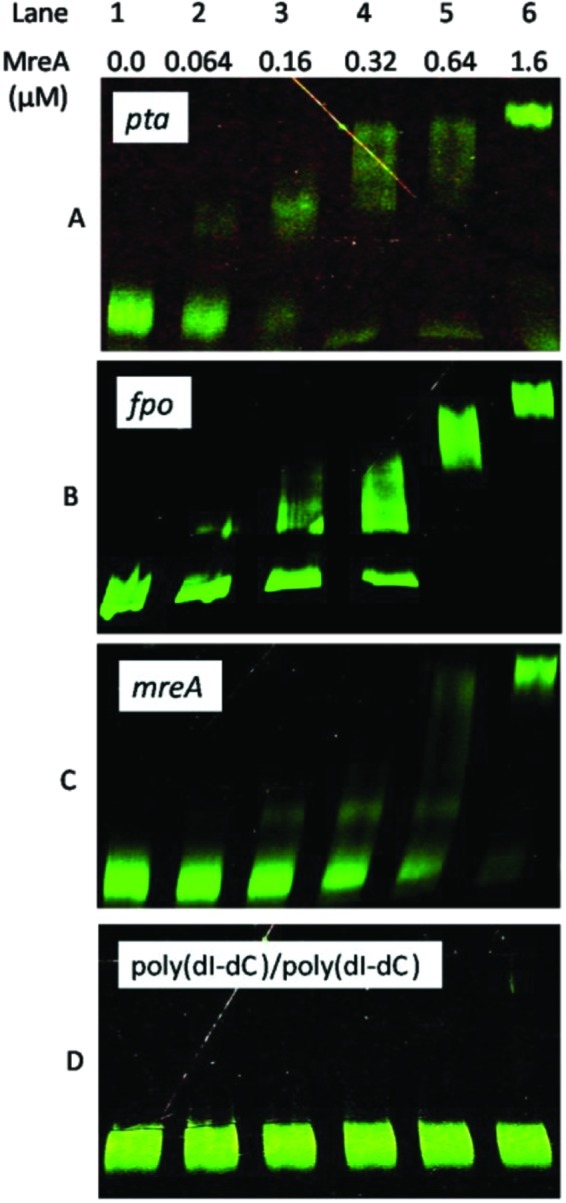
Electrophoretic mobility shift assays. The micromole concentrations of MreA are shown below the lane numbers. (A) The probe DNA encompassed 230 bp upstream of the M. acetivorans pta start codon which included the TATA box, BRE, and 210 bp upstream of a putative TSS deduced from the previously identified TSS for Methanosarcina thermophila pta (29) which is conserved in M. acetivorans. (B) The probe DNA encompassed 420 bp of DNA upstream of the start codon for the first gene in the fpo operon which included 355 bp upstream of the previously published TSS of the M. acetivorans fpo operon (36). (C) The probe DNA contained the 450-bp DNA fragment upstream of the mreA start codon shown in Fig. S1 in the supplemental material which included the TATA box, BRE, and 226 bp upstream of a putative TSS deduced from the previously identified TSS for Methanosarcina mazei mreA (35) which is conserved in M. acetivorans. (D) The probe was poly(dI-dC)/poly(dI-dC).
Characterization of the deletion mutant strain ΔmreA.
The potential role of MreA in gene expression was further investigated by deleting mreA in wild-type M. acetivorans strain WWM75, producing the ΔmreA strain. Growth parameters of the ΔmreA strain with methanol (Fig. 2) or TMA (not shown) were indistinguishable from those of WWM75. However, a significantly reduced growth rate, a prolonged lag phase, and lower growth yield were observed for the acetate-grown ΔmreA strain versus acetate-grown WWM75 (Fig. 2), suggesting a potential regulatory role for MreA specific to this substrate.
FIG 2 .
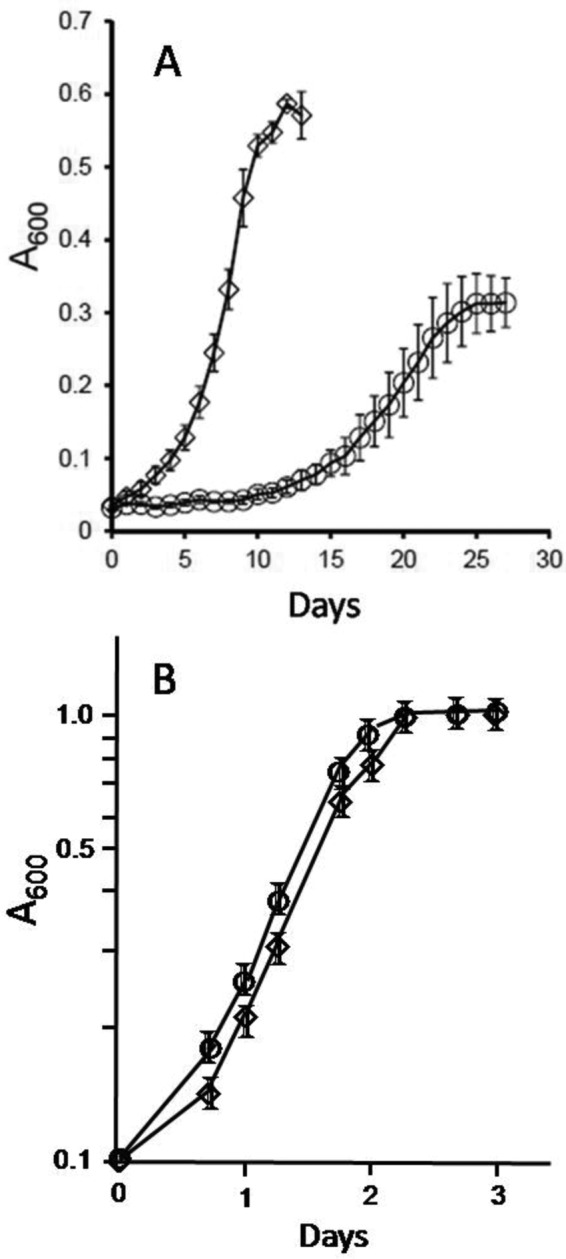
Growth of WWM75 and ΔmreA Methanosarcina acetivorans strains cultured with either acetate or methanol. (A) Growth with acetate. Values represent the averages and standard deviations of the results determined for five replicate cultures. (B) Growth with methanol. Values represent the averages and standard deviations of the results determined for three replicate cultures. Symbols: ◊, wild-type strain WWM75; ○, mutant strain ΔmreA.
The potential regulatory function of MreA was investigated by comparing the genome-wide expression levels of genes between acetate-grown M. acetivorans wild-type strain WWM75 and the ΔmreA mutant strain by means of RNA-Seq (see Table S1 in the supplemental material). A total of 280 genes were identified with a differential of transcript abundance of ≥3-fold between WWM75 and the ΔmreA strain (see Table S2 in the supplemental material). Of these 280 genes, 228 and 52 exhibited higher and lower transcript abundance, respectively, in the ΔmreA strain and WWM75 strains, indicating either direct or indirect roles for MreA in both positive and negative regulation of gene expression. A grouping of the 280 differentially expressed genes, several of which are essential for either the acetotrophic or methylotrophic pathways of methanogenesis (see Fig. S3 in the supplemental material), into their respective COG functional categories revealed a disproportionally high fraction (P ≤ 5.85 × 10−12) in the category “energy production and conversion” (see Table S3 in the supplemental material).
