

ABSTRACT
Respiratory syncytial virus (RSV) is a leading cause of infant mortality worldwide. Toll-like receptor 4 (TLR4), a signaling receptor for structurally diverse microbe-associated molecular patterns, is activated by the RSV fusion (F) protein and by bacterial lipopolysaccharide (LPS) in a CD14-dependent manner. TLR4 signaling by LPS also requires the presence of an additional protein, MD-2. Thus, it is possible that F protein-mediated TLR4 activation relies on MD-2 as well, although this hypothesis has not been formally tested. LPS-free RSV F protein was found to activate NF-κB in HEK293T transfectants that express wild-type (WT) TLR4 and CD14, but only when MD-2 was coexpressed. These findings were confirmed by measuring F-protein-induced interleukin 1β (IL-1β) mRNA in WT versus MD-2−/− macrophages, where MD-2−/− macrophages failed to show IL-1β expression upon F-protein treatment, in contrast to the WT. Both Rhodobacter sphaeroides LPS and synthetic E5564 (eritoran), LPS antagonists that inhibit TLR4 signaling by binding a hydrophobic pocket in MD-2, significantly reduced RSV F-protein-mediated TLR4 activity in HEK293T-TLR4–CD14–MD-2 transfectants in a dose-dependent manner, while TLR4-independent NF-κB activation by tumor necrosis factor alpha (TNF-α) was unaffected. In vitro coimmunoprecipitation studies confirmed a physical interaction between native RSV F protein and MD-2. Further, we demonstrated that the N-terminal domain of the F1 segment of RSV F protein interacts with MD-2. These data provide new insights into the importance of MD-2 in RSV F-protein-mediated TLR4 activation. Thus, targeting the interaction between MD-2 and RSV F protein may potentially lead to novel therapeutic approaches to help control RSV-induced inflammation and pathology.
IMPORTANCE
This study shows for the first time that the fusion (F) protein of respiratory syncytial virus (RSV), a major cause of bronchiolitis and death, particularly in infants and young children, physically interacts with the Toll-like receptor 4 (TLR4) coreceptor, MD-2, through its N-terminal domain. We show that F protein-induced TLR4 activation can be blocked by lipid A analog antagonists. This observation provides a strong experimental rationale for testing such antagonists in animal models of RSV infection for potential use in people.
Introduction
Human RSV (respiratory syncytial virus) is a major cause of severe lower respiratory tract disease in infants, adults, and immunocompromised patients (1-4). There is no long-lasting immunity to RSV, as evidenced by the fact that most adults are reinfected every few years (5). The RSV fusion (F) protein mediates fusion of the viral envelope with the target cell membrane during virus entry (6). Only membrane-associated protein is indispensable for viral replication in tissue culture (7), and this protein is the primary target for antiviral drug and vaccine development (1, 8, 9). At present, a monoclonal antibody directed against the RSV F protein (Synagis) is routinely administered in the United States prophylactically to high-risk infants. This treatment has led to a marked reduction in RSV-induced hospitalizations (10, 11).
Lipopolysaccharide (LPS) from Gram-negative bacteria is a potent agonist for cellular activation through TLR4 (12-16). Optimal LPS-induced TLR4 signaling requires soluble or membrane-associated CD14 (17), as well as MD-2, a non-membrane-spanning protein that associates with the TLR4 ectodomain (18, 19). However, TLR4 can be activated by other structurally unrelated, microbial structures, such as chlamydial Hsp60 (20), pneumolysin (21), DnaK from Francisella tularensis (22), and Ebola virus glycoprotein (23), as well as endogenous mammalian “danger signals,” such as fibrinogen (24), fibronectin (25), low-molecular-weight oligosaccharide fragments of hyaluronan (26), surfactant protein A (27), and HMGB1 (28). Kurt-Jones and colleagues first reported that the RSV F protein is also a TLR4 agonist and activates the innate immune response by driving NF-κB-mediated cytokine expression (29). Mice with mutations in tlr4 have a significantly impaired ability to clear RSV (30). While it is now clear that the monomeric LPS–MD-2 complex, and not LPS itself, is the ligand that specifies LPS-dependent activation of TLR4, a similar role and physical interaction of MD-2 with these other putative TLR4 ligands and agonists—including the RSV F protein—have not yet been demonstrated.
In this study, we provide compelling evidence to support a molecular requirement for MD-2 in RSV F-protein-mediated TLR4 signaling that includes direct interaction of RSV F protein with MD-2–TLR4. These findings provide significant new insights into the molecular basis of TLR4 activation by the RSV F protein that should help focus new therapeutic approaches that target and modulate immune responses against RSV.