Table 2 lists genes encoding enzymes essential to the acetotrophic pathway (see Fig. S3 in the supplemental material) for most of which the transcript abundance ratios were lower in the acetate-grown ΔmreA strain than in the WWM75 strain, indicating that MreA is necessary for induction in M. acetivorans. Acetate kinase (Ack) and phosphotransacetylase (Pta) are upregulated in acetate-grown versus methanol-grown M. acetivorans (17), Methanosarcina mazei (16), and Methanosarcina thermophila (29), where the enzymes function in concert to activate acetate. The pta and ack genes are adjacent on the Methanosarcina genomes and have been shown to be cotranscribed in M. thermophila (29). Thus, although the reduction in expression of ack was less than that of pta, it is likely that expression of both genes is dependent on MreA. Finally, the results in Table 2 indicate a role for MreA in regulation of carbonic anhydrase (Cam), which is upregulated in acetate-grown versus methanol-grown M. acetivorans (17), M. mazei (16), and M. thermophila (30, 31) and has been proposed to function in removal of CO2 from the cytoplasm during growth on acetate.
TABLE 2 .
Expression ratios of genes in the acetotrophic pathway for the acetate-grown ΔmreA strain versus the WWM75 strain of M. acetivorans
| Gene(s) | Transcript abundance (ΔmreA strain/WWM75)a |
|---|---|
| MA0658–MA0664 (rnfXCDGEAB) | 1.13–1.33 |
| MA0665 (cytC) | 1.18 |
| MA1016–MA1011 (cdhABCDE-1) | 0.06–0.15 |
| MA1016 (cdhA-1) | 0.03 ± 0.00b |
| MA2536 (cam) | 0.11 (0.10 ± 0.01)b |
| MA3606 (ack) | 0.4 |
| MA3607 (pta) | 0.18 (0.20 ± 0.06)b |
| MA3860–MA3865 (cdhABCDE-2) | 2.17–4.91 |
| MA3860 (cdhA-2) | 5.21 ± 0.82b |
| MA4399 (cdhA-3) | 6.95 |
| MA4566–MA4572 (mrpABCDEFG) | 0.55–0.67 |
Determined by RNA-Seq unless noted otherwise, as indicated in footnote b.
Determined by qRT-PCR, calculated using the ΔΔCT method with the 16s rRNA gene used as an invariant control. Values represent the averages and standard deviations of the results of two biological replicate experiments assayed in triplicate. Primer and probe sequences are listed in Table S4 in the supplemental material. Primers and probes for MA2536 were those used previously (40).
The reduced expression of the acetate pathway genes cam, pta, ack, and cdhABCDE-1 in the ΔmreA strain versus WWM75 alone (Table 2) explains the growth defect (Fig. 2) of the ΔmreA strain. The genome of M. acetivorans is annotated with duplicate gene clusters (MA1011–MA1016 and MA3860–MA3865), each encoding five subunits of the Cdh complex (CdhABCDE) which is central to the acetate pathway. Although proteomics analysis revealed upregulation of both Cdh complexes (17), the complex encoded by MA1011–MA1016 (cdhABCDE-1) was down regulated and the complex encoded by MA3860–MA3865 (cdhABCDE-2) was upregulated in the ΔmreA strain versus WWM75 (Table 2). This result indicates that expression of cdhABCDE-2 is independent of MreA and that expression is upregulated in the ΔmreA strain to compensate for decreased expression of cdhABCDE-1. Rnf and Mrp complexes are upregulated in acetate-grown versus methanol-grown M. acetivorans and proposed to be essential for acetate-dependent growth (28) (see Fig. S3 in the supplemental material); however, the results shown in Table 2 indicate no significant difference in expression between the ΔmreA strain and WWM75 of genes encoding either complex. It is noteworthy that rnf and mrp genes are absent in M. mazei and M. barkeri that synthesize alternative energy-converting complexes absent in M. acetivorans. Therefore, it is tempting to speculate that separate regulatory control mechanisms evolved to coordinate expression of the different energy-converting complexes in Methanosarcina species.
There was no significant change (see Table S1 in the supplemental material) for the acetate-grown ΔmreA strain versus WWM75 in the expression of genes common to both the acetotrophic and methylotrophic pathways (see Fig. S3 in the supplemental material). This result is consistent with a role for MreA in the global regulation of pathway-specific genes, thereby facilitating the switch between growth substrates. An exception was the gene cluster encoding the tetrahydromethanopterin S-methyltransferase complex (Mtr), with a 4-fold increase in transcript abundance for the ΔmreA strain versus WWM75 (see Table S2 in the supplemental material). This result was unexpected, considering that the acetotrophic pathway has a higher demand for Mtr than methylotrophic pathways supported by upregulation of mtr in acetate-grown versus methanol-grown M. acetivorans (28). Thus, it appears that the role for MreA in the regulation of mtr is complex.
Table 3 lists genes encoding enzymes unique to the pathways of methanogenesis from methylotrophic substrates (see Fig. S3 in the supplemental material), among which methanol-specific genes are upregulated in methanol-grown versus acetate grown M. acetivorans (17, 21, 22) (see Table S2 in the supplemental material) and M. mazei (16). In the methyl transfer branch specific to methanol, the methyl group is transferred to coenzyme M catalyzed by MtaBC and MtaA, for which the genome contains three and two copies, respectively, of the encoding genes (32). In the oxidative branch, a portion of methyl-coenzyme M is oxidized to CO2, providing electrons for reduction of the methyl group of the remaining methyl-coenzyme M to CH4 via reactions common to all methanogenic pathways (33). Three genes (fmd, ftr, and mer) of the five encoding enzymes in the oxidative branch showed >3-fold increases in expression in the ΔmreA strain versus WWM75 and the other two (mch and mtd) nearly 2-fold increases, indicating involvement of MreA in repressing genes of this branch during growth on acetate (Table 3). Furthermore, all 14 genes encoding the Fpo complex involved in electron transport and energy conversion of all methylotrophic pathways showed increased expression in the ΔmreA strain versus WWM75, supporting a role for MreA in repression during growth on acetate. Although proteins regulating the oxidative branch were not previously reported, methanol-specific regulators MsrA–MsrE that either activate or repress genes of the methyl transfer branch in M. acetivorans have previously been described (22). The expression of all three copies of mtaBC showed no significant change in the ΔmreA strain versus WWM75 strains (Table 3), indicating no direct role for MreA in activation of these genes, consistent with roles for MsrA–MsrE. No transcriptional regulators are reported for genes encoding enzymes specific for methylamines in the methyl transfer branch of methylotrophic pathways. However, the results in Table 3 indicate that MreA is involved in repression of methyltransferases specific for monomethylamine (MtmBC-1 and MtbA), dimethylamine (MtbBC-2 and MtbA), and trimethylamine (MttBC-2 and MtbA), for which expression of the encoding genes was greater in the acetate-grown ΔmreA strain versus WWM75. Yet genes encoding duplicate methyltransferases (MtmBC-2, MtbBC-1, MtbBC-3, and MttBC-1) showed no significant difference in expression levels (Table 3), indicating a different mode of regulation. Likewise, genes encoding methyltransferases (MtsD, MtsF and MtsH) showed no significant difference in expression levels for the ΔmreA strain versus WWM75, consistent with previously published roles for MsrF, MsrC, and MsrG in regulation of these genes in M. acetivorans (21).