RESULTS
RSV F protein requires MD-2 for the induction of the TLR4-mediated inflammatory response.
LPS, the prototype TLR4 agonist, is among the most potent of inflammatory stimuli in vivo and in vitro and is ubiquitous. Therefore, when other structurally unrelated molecules are assessed for their capacity to induce a TLR4-mediated proinflammatory response, it is imperative that they are LPS free. To ensure that our purified RSV F protein preparations were free of LPS contamination, induction of NF-κB-luciferase activity in HEK293T cells expressing the TLR4–CD14–MD-2 complex was compared after pretreatment of RSV F protein with medium only, polymyxin B, anti-F antibodies, or proteinase K. Similar treatments of LPS were included as controls. As expected, LPS-induced NF-κB was inhibited only by polymyxin B that has been shown previously to bind and neutralize LPS (31) but not by anti-F antibodies or proteinase K treatment. In contrast, TLR4 signaling induced by purified native RSV F protein was inhibited by both anti-F antibodies and proteinase K but not by polymyxin B (Fig. 1). Therefore, this result indicates that the activity induced by purified RSV F protein is not attributable to contaminating LPS. As previously reported, we observed in these experiments that LPS is a stronger inducer of TLR4 activity than the purified F protein (32).
FIG 1 .

TLR4 signaling induced by LPS, but not by F protein, was inhibited only by polymyxin B, while signaling induced by F protein, but not by LPS, was inhibited only by anti-F antibodies and proteinase K. HEK293T cells transfected with TLR4–CD14–MD-2 complex and β-galactosidase expression vectors, along with the NF-κB-luc reporter construct, were treated for 5 h with medium only or with LPS (5 ng/ml) or purified F protein (3 ng/ml) after pretreatment of the transfectants for 1 h with medium alone or with polymyxin B (10 µg/ml), anti-F antibodies, or proteinase K (5 µg/ml). After the 5-h treatment, luciferase and β-galactosidase activities were measured in cell lysates. Relative luciferase activity (RLU) was calculated by normalizing luciferase to β-galactosidase activity for each sample. Data are means ± standard errors of the means (SEM) for triplicate samples (n = 5). *, P < 0.001.
RSV F-protein-induced TLR4 activation has been shown to be CD14 dependent (29), but the requirement for MD-2 was not previously investigated. To determine if MD-2 is also required for F-protein-induced TLR4 activation, we first assessed NF-κB activation in HEK293T cells transiently transfected with TLR4 and CD14 expression vectors, in the absence or presence of the MD-2 expression. HEK293T cells expressing TLR4 and CD14 exhibited robust NF-κB luciferase activity when stimulated by RSV F protein, but only when MD-2 was coexpressed (Fig. 2). To confirm these findings, primary peritoneal macrophages from WT (wild-type) C57BL/6J, and MD-2−/− mice were stimulated with medium alone, with increasing concentrations of RSV F protein, or with LPS (as a positive control) for 2 h. In WT macrophages, RSV F protein induced a significant, dose-dependent upregulation of IL-1β mRNA (Fig. 3), but, as seen with the NF-κB-mediated reporter activity in HEK293T TLR4–MD-2–CD14 transfectants (32) (Fig. 2), F protein was again shown to be a less potent TLR4 agonist than LPS (Fig. 3). In contrast, neither RSV F protein nor LPS stimulated IL-1β mRNA expression in macrophages derived from MD-2−/− mice (Fig. 3). Taken together, the data indicate that MD-2, an extracellular protein that binds both TLR4 and LPS for LPS-induced TLR4 signaling (33-35), is also required for RSV F-protein-mediated TLR4 signaling.
FIG 2 .

F protein activation of NF-κB-luc requires MD-2. HEK293T cells cotransfected with TLR4-CD14 without or with the MD-2 construct, along with NF-κB-luc and β-galactosidase reporter constructs, were treated for 5 h with medium (M) only or with LPS (5 ng/ml) or purified F protein (3 ng/ml). Luciferase and β-galactosidase activities were measured in cell lysates, and RLU were calculated. Data are means ± SEM for triplicate samples (n = 5). *, P < 0.001.
FIG 3 .