TABLE 3 .
Expression ratios of genes in methylotrophic pathways for the acetate-grown ΔmreA strain versus the WWM75 strain of M. acetivorans
| Gene | Transcript abundance (ΔmreA strain/WWM75)a |
|---|---|
| Methyl transfer branch | |
| MA0144–MA0145 (mtmBC-1) | 3.95–3.13 |
| MA0146 (mtbA) | 4.24 |
| MA0455–MA0456 (mtaBC-1) | 0.88–0.49 |
| MA0527, MA0532 (mtbBC-1) | 0.68–1.70 |
| MA0528–MA0529 (mttBC-1) | 0.53–0.57 |
| MA0859 (mtsD) | 1.36 |
| MA0931–MA0932 (mttBC-2) | 6.12–9.27 |
| MA0933–MA0934 (mtbBC-2) | 8.2–10.60 |
| MA1615 (mtaA2) | 0.92 |
| MA1616–MA1617 (mtaBC-3) | 1.21–1.22 |
| MA2424–MA2425 (mtbBC-3) | 0.78–0.95 |
| MA2971–MA2972 (mtmBC-2) | 1.37–1.44 |
| MA4379 (mtaA-1) | 7.83 |
| MA4384 (mtsF) | 1.00 |
| MA4558 (mtsH) | 0.93 |
| Oxidative branch | |
| MA0010 (ftr) | 4.98 (7.07 ± 1.52)b |
| MA0304–MA0309 (fmdEFADCB) | 7.81–10.09 |
| MA0304 (fmdE) | 14.8 ± 4.0b |
| MA0309 (fmdB) | 11.3 ± 2.4b |
| MA1710 (mch) | 1.85 |
| MA3733 (mer) | 6.55 (8.70 ± 2.11)b |
| MA4430 (mtd) | 1.97 |
| Energy conversion | |
| MA1494–MA1507 (fpoPABCDHIJJKLMNO) | 6.35–13.64 |
| MA1495 (fpoA) | 9.34 ± 0.92b |
| MA1498 (fpoD) | 11.9 ± 1.5b |
| MA3732 (fpoF) | 3.91 |
Determined by RNA-Seq unless noted otherwise, as indicated in footnote b.
Determined by qRT-PCR, calculated using the ΔΔCT method with the 16s rRNA gene used as an invariant control. Values represent the averages and standard deviations of the results of two biological replicate experiments assayed in triplicate. Primer and probe sequences are listed in Table S4 in the supplemental material.
Differential expression of regulatory proteins.
In addition to metabolic genes, several genes encoding putative regulatory proteins showed elevated expression in the acetate-grown ΔmreA strain versus WWM75, indicating that MreA is somehow involved in repression of these putative regulators during growth on acetate (see Table S2 in the supplemental material). The results suggest that, at least in some cases, the mreA deletion is indirectly modulating gene expression by affecting the expression of other regulators.
The genes are annotated (21) as encoding nine putative sensory transduction histidine kinases (MA0490, MA0551, MA0619, MA0759, MA0863, MA1645, MA1646, MA1800, and MA2397), one putative response regulator (MA2445), and eight DNA-binding regulatory proteins (Table 4). Five of the DNA-binding proteins are members of the previously described methanol-specific regulatory (Msr) family (21, 22), consistent with a direct or indirect role for MreA in repression of the encoding genes during growth on acetate (Table 4). Locus MA0459 encodes MsrA, which, together with MsrB (MA0460), activates methanol-specific mtaBC1 (22). Although msrB was elevated less than the 3.0-fold cutoff (Table 4), it is reasonable to expect that both expression of msrB and expression of msrA are directly or indirectly regulated by MreA since the genes are cotranscribed (22). However, the expected increase in expression of mtaBC1 by the ΔmreA strain was not observed (Table 3), indicating a requirement of other regulatory factors during growth on methanol. Loci MA0862 and MA4383 encode MsrF and MsrC, which activate transcription of mtsD and mtsF, respectively, previously reported to be involved in the pathway of methanogenesis from dimethylsulfide (21). Thus, the finding that genes encoding MsrF and MsrC were upregulated in the ΔmreA strain versus WWM75 predicted increased expression of mtsD and mtsF in the ΔmreA strain versus WWM75. However, the expected increase in expression was not observed (Table 3), indicating that the regulation of mtsD and mtsF is more complex. Expression of proteins encoded by the mtsF and mtsD genes is elevated 50-fold in CO-grown versus acetate-grown or methanol-grown M. acetivorans, suggesting a role in addition to metabolism of dimethylamine (36). Indeed, the protein encoded by mtsF (renamed cmtA) catalyzes methyl tetrahydromethanopterin-coenzyme M methyltransferase activity that functions in the CO-dependent CO2 reduction pathway (34). Finally, the deduced sequence of locus 4167 was found to have 33% to 40% identity, with conservation of the helix-turn-helix motif, to previously described Msr family members (21), identifying it as a new member of the Msr family that we designate MsrH.
TABLE 4 .
Expression ratios of genes encoding Msr (methanol-specific regulatory) family and putative Mre family regulatory proteins in the acetate-grown ΔmreA strain versus the WWM75 strain of M. acetivorans
| Gene | Transcript abundance (ΔmreA strain/WWM75)a |
|---|---|
| MA0459 (msrA) | 3.06 |
| MA0460 (msrB) | 1.93 |
| MA0862 (msrF) | 8.66 |
| MA1671 (mreB) | 8.64 (10.2 ± 2.6)b |
| MA3129 (mreC) | 8.67 (13.0 ± 3.8)b |
| MA3130 (mreD) | 363.95 (1440 ± 430)b |
| MA4167 (msrH) | 11.77 (19.0 ± 5.2)b |
| MA4383 (msrC) | 27.93 |
Determined by RNA-Seq (see Table S2 in the supplemental material) unless noted otherwise, as indicated in footnote b.
Determined by qRT-PCR, calculated using the ΔΔCT method with the 16s rRNA gene used as an invariant control. Values represent the averages and standard deviations of the results of two biological replicate experiments assayed in triplicate. Primer and probe sequences are listed in Table S4 in the supplemental material.
The three other putative DNA-binding proteins (MA1671, MA3129, and MA3130) with elevated gene expression in the ΔmreA strain versus WWM75 (Table 4) had deduced sequence identity to MreA ranging from 43% to 47% with conservation of the helix-turn-helix motif. Transcript abundance ratios indicated that each gene is upregulated in either methanol-grown or TMA-grown versus acetate-grown wild-type WWM75 (Table 5), a result in contrast to that seen with mreA, which was upregulated in acetate-grown versus methanol-grown or TMA-grown M. acetivorans (Table 1). The results suggest that the genes encode the putative homologs of MreA that we designate MreB (MA1671), MreC (MA3129), and MreD (MA3130) with potential to activate the expression of genes unique to methylotrophic pathways. Despite robust increases in expression for all three homologs in the acetate-grown ΔmreA strain versus the WWM75 strain, there was no apparent restoration of expression of acetate-specific genes (Table 2), indicating that induction of the homologs in the acetate-grown ΔmreA strain does not function to compensate for loss of MreA. This result further supports the idea of a functional role for the homologs during methylotrophic growth.
TABLE 5 .
Relative transcript abundance ratios for genes encoding putative Mre family regulatory proteins in methanol-grown or trimethylamine-grown versus acetate-grown wild-type M. acetivorans strain WWM75
| Gene | Transcript abundancea |
|
|---|---|---|
| MeOHb/HOAcc | TMAd/HOAcc | |
| MA1671 (mreB) | 5.09 ± 0.91 | 17.7 ± 1.9 |
| MA3129 (mreC) | 1.89 ± 0.21 | 6.51 ± 0.51 |
| MA3130 (mreD) | 85.7 ± 13.2 | 393 ± 17 |
Determined by qRT-PCR. The ΔCT values for acetate-grown cells were used as the calibrator. Values represent the averages and standard deviations of the results of two biological replicate experiments assayed in triplicate. Primer and probe sequences are listed in Table S4 in the supplemental material.
Methanol grown.
Acetate grown.
Trimethylamine grown.
The overall results establish a role for MreA in the global regulation of energy-converting pathways of M. acetivorans as summarized in Fig. 3.
FIG 3 .
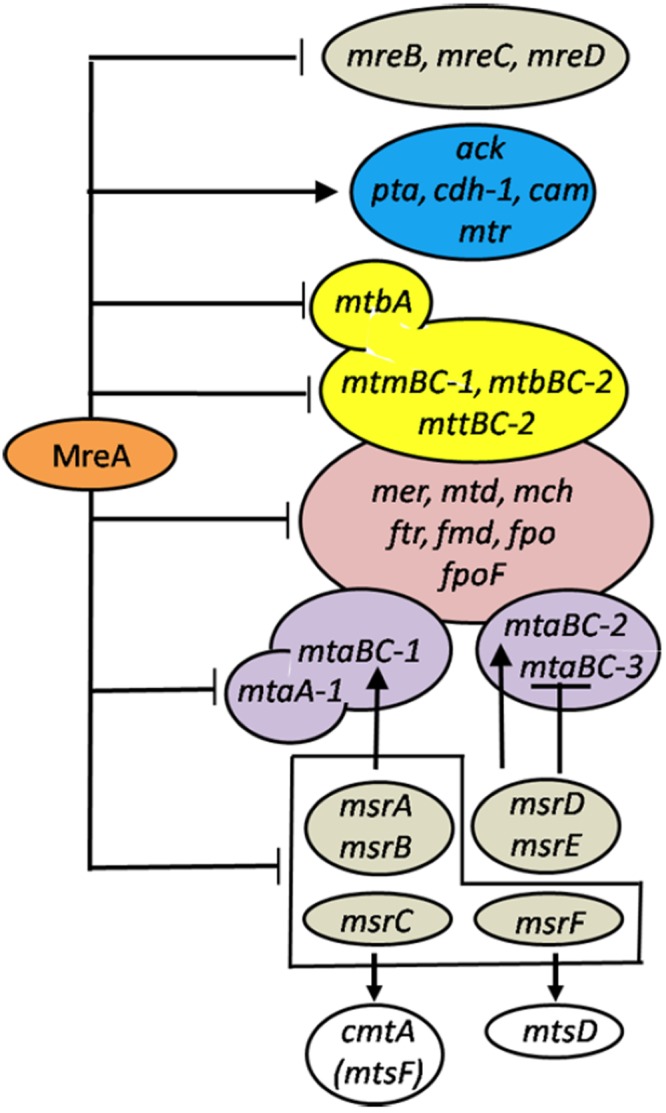
Global regulatory network model of energy-converting pathways. Genes mreB, mreC, and mreD (Methanosarcina regulator of energy-converting metabolism) are homologs of mreA postulated to encode proteins involved in regulation of genes required for conversion of methylamines and methanol to methane. Enzymes for conversion of acetate to methane are encoded by ack (acetate kinase), pta (phosphotransacetylase), cdh-1 (carbon monoxide dehydrogenase), cam (carbonic anhydrase), and mtr (membrane-bound tetrahydromethanopterin S-methyltransferase complex). Enzymes required for conversion of methylamines to methane are encoded by mtbA (methylamine-specific methylcobamide:CoM methyltransferase), mtmBC-1 (B, monomethylamine-specific methyltransferase; C, cognate corrinoid protein), mtbBC-2 (B, dimethylamine-specific methyltransferase; C, cognate corrinoid protein), and mttBC-2 (B, trimethylamine-specific methyltransferase; C, cognate corrinoid protein). Enzymes required for conversion of methanol to methane are encoded by mtaBC-1, mtaBC-2, mtaBC-3 (B, methanol-specific methyltransferase; C, cognate corrinoid protein), and mtaA-1 (methanol-specific methylcobamide:CoM methyltransferase). Enzymes common to the pathways for conversion of methanol or methylamines to methane are encoded by mer (F420-dependent methylene-H4MPT reductase), mtd (F420-dependent methylene-H4MPT dehydrogenase), mch (methenyl-H4MPT cyclohydrolase), ftr (formylmethanofuran; H4MPT formyltransferase), fmd (molybdenum-dependent formyl-methanofuran dehydrogenase), fpo (coenzyme F420 dehydrogenase complex), and fpoF (a subunit of the coenzyme F420 dehydrogenase complex). Proteins regulating genes specific to the conversion of methanol are encoded by msrA, msrB, msrD, msrE, msrC, and msrF (methanol-specific regulators). An enzyme proposed to be specific for conversion of dimethylsulfide to methane is encoded by mtsD (methyltransferase). An enzyme in the pathway for conversion of carbon monoxide to methane is encoded by cmtA (cytoplasmic methyltransferase) (formerly mtsF).
Conservation of MreA and homologs across the genus Methanosarcina.
Homologs of MreA with >97% amino acid sequence identity were identified in both M. mazei and M. barkeri (see Fig. S4 in the supplemental material). In addition, the gene context of mreA in both species was found to be similar to that for M. acetivorans strain C2A, as was the promoter DNA sequence, including the putative TATA box and BRE previously mapped for M. mazei mreA (35) (see Fig. S1 in the supplemental material). Together, these data are consistent with similar functions for mreA in all three species. M. barkeri has a second MreA homolog designated MreA-2 with 88% sequence identity to MreA of M. acetivorans (see Fig. S4 in the supplemental material). However, unlike mreA-1, the promoter region and gene context for M. barkeri mreA-2 were found to be dissimilar to those of the other mreA homologs. Thus, it is unclear whether the functions for M. barkeri mreA-1 and mreA-2 are similar. MreB, MreC, and MreD are also conserved in both M. mazei and M. barkeri with >80% identity. Conservation of MreA, MreB, MreC, and MreD indicates an important role for the Mre family consistent with regulation of energy metabolism across the genus Methanosarcina.