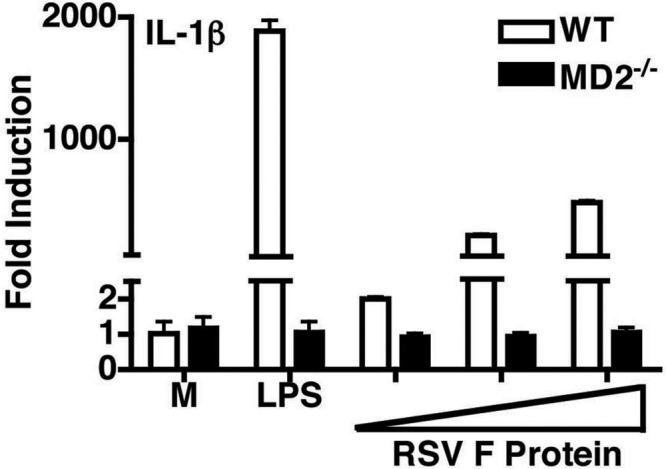
MD-2−/− macrophages fail to respond to purified F protein to produce IL-1β mRNA. Macrophages from C57BL/6J (WT) or MD-2−/− mice were stimulated with E. coli LPS (10 ng/ml) or increasing concentrations of purified RSV F protein (0.75, 1.5, and 3 ng/ml) for 2 h, and steady-state IL-1β mRNA was measured by quantitative real-time PCR. Results are means ± SEM (n = 2).
RSV F-protein-mediated TLR4 signaling is inhibited by LPS analog antagonists.
Rhodobacter sphaeroides LPS (RsLPS) and its analog and synthetic TLR4 inhibitor, eritoran (E5564), are potent LPS antagonists that inhibit LPS-triggered signaling and cytokine production in a dose-dependent manner (36, 37). Recent crystallographic analyses revealed that eritoran’s four acyl groups bind within the deep hydrophobic pocket of MD-2 and thereby block the binding of the lipid A region of LPS, leading to the inhibition of LPS-mediated TLR4 activation (38, 39). Since RSV F protein requires MD-2 for TLR4 activation (Fig. 2 and 3), we hypothesized that both RsLPS and eritoran would also inhibit RSV F-protein-induced activation of TLR4. To test this hypothesis, HEK293T cells expressing TLR4–CD14–MD-2 complex were pretreated for 1 h with the indicated concentrations of RsLPS or eritoran, and then cells were stimulated with RSV F protein for an additional 5 h. As shown in Fig. 4A, RsLPS (1 µg/ml) significantly inhibited F-protein-induced NF-κB activation at all concentrations of F protein tested. As expected, control reactions that tested the ability of RsLPS to inhibit TNF-α-mediated activation of reporter activity show no such inhibition. A similar inhibition of TLR4 activation was observed when the TLR4–CD14–MD-2-expressing transfectants were pretreated with eritoran and subsequently treated with RSV F protein or purified E. coli LPS (Fig. 4B). Both RSV F-protein- and LPS-induced TLR4 signaling was inhibited by eritoran under conditions where TNF-α-induced NF-κB activation was unaffected.
FIG 4 .

RSV F protein activation of NF-κB through TLR4–CD-14–MD-2 is inhibited by the MD-2 antagonists RsLPS (A) and eritoran (B). HEK293T cells were cotransfected with TLR4–CD14–MD-2 complex, along with pELAM-luc and pCMV1-β-galactosidase reporter constructs. After an overnight incubation, the transfectants were pretreated with medium only or RsLPS (1 µg/ml) (A) or with eritoran (5 and 10 ng/ml) (B) for 1 h and then stimulated with medium only, recombinant TNF-α (50 ng/ml), or decreasing concentrations of RSV F protein [4.5, 3, 1.5, and 0.75 ng/ml (A) or 3 and 1.5 ng/ml (B)] for 5 h. Cell lysates were prepared, β-galactosidase and luciferase activities were analyzed within the same sample, and the results are presented as RLU. Data are means ± SEM for triplicate samples from a representative experiment (n = 5). *, P < 0.01 compared to medium-pretreated controls (M).
Interaction of RSV F protein with human MD-2.
The ability of eritoran to inhibit LPS-triggered activation of TLR4 has been reported to stem from the ability of this TLR4 antagonist to occupy the hydrophobic pocket of MD-2, inhibiting interactions of LPS with MD-2 that are necessary for TLR4 activation. Since both RsLPS and eritoran blocked F-protein-mediated TLR4 signaling, we speculated that F-protein-mediated TLR4 signaling, like LPS, required a physical interaction of the F protein with MD-2. To test this hypothesis, we examined potential physical interactions between MD-2 and F protein by assaying coprecipitation of purified native F protein with nickel nitrilotriacetic acid (Ni-NTA) beads after incubation of F protein with insect cell conditioned medium containing baculovirus-encoded, recombinant His6 MD-2 versus conditioned medium from infected cells not expressing His6–MD-2. As shown in Fig. 5A and 5F, protein was coprecipitated after incubation with insect cell conditioned medium containing His-tagged MD-2 but not control conditioned medium, indicating a direct interaction between the F protein and human MD-2.
FIG 5 .

Physical interaction of RSV F protein with MD-2. (A) Interaction of native F protein with MD-2. Native purified F was incubated in the absence (lane 1) or presence (lane 2) of His-tagged MD-2. Complexes were precipitated using Ni-NTA beads targeting His-tagged MD-2. Bound proteins were subjected to 12% SDS-PAGE and Western blotting for F protein (top) and MD-2 (bottom) (anti-His antibodies). (B) Diagram of fragments of the fusion protein of RSV showing the fusion peptide (FP), heptad repeats (HR) A and B, transmembrane domain (TM), and intracellular domain (CT) of the F1 fragment. Polypeptides N1, N2, C1, and C2 represent the fragments used for interaction analysis with MD-2. (C to E) Pulldown assay for the analysis of the interaction between the different F protein peptides and MD-2. (C) Represents the loading of the input amounts of each His-tagged polypeptide. The arrow indicates the position of the F protein polypeptides. (D) Western blot using anti-His antibodies to detect coprecipitation reactions of His-tagged F protein polypeptides captured by anti-FLAG antibody beads following incubation of the His-tagged polypeptides with conditioned medium containing MD-2–FLAG-TLR4ecd complex, FLAG-TLR4ecd alone, or neither MD-2 nor TLR4ecd (control). The arrow indicates the position of the F-protein polypeptides. (E) Graph representing the capture of the four F-protein-derived polypeptides under the conditions described for panel D. The results are capture of the indicated protein under the indicated conditions (means ± SD), expressed as percentage of capture measured in the absence of MD-2 and FLAG-TLR4ecd (control). Protein capture was quantified by densitometric analysis of the various immunoblots, representing five independent experiments for N1 and three independent experiments for N2, C1, and C2. There is statistically significant greater capture of the N1 peptide after incubation with MD-2–TLR4ecd than with TLR4ecd alone (P < 0.05 [= 0.03]) or incubation alone (i.e., without MD-2 and TLR4ecd) (P < 0.01 [= 0.002]) (Student’s t test). (F) N1 peptide inhibits LPS-induced activation of NF-κB-luc. HEK293T cells cotransfected with TLR4-CD14 and MD-2 constructs, along with NF-κB-luc and β-galactosidase reporter constructs, were treated for 5 h with medium (M) only or with LPS (0.5 ng/ml) in the presence or absence of purified N1 or N2 peptides (40 ng/ml). Luciferase and β-galactosidase activities were measured in cell lysates, and RLU were calculated. Data are means ± SEM for duplicate samples (n = 2). *, P < 0.05.
To identify the subregion(s) within the F protein that mediates this interaction, overlapping His-tagged polypeptides (N1, N2, C1, and C2) (Fig. 5B) that encompass the F1 subunit of the RSV F protein were engineered and expressed in a baculovirus expression system. Lysates of the baculovirus-infected insect cells expressing the various His-tagged F1 protein-derived polypeptides (Fig. 5C) were preincubated with conditioned medium containing FLAG-tagged TLR4 ectodomain-vlr-Fc protein (FLAG-TLR4ecd) with or without MD-2 (or control conditioned medium lacking FLAG-TLR4ecd and MD-2), followed by incubation with anti-FLAG agarose beads. After extensive washing of the beads to remove unbound proteins, bound His-tagged proteins (i.e., N1, N2, C1, and C2) were eluted from the beads and detected by SDS-PAGE and immunoblotting using anti-His4 antibody (Fig. 5D). As shown in Fig. 5D and E, only the N1 polypeptide bound to the beads in an MD-2–TLR4ecd-dependent fashion. This polypeptide also showed some nonspecific binding to the beads (i.e., in the absence of FLAG-TLR4ecd) (Fig. 5D), but binding of N1 to the beads was significantly (P < 0.01) increased after preincubation of N1 with FLAG-TLR4ecd plus MD-2 but not with FLAG-TLR4ecd alone (Fig. 5E). These results suggest a direct interaction between the F protein and MD-2–TLR4 that is mediated by the N-terminal domain of the F1 protein and MD-2. Finally, to extend this observation, we hypothesized that the N1 polypeptide would be able to compete with LPS for binding to MD-2 and thereby inhibit signaling. In two separate experiments, we observed ~30% inhibition of LPS-induced signaling in the HEK293T TLR4–CD14–MD-2 transfection system induced by preincubation of cells with N1 but not the other polypeptides (Fig. 5F). Taken together, these data strongly support the hypothesis that TLR4 signaling by RSV F protein, like LPS, requires binding of F protein to MD-2 and that the N1 region of the F protein appears to compete for the same binding site on MD-2. This binding likely occurs at a site in MD-2 that is affected by the LPS antagonists RsLPS and eritoran, i.e., the hydrophobic pocket of MD-2. Of note, the N-terminal domain of the N1 peptide corresponds to the fusion peptide of the F protein, which is highly hydrophobic.