Conclusions.
The results presented here have identified a novel archaeal regulatory protein (MreA) which was previously annotated as a hypothetical protein. MreA is the first global regulatory protein described for methanogenic species. Overall, the results presented are consistent with MreA functioning in the global regulation of energy-converting metabolism based on the finding that MreA is involved in regulation of (i) genes unique to the acetotrophic and methylotrophic pathways, (ii) Msr family regulatory proteins, and (iii) homologs of MreA that are upregulated in methanol-grown or TMA-grown versus acetate-grown M. acetivorans (Fig. 3).
Comparative transcriptomic analyses revealed a dual role for MreA in both the transcriptional activation of genes specific for the metabolism of acetate and repression of genes unique to methylotrophic pathways. Furthermore, the results indicate MreA is involved in the transcriptional repression of genes encoding Msr family regulators and MreA homologs with the potential for regulating genes unique to methylotrophic pathways. However, a direct versus indirect role for MreA in regulating gene expression has yet to be determined. MreA joins a growing number of archaeal proteins, including TrmBL1, Tgr, and SurR (2, 3, 5), that function in both the repression and activation of gene expression for which the mechanism has not been reported. The similarity of MreA to TrmBL2 warrants more-extensive investigations to determine other proteins required to interact with MreA to sense effector molecules and the manner in which MreA and homologs bind DNA contributing to chromosome architecture similar to that of TrmBL2.
MATERIALS AND METHODS
Cell growth.
Growth and harvesting of M. acetivorans C2A (DSM 800) in a single-cell morphology cultured with high-salt (HS) broth medium with acetate, methanol, or TMA were performed as previously described (36). Growth of all strains was monitored by measuring the optical density at 600 nm.
RNA isolation.
All M. acetivorans RNA samples were isolated with an RNeasy Total RNA Minikit (Qiagen). Purified RNA was treated twice with RNase-free DNase I (Qiagen) and once with RQ1 DNase (Promega) to remove contaminating DNA.
TaqMan quantitative RT-PCR.
TaqMan assays were performed as previously described (36) with total RNA isolated from the mid-exponential phase (A600 ~ 0.15 to 0.2, 0.4 to 0.5, or 0.4 to 0.5 for acetate-, TMA-, or methanol-grown cells, respectively). Primer and probe sequences are listed in Table S4 in the supplemental material.
Construction of the M. acetivorans mutant strain.
Liposome-mediated transformation and homologous recombination-mediated gene replacement were performed as previously described (37, 38) to generate an M. acetivorans ΔmreA::pac-hpt strain (here designated the ΔmreA strain) and PmreA-uidA. The ΔmreA mutant strain was generated in an M. acetivorans WWM75 background, and a PmreA-uidA promoter fusion strain was constructed in an M. acetivorans WWM73 background. Cells were transformed with 2 µg appropriate shuttle vector DNA (see Table S4 in the supplemental material) (linearized by restriction digestion in the case of the gene knockout shuttle vector), and transformants were selected on HS agar media with 1.5% (wt/vol) agar, with 50 mM TMA and 2 µg ml−1 puromycin (Sigma, St. Louis, MO) added from sterile, anaerobic 100× stock solution. 8-Aza-2,6-diaminopurine (8ADP) (Sigma, St. Louis, MO) sensitivity was tested in media containing 20 µg ml−1 8ADP added from sterile, anaerobic 200× stock solution. Removal of pac-hpt cassettes in mutant strains by markerless exchange was not performed in this study.
Gus assays.
β-glucuronidase (Gus) assays were performed essentially as previously described (23, 39, 40). An M. acetivorans strain carrying the PmreA-uidA reporter gene was grown to mid-exponential phase on different methanogenic substrates (A600 ~ 0.15 to 0.2, 0.4 to 0.5, or 0.4 to 0.5 for acetate-, TMA-, or methanol-grown cells, respectively) and harvested by centrifugation for 10 min at 5,000 × g. Cells were osmotically lysed by addition of 50 mM Tris-HCl (pH 8.0) containing 1 mM dithiothreitol (DTT) and 0.1 µg ml–1 DNase. Cell debris was cleared by recentrifugation for 15 min at 16,000 × g. Cleared lysate was held at 4°C or frozen at −80°C until use. For each assay, cleared lysate was diluted 1:20 into 50 mM Tris-Cl (pH 8.0) prechilled to 4°C and brought to room temperature. Assays were initiated by addition of 4 mM p-nitrophenyl-β-d-glucuronide, and activity was monitored by measuring absorbance at 415 nm with a Beckman DU 640 spectrophotometer. The specific activity (in nanomoles per minute per milligram of protein) was based on a previously reported molar extinction coefficient (39). The protein concentration of the cell lysate was determined using a bicinchoninic acid (BCA) protein assay kit (Pierce) with bovine serum albumin (BSA) as the standard.
RNA-Seq.
Total RNA from acetate-grown wild-type and ΔmreA M. acetivorans was collected at the mid-exponential phase (A600 ~ 0.15 to 0.2). Transcriptome libraries were prepared by following the Whole Transcriptome Library Preparation for SOLiD Sequencing Protocol (Rev. B) (Applied Biosystems). The libraries were constructed with reagents from a SOLiD small RNA expression kit (Applied Biosystems). Briefly, total RNA samples (1 µg) were fragmented with RNase and purified. Fragmented RNA (100 ng) was ligated overnight to adaptors and subsequently reverse transcribed. Purified cDNA was size selected and then amplified from bar-coded primers. The resulting material was purified and assessed for proper amplification using an Agilent Bioanalyzer DNA 1000 chip (Agilent Technologies). Bar-coded libraries were combined and prepared for sequencing according to the Applied Biosystems SOLiD 3 Plus System Templated Bead Preparation Guide (Applied Biosystems). Fragment library sequencing (50 bp) was performed on a SOLiD 3 Plus system according to the Applied Biosystems SOLiD 3 Plus System Instrument Operation Guide (Applied Biosystems). Sequenced library fragments were mapped to the M. acetivorans reference genome sequence with the Qseq program in ArrayStar 4.0. Noncoding RNAs (e.g., rRNA, tRNA, miscellaneous RNAs) as well as nonunique reads were excluded from mapping. Expression values for each gene were normalized by RPKM (assigned reads per kilobase of feature per million mapped reads). Statistical analyses of the normalized data were performed using a moderated t test with false-discovery-rate (FDR) multiple-testing correction (Benjamini-Hochberg) to determine differential transcript abundance. Changes in transcript abundance were considered significant if they were ≥3-fold (P values < 0.05). Functional categories represented by genes exhibiting differential transcript abundances in the ΔmreA strain were evaluated using a hypergeometric distribution to determine if any particular categories were statistically over- or underrepresented.