DISCUSSION
The major finding presented herein is that MD-2 interaction with RSV F protein is necessary for F protein to mediate signaling through the TLR4 axis. The physical interaction between the RSV F protein and MD-2 is direct and the resulting activation is altered by the presence of natural or synthetic TLR4–MD-2 antagonists.
RSV G and F proteins comprise the major glycoprotein spikes on the viral surface and the major targets of neutralizing antibodies. RSV F mediates fusion of virus with the target cell membrane during virus entry, and unlike the G protein, F is essential for viral replication in vitro and in vivo (7, 40, 41). Hence, it is considered the primary target for vaccine and antiviral drug development. Monoclonal antibodies to the F glycoprotein, which is highly conserved among the two antigenic groups of human RSV, passively protect against human RSV challenge in cotton rats (42) and reduce the severity of disease in premature and newborn babies (11). Thus, the use of the RSV F protein as an antigenic component for an RSV vaccine became the most logical and popular strategy. However, the failed “lot 100” formalin-inactivated RSV vaccine of the 1960s, which led to enhanced disease in infants and children who were infected by RSV postvaccination (43), raised many safety concerns about subunit vaccines, particularly those that involve F protein (44-47). Moghaddam and collaborators showed that carbonyl groups on formaldehyde-treated vaccine RSV antigens produce enhanced RSV disease in the mouse model (46). Recently, another group demonstrated that the same treatment of purified F protein generated an immunological response that lacks affinity maturation of anti-F antibodies, leading to RSV vaccine-enhanced disease (47). Furthermore, we previously demonstrated that RSV vaccine enhanced disease resulting from vaccination using the original lot 100 RSV vaccine used in the failed clinical trials of the 1960s could be overcome by the addition of the TLR4 agonist monophosphoryl lipid A (MPL) (48, 49). Altogether, these studies illustrate the importance of the molecular integrity of the F protein when used as a vaccine to avoid vaccine enhancement of RSV disease. At the same time, these findings suggest that F protein and TLR4 signaling can be manipulated to overcome some of the molecular alteration of the F proteins caused by formalin inactivation.
The TLR4 coreceptors CD14 and MD-2 are essential in the initiation of signaling induced by LPS (18, 19, 50) and have been suggested to be important for activation of TLR4 by other immunomodulatory proteins from human pathogens such as chlamydial Hsp70 (20) and Francisella tularensis DnaK (22). A role for CD14 has been shown for RSV F-protein-induced TLR4 activation (29, 32), but a requirement for MD-2 had not been formally demonstrated. In this study, we provide evidence that RSV F protein requires MD-2 as an essential coreceptor for TLR4 activation. In HEK293T transfectants carrying an NF-κB-regulated reporter gene, expression of MD-2, in addition to TLR4 and CD14, was necessary in F-protein-mediated NF-κB activation, paralleling that of E. coli LPS (Fig. 2). In mouse peritoneal macrophages, MD-2 expression is required for F-mediated cytokine induction, as upregulation of IL-1β gene expression is not detected in MD-2−/− primary peritoneal macrophages. These data further establish that RSV F protein, like LPS, requires MD-2 to induce functional TLR4 activation.
Previous studies have shown that certain LPS molecules, such as Rhodobacter sphaeroides LPS (RsLPS, containing penta-acylated lipid A) and eritoran (E5564), a synthetic tetra-acylated lipid A molecule whose structure is based on that of RsLPS, are potent LPS antagonists (36, 37). Recent crystallographic analyses of TLR4–MD-2 complexed with eritoran (E5564) revealed that MD-2 binds to the concave surface of the N-terminal and central domains of TLR4 and that the four acyl chains of eritoran occupy nearly 90% of the solvent-accessible volume of a hydrophobic internal pocket in MD-2, precluding the binding of LPS to MD-2 that is required for LPS-mediated TLR4 activation (38, 39). In this study, we have shown that both RsLPS and eritoran also inhibit F-protein-induced TLR4 dependent activation but not TLR4-independent, TNF-α-mediated signaling in the transfectants (Fig. 4A and B). The most likely explanation for this result is that F-protein-induced TLR4 activation depends on F-protein–MD-2 interactions that are inhibited by prior occupation of the hydrophobic pocket of MD-2 by either RsLPS or eritoran.
We have demonstrated human MD-2-dependent interactions of F protein in two independent ways: (i) coprecipitation of native F protein with recombinant His6–MD-2 to Ni-NTA beads (Fig. 5A) and (ii) increased cocapture of recombinant N1 polypeptide to anti-FLAG agarose following incubation with MD-2–FLAG-TLR4ecd (but not with FLAG-TLR4ecd alone) (Fig. 5D and E). The RSV F protein is a type I glycoprotein that becomes active only after intracellular proteolytic cleavage, forming two disulfide-linked subunits, F1 and F2. The N-terminal portion of the major subunit, F1, contains a highly hydrophobic peptide segment named fusion peptide (FP) (Fig. 5B) that is required for triggering the fusion process and thus becomes exposed during RSV-host cell interactions. Our findings suggest that among the various regions of exposed portions of the F1 protein, it is preferentially the region corresponding to the recombinant N1 polypeptide that engages TLR4 in an MD-2-dependent fashion (Fig. 5E), inhibiting TLR4–MD-2-dependent induction by LPS (Fig. 5F). The hydrophobic nature and size of the FP are consistent with the possibility that this region of the F protein interacts with MD-2 similarly to the acyl groups of LPS or eritoran, binding within the deep hydrophobic pocket of MD-2. Based on the ability of both RsLPS and eritoran to inhibit F protein signaling, it seems likely that the hydrophobic pocket of MD-2 must be occupied, associated with, or affected by the presence of the FP of F protein for TLR4 signaling. Whatever the precise mechanism, our data clearly indicate that there is a functional competition between RsLPS or eritoran and the F protein of RSV that renders the MD-2–TLR4 complex refractory to F-protein-dependent signaling.
Together, the data presented in this report suggest that the N-terminal region of F1 plays not only a fusogenic role in RSV but also an immune-regulatory one. Receptor-mediated cell signaling involves a highly integrated series of protein-protein interactions. Generally, there exists a physical interaction, whether it is between the cellular receptor and its coreceptors (e.g., CD14 and MD-2 coreceptors and TLR4) or between the agonists and the components of the signaling complex, or both, to bring the signalosome together to achieve an optimal biological response. Collectively, our data strongly demonstrate that TLR4 signaling by RSV F protein requires MD-2 and that F protein physically and functionally interacts with MD-2. The involvement of the TLR4-F protein interaction in the mechanism associated with vaccine enhanced disease was previously demonstrated (48, 49). However, up until now, there was no description of this interaction at the molecular level. The information presented here provides the first direct evidence for this interaction and also delineates the subregion in the F protein that drives this interaction. Although we are unable to conclude at this time that this interaction has a role in the life cycle of RSV, we hypothesize that targeting MD-2 and RSV F-protein interactions, and possibly disrupting them, may potentially lead to novel therapeutic approaches to help control RSV-induced inflammatory symptoms and pathology; also, based on this knowledge, it should be possible to define vaccine components that can be used to generate safe vaccines against RSV. Studies are under way to test these possibilities.
MATERIALS AND METHODS
Reagents and cell culture.
Human embryonic kidney (HEK) 293T cells (ATCC, Rockville, MD) were cultured in Dulbecco’s modified Eagle medium (DMEM) (BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. RsLPS was obtained from InvivoGen (San Diego, CA). Eritoran (E5564) was kindly provided by Eisai Inc. (Andover, MA). Recombinant human TNF-α was obtained from Cetus Corporation (Emeryville, CA). Protein-free, phenol-water-extracted Escherichia coli K235 LPS was prepared as described elsewhere (51), and F protein was prepared and purified as described elsewhere (32, 52). Polymyxin B sulfate was purchased from Sigma-Aldrich (St. Louis, MO), and proteinase K was obtained from Fermentas, Inc. (Glen Burnie, MD). The RNeasy total RNA extraction kit and Superfect transfection reagent were purchased from Qiagen (Valencia, CA). The RT-PCR kit was purchased from Promega Corporation (Madison, WI). All expression plasmid constructs were prepared using an EndoFree plasmid maxikit (Qiagen). Thioglycolate-elicited peritoneal macrophages from C57BL/6J mice (Jackson Laboratories) and MD-2−/− mice on a C57BL/6 background (n ≥ 8) (kindly provided by Katherine Fitzgerald, University of Massachusetts Medical School, Worcester, MA) were cultured for in vitro studies as described elsewhere (53-55). All animal experiments were carried out with institutional approval.
Plasmid constructs.