There were 4,542,508 and 2,099,564 unique reads assigned for the two respective biological replicates of the wild-type strain and 2,704,850 and 2,941,777 unique reads assigned for the two respective biological replicates for the mutant strain.
Cloning, expression, and purification of MreA.
The gene encoding MreA was amplified from M. acetivorans genomic DNA by PCR. The PCR-amplified DNA fragment was cloned into pSUMO vector (LifeSensors, Malvern, PA), generating plasmid pSUMO-MreA. pSUMO-MreA contains the N-terminally His-tagged Sumo domain-MreA fusion.
The His6-Sumo-MreA fusion was overproduced in E. coli BL21(DE3) cells transformed with pSUMO-MreA. Cells were grown in LB at 37°C with shaking at 250 rpm until an optical density at 600 nm of 0.5 was reached, after which the culture was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and then harvested by centrifugation 16 h after induction. Approximately 5 g (wet weight) of cells was suspended in 15 ml of 20 mM Tris-Cl (pH 8) containing 150 mM NaCl, 20 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (PMSF), 20 µg ml−1 DNase, and 10% (wt/vol) glycerol (buffer A). The cells were lysed by two passages through a French pressure cell at 138 MPa. The lysate was centrifuged at 74,000 × g for 30 min at 4°C. The supernatant solution containing the His6-Sumo-MreA fusion protein was filtered (pore size, 0.45 µm) and applied at a flow rate of 0.5 ml/min to a column containing 25 ml of Ni Sepharose 6 fast-flow resin (GE Healthcare). The column was then washed with 200 ml of buffer A a flow rate of 2 ml/min. His6-Sumo-MreA was then eluted from the column with 60 ml of 20 mM Tris-Cl (pH 8) containing 150 mM NaCl, 500 mM imidazole, and 10% (wt/vol) glycerol (buffer B). The eluate was concentrated to 2.5 ml with a Vivacell concentrator fitted with a 10,000-molecular-weight-cutoff filter. The concentrated His6-Sumo-MreA was desalted with a PD-10 gel filtration column (Amersham Biosciences) developed with buffer A. MreA was cleaved from His6-Sumo by incubating the full-length fusion protein at 30°C for 2 h with Sumo protease 1 (LifeSensors, Malvern, PA) (10 units added per 100 µg protein) plus 2 mM DTT. The cleaved protein was applied at a flow rate of 0.5 ml/min to a second column containing 25 ml of Ni Sepharose 6 fast-flow resin (New England Biolabs). The flowthrough containing MreA was collected and concentrated to 2.5 ml with a Vivacell concentrator fitted with a 10,000-molecular-weight-cutoff filter. The potein concentration was determined with a BCA protein assay kit (Pierce) and bovine serum albumin as the standard.
Electrophoretic mobility shift assay.
Probe DNA (100 nM final concentration) was titrated with the indicated increasing concentrations of MreA in 10 µl binding buffer (10 mM Tris-Cl [pH 8.0], 100 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 15% glycerol) containing 0.6 µg bovine serum albumin (BSA) and 2.5 µg heparin. Poly(dI-dC)/poly(dI-dC) (Sigma-Aldrich) was used at 100 nM (final concentration). Reaction mixtures were incubated 30 min at 21°C and loaded onto prerun 6% native polyacrylamide gel electrophoresis (PAGE) gels containing 15% glycerol. Gels were developed with 0.5× TBE buffer (45 mM Tris-Cl, 45 mM borate, 1 mM EDTA, 15% glycerol) until the desired migration was achieved. Following electrophoresis, gels were stained for 20 min in 1× SYBR green stain diluted in 0.5× TBE. Gels were visualized by excitation at 254 nm with a UV epi-illumination lamp and a 490-nm Longpass SYBR photographic filter.
SUPPLEMENTAL MATERIAL
Promoter alignment for Methanosarcina acetivorans mreA with Methanosarcina mazei and Methanosarcina barkeri orthologs. Alignments were performed using ClustalW. Conserved bases are denoted with asterisks. The TSS previously published for the M. mazei gene is denoted with a hooked arrow above the corresponding base (Jager D, et al., Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability; Proceedings of the National Academy of Sciences of the United States of America, 106:21878-21882, 2009). The putative start codon for the corresponding proteins is denoted with a boxed arrow. The putative TATA boxes, BRE’s, and ribosome binding sites are boxed. Brackets denote the boundaries for the DNA fragment representing the mreA promoter regions used in electrophoretic mobility shift assays. A potential regulatory motif with an invert repeat centered 22 bp upstream of the putative TATA box/BRE is underlined. Both the M. acetivorans and M. mazei MreA proteins have annotations for 7 and 21 additional amino acids at their respective N termini which are not conserved in M. barkeri MreA-1 or MreA-2. Annotated TTG start codons for M. acetivorans and M. mazei genes are also denoted with boxed arrows. However, these additional residues are likely incorrectly annotated portions of the M. acetivorans and M. mazei mreA 5′ untranslated region (UTR) based on the following: (i) the sequence conservation of these N-terminal extensions is poor in relation to the remainder of the protein; (ii) both annotated ORF’s initiate translation from a TTG start codon which is utilized infrequently in Methanosarcina species (Torarinsson E, Klenk HP, Garrett RA, Divergent transcriptional and translational signals in Archaea; Environ. Microbiol. 7:47-54, 2005); (iii) the putative ATG start codon for M. barkeri mreA-1 and mreA-2 is conserved in frame in M. acetivorans and M. mazei mreA, as is the sequence upstream corresponding to a putative ribosome binding site; and (iv) no sequences resembling a putative ribosome binding site are present upstream of the annotated TTG start codon for either M. acetivorans or M. mazei mreA. Download FIG S1, PDF file, 0.2 MB.
Methanosarcina acetivorans MreA aligned against the N-terminal DNA-binding domain of Thermococcus kodakaraensis TrmB. Alignments were performed with Clustal W. The first 111 N-terminal residues of the 341-residue TrmB derived from the Comprehensive Microbial Resource (The Comprehensive Microbial Resource, J. Craig Venter Institute; <http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi>, 2011) are shown. Asterisks denote identical residues, and similar residues are denoted with dots. Residues forming predicted α-helices and β-sheets are depicted in red and green italics, respectively. The secondary structure prediction was performed using Jpred 3 (Cuff JA, Barton GJ, Application of multiple sequence alignment profiles to improve protein secondary structure prediction; Proteins 40:502-511, 2000). The residues forming the putative helix-turn-helix (HTH) motif from the T. kodakaraensis TrmB are denoted with a solid red line. Brackets denote the region comprising the COG4738 domain in MreA. Download FIG S2, PDF file, 0.1 MB.
Acetotrophic (A) and methylotrophic (B) pathways. Shaded shapes indicate enzymes for which transcription of the encoding genes decreased (A) or increased (B) in the ΔmreA mutant strain versus the WWM75 wild-type strain. Enzymes without asterisks are unique to each pathway. Asterisks indicate enzymes common to both pathways. Download FIG S3, TIF file, 0.2 MB.