Expression plasmids, pcDNA3-huTLR4, pcDNA3-huCD14, pEFBOS-HA-huMD-2, and pELAM-1 luciferase (pELAM-luc; an NF-κB reporter) and pCMV1-β-galactosidase reporter plasmids have been described elsewhere (32, 56). The coding sequence of human MD-2 was cloned in pBAC3 and contains a gp64 signal sequence for secretion and an N terminus hexapolyhistidine tag (His6). MD-2 expression was achieved by infection of High Five cells (Invitrogen) (MD-2–His6). Medium containing secreted FLAG-tagged TLR4 ectodomain-vlr-Fc protein (FLAG-TLR4ecd)used in capture assays was obtained from Freestyle HEK293F cells stably transfected with TLR4 (amino acids 27 to 527) in a cytomegalovirus (CMV) vector containing additional hagfish amino acid sequence at the C-terminal end followed by the Fc domain. The stable transfected cells were a generous gift from Richard Tapping, University of Illinois, Urbana, IL.
Reporter assay.
HEK293T cells were seeded in 12-well Costar plates (Corning Inc., Corning, NY) at 2 × 105 cells/well and incubated overnight at 37°C in a 5% CO2 atmosphere. Cells were transfected by using the Superfect transfection reagent for 4 h with TLR4–CD14–MD-2 complex (pcDNA3-huTLR4, 300 ng/well; pEFBOS-HA-huMD-2, 3 ng/well; and pcDNA3-huCD14, 30 ng/well), together with the ELAM-luc reporter (500 ng/well) and pCMV1-β-galactosidase (100 ng/well). The final input DNA was adjusted to 1.5 µg/well with the pcDNA3 blank vector (Invitrogen, Carlsbad, CA). After overnight recovery, cells were washed with 1× PBS and stimulated with F protein or LPS at various concentrations for 5 h. Cells were lysed in 1× reporter assay lysis buffer (Promega). β-Galactosidase (Tropix; Galacto-Light System, Bedford, MA) and luciferase (luciferase assay system; Promega) activities were analyzed using a Berthold LB 9507 luminometer (Berthold Technologies, Bad Wildbad, Germany). Relative luciferase activity, given in relative luciferase units (RLU), was calculated by normalizing each sample’s luciferase activity to the constitutive β-galactosidase activity measured within the same sample.
To ensure that RSV F protein used in this study was LPS free, F protein was left untreated or was preincubated with polymyxin B (10 µg/ml) for 15 min at RT, with anti-F-protein antibodies for 1 h at 37°C, or with proteinase K (5 µg/ml) for 1 h at 37°C and subsequently at 95°C for 5 min to inactivate proteinase K, before the HEK293T TLR4–CD14–MD-2 transfectants were treated.
Quantitative real-time reverse transcriptase PCR (qRT-PCR).
Murine thioglycolate-elicited peritoneal macrophages were plated in 12-well plates (2 × 106 cells/well) and cultured as previously described (54, 55). Macrophages were stimulated with medium alone, LPS (10 ng/ml), or purified RSV F protein at various concentrations and incubated at 37°C for 2 h. Total RNA isolation and real-time PCR were performed as described previously (54, 55). Levels of IL-1β mRNA were reported as relative gene expression normalized to that in culture medium-treated samples.
Physical interaction between F protein and MD-2.
Ni-NTA beads were incubated with serum-free insect cell medium containing baculovirus-expressed, secreted, and His-tagged human MD-2 protein (His6–MD-2; 200 ng in 250 µl) or with insect media from mock-infected cells. Each set of beads was washed and equilibrated in buffer containing 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 0.1% Triton X-100. Beads were incubated with native RSV F protein for 2 h at 37°C. Both bead preparations were washed three times in buffer, mixed with an equal volume of 2× SDS loading buffer, and resolved on a 12% Tris-glycine SDS-PAGE gel, followed by Western analysis. The membrane was cut into two parts; the upper part was immunoblotted with an antibody against RSV F protein, and the lower part was probed with antibodies raised against the His tag on MD-2.
RSV F-protein-encoding gene segments (from GenBank sequence AY911262.1) were cloned into the pVL1393 (BD BaculoGold). The polypeptides were engineered to contain a C-terminal His tag and expressed in High Five (insect) cells. Cell lysates were prepared with RTL buffer (Qiagen), according to the manufacturer’s instructions. Supernatants from the cell lysates were used as a source of the His-tagged F-protein-derived recombinant polypeptides for the cocapture experiments described below. Insect cell-derived N-terminal His6-tagged MD-2 (His6–MD-2) and the TLR4 ectodomain (FLAG-TLR4ecd) were described above (57).