Phylogenetic tree for MreA family members in Methanosarcina acetivorans, Methanosarcina mazei, and Methanosarcina barkeri. The tree was generated in MEGA v 3.83 bootstrapped with the neighbor-joining algorithm following alignment by ClustalW. Clades denoting orthologs to MreA (MA3302, MM0138, MbarA3549, MbarA3027), MreB (MA1671, MM2835, MbarA2849), MreC (MA3129, MM0386, MbarA1951), and MreD (MA3130, MM0385, MbarA1949) in each organism are outlined. Sequences were derived from the Comprehensive Microbial Resource (The Comprehensive Microbial Resource, J. Craig Venter Institute; <http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi>, 2011). Download FIG S4, PDF file, 0.2 MB.
Genome-wide expression values for wild-type versus ΔmreA Methanosarcina acetivorans strains determined by RNA-Seq.
All genes with a significant change in expression in the ∆mreA strain versus the wild-type Methanosarcina acetivorans strain.
Grouping of differentially expressed genes in the ∆mreA strain versus the wild-type Methanosarcina acetivorans strain into COG functional categories.
Microbial strains, primers, and plasmids used.
ACKNOWLEDGMENTS
We thank W. W. Metcalf for the donation of all plasmids and strains used for the genetic manipulation of M. acetivorans, D. Grove (Penn State Genomics Core Facility—University Park, University Park, PA) for optimization of TaqMan assays, and Craig Praul and Candace Price for whole-transcript library preparation.
This work was funded by grants from the Department of Energy Energy Biosciences Program (DE-FG02-95ER20198) to J.G.F. and the National Institutes of Health (GM087350) to K.S.M.
Footnotes
Citation Reichlen MJ, Vepachedu VR, Murakami KS, and Ferry JG. 2012. MreA functions in the global regulation of methanogenic pathways in Methanosarcina acetivorans. mBio 3(4):e00189-12 doi:10.1128/mBio.00189-12.
REFERENCES
- 1. Bell SD. 2005. Archaeal transcriptional regulation—variation on a bacterial theme? Trends Microbiol. 13:262–265 [DOI] [PubMed] [Google Scholar]
- 2. Kanai T, et al. 2007. A global transcriptional regulator in Thermococcus kodakaraensis controls the expression levels of both glycolytic and gluconeogenic enzyme-encoding genes. J. Biol. Chem. 282:33659–33670 [DOI] [PubMed] [Google Scholar]
- 3. Lee SJ, Surma M, Hausner W, Thomm M, Boos W. 2008. The role of TrmB and TrmB-like transcriptional regulators for sugar transport and metabolism in the hyperthermophilic archaeon Pyrococcus furiosus. Arch. Microbiol. 190:247–256 [DOI] [PubMed] [Google Scholar]
- 4. Lee SJ, et al. 2007. Characterization of the TrmB-like protein, PF0124, a TGM-recognizing global transcriptional regulator of the hyperthermophilic archaeon Pyrococcus furiosus. Mol. Microbiol. 65:305–318 [DOI] [PubMed] [Google Scholar]
- 5. Lipscomb GL, et al. 2009. SurR: a transcriptional activator and repressor controlling hydrogen and elemental sulphur metabolism in Pyrococcus furiosus. Mol. Microbiol. 71:332–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ochs SM, et al. 2012. Activation of archaeal transcription mediated by recruitment of transcription factor B. J. Biol. Chem. 287:18863–18871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teufel K, Pfeifer F. 2010. Interaction of transcription activator GvpE with TATA-box-binding proteins of Halobacterium salinarum. Arch. Microbiol. 192:143–149 [DOI] [PubMed] [Google Scholar]
- 8. Weidenbach K, Ehlers C, Kock J, Schmitz RA. 2010. NrpRII mediates contacts between NrpRI and general transcription factors in the archaeon Methanosarcina mazei Go1. FEBS J. 277:4398–4411 [DOI] [PubMed] [Google Scholar]
- 9. Pérez-Rueda E, Janga SC. 2010. Identification and genomic analysis of transcription factors in archaeal genomes exemplifies their functional architecture and evolutionary origin. Mol. Biol. Evol. 27:1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karr EA. 2010. The methanogen-specific transcription factor MsvR regulates the fpaA-rlp-rub oxidative stress operon adjacent to msvR in Methanothermobacter thermautotrophicus. J. Bacteriol. 192:5914–5922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lie TJ, Leigh JA. 2003. A novel repressor of nif and glnA expression in the methanogenic archaeon Methanococcus maripaludis. Mol. Microbiol. 47:235–246 [DOI] [PubMed] [Google Scholar]
- 12. Xie Y, Reeve JN. 2005. Regulation of tryptophan operon expression in the archaeon Methanothermobacter thermautotrophicus. J. Bacteriol. 187:6419–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ouhammouch M, Dewhurst RE, Hausner W, Thomm M, Geiduschek EP. 2003. Activation of archaeal transcription by recruitment of the TATA-binding protein. Proc. Natl. Acad. Sci. U. S. A. 100:5097–5102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun J, Klein A. 2004. A LysR-type regulator is involved in the negative regulation of genes encoding selenium-free hydrogenases in the archaeon Methanococcus voltae. Mol. Microbiol. 52:563–571 [DOI] [PubMed] [Google Scholar]
- 15. Schlesinger WH. 2000. The global carbon cycle, p. 308–321 Biogeochemistry. Academic Press, San Diego, CA. [Google Scholar]
- 16. Hovey R, et al. 2005. DNA microarray analysis of Methanosarcina mazei Go1 reveals adaptation to different methanogenic substrates. Mol. Genet. Genomics 273:225–239 [DOI] [PubMed] [Google Scholar]
- 17. Li L, et al. 2007. Quantitative proteomic and microarray analysis of the archaeon Methanosarcina acetivorans grown with acetate versus methanol. J. Proteome Res. 6:759–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rother M, Metcalf WW. 2005. Genetic technologies for Archaea. Curr. Opin. Microbiol. 8:745–751 [DOI] [PubMed] [Google Scholar]
- 19. Rother M, Sattler C, Stock T. 2011. Studying gene regulation in methanogenic archaea. Methods Enzymol. 494:91–110 [DOI] [PubMed] [Google Scholar]
- 20. Sowers KR. 2007. Genetics of Archaea, p 800–824 In Reddy CA, Beveridge TJ, Breznak JA, Marzluf GA, Schmidt TM, Snyder LR, Methods for general and molecular microbiology, 3rd ed. American Society for Microbiology, Washington, DC: [Google Scholar]
- 21. Bose A, Kulkarni G, Metcalf WW. 2009. Regulation of putative methyl-sulphide methyltransferases in Methanosarcina acetivorans C2A. Mol. Microbiol. 74:227–238 [DOI] [PubMed] [Google Scholar]
- 22. Bose A, Metcalf WW. 2008. Distinct regulators control the expression of methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol. Microbiol. 67:649–661 [DOI] [PubMed] [Google Scholar]
- 23. Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. 2008. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anonymous. 2011. The comprehensive microbial resource. J. Craig Venter Institute, Rockville, MD: http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi [Google Scholar]
- 25. Tatusov RL, et al. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee SJ, et al. 2005. TrmB, a sugar sensing regulator of ABC transporter genes in Pyrococcus furiosus exhibits dual promoter specificity and is controlled by different inducers. Mol. Microbiol. 57:1797–1807 [DOI] [PubMed] [Google Scholar]
- 27. Maruyama H, et al. 2011. Histone and TK0471/TrmBL2 form a novel heterogeneous genome architecture in the hyperthermophilic archaeon Thermococcus kodakarensis. Mol. Biol. Cell 22:386–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Q, et al. 2006. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J. Bacteriol. 188:702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh-Wissmann K, Ferry JG. 1995. Transcriptional regulation of the phosphotransacetylase-encoding and acetate kinase-encoding genes (pta and ack) from Methanosarcina thermophila. J. Bacteriol. 177:1699–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jablonski PE, DiMarco AA, Bobik TA, Cabell MC, Ferry JG. 1990. Protein content and enzyme activities in methanol- and acetate-grown Methanosarcina thermophila. J. Bacteriol. 172:1271–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimmerman SA, Tomb JF, Ferry JG. 2010. Characterization of CamH from Methanosarcina thermophila, founding member of a subclass of the {gamma} class of carbonic anhydrases. J. Bacteriol. 192:1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galagan JE, et al. 2002. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12:532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ferry JG. 2010. How to make a living by exhaling methane. Annu. Rev. Microbiol. 64:453–473 [DOI] [PubMed] [Google Scholar]
- 34. Vepachedu VR, Ferry JG. 25 May 2012. Role of the fused corrinoid/methyl transfer protein CmtA during co-dependent growth of Methanosarcina acetivorans. J. Bacteriol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jäger D, et al. 2009. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc. Natl. Acad. Sci. U. S. A. 106:21878–21882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lessner DJ, et al. 2006. An unconventional pathway for reduction of CO2 to methane in co-grown Methanosarcina acetivorans revealed by proteomics. Proc. Natl. Acad. Sci. U. S. A. 103:17921–17926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boccazzi P, Zhang JK, Metcalf WW. 2000. Generation of dominant selectable markers for resistance to pseudomonic acid by cloning and mutagenesis of the ileS gene from the archaeon Methanosarcina barkeri fusaro. J. Bacteriol. 182:2611–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. 1997. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl. Acad. Sci. U. S. A. 94:2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pritchett MA, Zhang JK, Metcalf WW. 2004. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl. Environ. Microbiol. 70:1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rother M, Boccazzi P, Bose A, Pritchett MA, Metcalf WW. 2005. Methanol-dependent gene expression demonstrates that methyl-coenzyme M reductase is essential in Methanosarcina acetivorans C2A and allows isolation of mutants with defects in regulation of the methanol utilization pathway. J. Bacteriol. 187:5552–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Promoter alignment for Methanosarcina acetivorans mreA with Methanosarcina mazei and Methanosarcina barkeri orthologs. Alignments were performed using ClustalW. Conserved bases are denoted with asterisks. The TSS previously published for the M. mazei gene is denoted with a hooked arrow above the corresponding base (Jager D, et al., Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability; Proceedings of the National Academy of Sciences of the United States of America, 106:21878-21882, 2009). The putative start codon for the corresponding proteins is denoted with a boxed arrow. The putative TATA boxes, BRE’s, and ribosome binding sites are boxed. Brackets denote the boundaries for the DNA fragment representing the mreA promoter regions used in electrophoretic mobility shift assays. A potential regulatory motif with an invert repeat centered 22 bp upstream of the putative TATA box/BRE is underlined. Both the M. acetivorans and M. mazei MreA proteins have annotations for 7 and 21 additional amino acids at their respective N termini which are not conserved in M. barkeri MreA-1 or MreA-2. Annotated TTG start codons for M. acetivorans and M. mazei genes are also denoted with boxed arrows. However, these additional residues are likely incorrectly annotated portions of the M. acetivorans and M. mazei mreA 5′ untranslated region (UTR) based on the following: (i) the sequence conservation of these N-terminal extensions is poor in relation to the remainder of the protein; (ii) both annotated ORF’s initiate translation from a TTG start codon which is utilized infrequently in Methanosarcina species (Torarinsson E, Klenk HP, Garrett RA, Divergent transcriptional and translational signals in Archaea; Environ. Microbiol. 7:47-54, 2005); (iii) the putative ATG start codon for M. barkeri mreA-1 and mreA-2 is conserved in frame in M. acetivorans and M. mazei mreA, as is the sequence upstream corresponding to a putative ribosome binding site; and (iv) no sequences resembling a putative ribosome binding site are present upstream of the annotated TTG start codon for either M. acetivorans or M. mazei mreA. Download FIG S1, PDF file, 0.2 MB.
Methanosarcina acetivorans MreA aligned against the N-terminal DNA-binding domain of Thermococcus kodakaraensis TrmB. Alignments were performed with Clustal W. The first 111 N-terminal residues of the 341-residue TrmB derived from the Comprehensive Microbial Resource (The Comprehensive Microbial Resource, J. Craig Venter Institute; <http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi>, 2011) are shown. Asterisks denote identical residues, and similar residues are denoted with dots. Residues forming predicted α-helices and β-sheets are depicted in red and green italics, respectively. The secondary structure prediction was performed using Jpred 3 (Cuff JA, Barton GJ, Application of multiple sequence alignment profiles to improve protein secondary structure prediction; Proteins 40:502-511, 2000). The residues forming the putative helix-turn-helix (HTH) motif from the T. kodakaraensis TrmB are denoted with a solid red line. Brackets denote the region comprising the COG4738 domain in MreA. Download FIG S2, PDF file, 0.1 MB.
Acetotrophic (A) and methylotrophic (B) pathways. Shaded shapes indicate enzymes for which transcription of the encoding genes decreased (A) or increased (B) in the ΔmreA mutant strain versus the WWM75 wild-type strain. Enzymes without asterisks are unique to each pathway. Asterisks indicate enzymes common to both pathways. Download FIG S3, TIF file, 0.2 MB.
Phylogenetic tree for MreA family members in Methanosarcina acetivorans, Methanosarcina mazei, and Methanosarcina barkeri. The tree was generated in MEGA v 3.83 bootstrapped with the neighbor-joining algorithm following alignment by ClustalW. Clades denoting orthologs to MreA (MA3302, MM0138, MbarA3549, MbarA3027), MreB (MA1671, MM2835, MbarA2849), MreC (MA3129, MM0386, MbarA1951), and MreD (MA3130, MM0385, MbarA1949) in each organism are outlined. Sequences were derived from the Comprehensive Microbial Resource (The Comprehensive Microbial Resource, J. Craig Venter Institute; <http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi>, 2011). Download FIG S4, PDF file, 0.2 MB.
Genome-wide expression values for wild-type versus ΔmreA Methanosarcina acetivorans strains determined by RNA-Seq.
All genes with a significant change in expression in the ∆mreA strain versus the wild-type Methanosarcina acetivorans strain.
Grouping of differentially expressed genes in the ∆mreA strain versus the wild-type Methanosarcina acetivorans strain into COG functional categories.
Microbial strains, primers, and plasmids used.