Coprecipitation experiments were performed using FLAG-TLR4ecd with and without His6–MD-2 (57). The MD-2–FLAG-TLR4ecd complex and FLAG-TLR4ecd alone were precipitated using anti-FLAG M2 affinity gel (Sigma-Aldrich, St. Louis, MO). Briefly, His6–MD-2 (25 ng) was added to medium containing FLAG-TLR4ecd (40 to 50 ng) and incubated for 30 min at 37°C in PBS supplemented with 1% human serum albumin (HSA) to permit TLR4–MD-2 complex formation. The TLR4–MD-2 complex was then incubated with an approximately 5-fold molar excess (~125 ng of each of the His-tagged polypeptides derived from the sequence of F1) at 37°C for 30 min. Forty microliters of anti-FLAG agarose beads equilibrated in PBS–1% HSA was incubated with the mixture of FLAG-TLR4ecd–His6–MD-2 complex and F1 polypeptide for 1 h at RT on a rotator. Beads were washed three times with PBS–1% HSA and then eluted with 30 µl of 0.5 M NaCl or 0.2 M glycine, pH 2.5. Eluate was treated with SDS-PAGE sample buffer, followed by electrophoresis on a 4-to-20% PAGE gel (Pierce) and Western blotting using an anti-His antibody (Qiagen) for the detection of the F1 polypeptides. Note that under these conditions, no immunoreactive signal from MD-2 was detected.
Statistical analysis.
Data are presented as means ± standard errors and were analyzed using a one-way analysis of variance (ANOVA) with repeated measures, followed by post hoc comparisons using Tukey’s multiple paired comparison test included in GraphPad PRISM 4.0 (GraphPad Software, Inc., San Diego, CA).
ACKNOWLEDGMENTS
This work was supported by NIAID, NIH, grants AI-057575 to J.C.G.B., AI-18797 to S.N.V., and AI05732 to J.P.W. and by a VA Merit grant to T.L.G.
Footnotes
Citation Rallabhandi P, et al. 2012. Respiratory syncytial virus fusion protein-induced Toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. mBio 3(4):e00218-12. doi:10.1128/mBio.00218-12.
REFERENCES
- 1. Collins PL, Crowe JE. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759 [DOI] [PubMed] [Google Scholar]
- 3. Welliver RC. 2003. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatr. 143:S112–S117 [DOI] [PubMed] [Google Scholar]
- 4. Nair H, et al. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henderson FW, Collier AM, Clyde WA, Jr, Denny FW. 1979. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N. Engl. J. Med. 300:530–534 [DOI] [PubMed] [Google Scholar]
- 6. Kahn JS, Schnell MJ, Buonocore L, Rose JK. 1999. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology 254:81–91 [DOI] [PubMed] [Google Scholar]
- 7. Karron RA, et al. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U. S. A. 94:13961–13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson PR, Jr., et al. 1987. Antigenic relatedness between glycoproteins of human respiratory syncytial virus subgroups A and B: evaluation of the contributions of F and G glycoproteins to immunity. J. Virol. 61:3163–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao X, Singh M, Malashkevich VN, Kim PS. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. U. S. A. 97:14172–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pollack P, Groothuis JR. 2002. Development and use of palivizumab (Synagis): a passive immunoprophylactic agent for RSV. J. Infect. Chemother. 8:201–206 [DOI] [PubMed] [Google Scholar]
- 11. The IMpact-RSV Study Group 1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537 [PubMed] [Google Scholar]
- 12. Poltorak A, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088 [DOI] [PubMed] [Google Scholar]
- 13. Hoshino K, et al. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–3752 [PubMed] [Google Scholar]
- 14. Qureshi ST, et al. 1999. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 189:615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vogel SN, Bhat N, Qureshi ST, Malo D. 1999. The LPS gene revisited, p 735–750 In Brade H, Opal SM, Vogel SN, Morrison DC, Endotoxin in health and disease. Marcel Dekker, New York, NY. [Google Scholar]
- 16. Perera PY, et al. 2001. CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J. Immunol. 166:574–581 [DOI] [PubMed] [Google Scholar]
- 17. Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433 [DOI] [PubMed] [Google Scholar]
- 18. Shimazu R, et al. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. 2003. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J. Biol. Chem. 278:48313–48320 [DOI] [PubMed] [Google Scholar]
- 20. Bulut Y, et al. 2002. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168:1435–1440 [DOI] [PubMed] [Google Scholar]
- 21. Malley R, et al. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ashtekar AR, et al. 2008. TLR4-mediated activation of dendritic cells by the heat shock protein DnaK from Francisella tularensis. J. Leukoc. Biol. 84:1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okumura A, Pitha PM, Yoshimura A, Harty RN. 2010. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 84:27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smiley ST, King JA, Hancock WW. 2001. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167:2887–2894 [DOI] [PubMed] [Google Scholar]
- 25. Okamura Y, et al. 2001. The extra domain A of fibronectin activates toll-like receptor 4. J. Biol. Chem. 276:10229–10233 [DOI] [PubMed] [Google Scholar]
- 26. Taylor KR, et al. 2004. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 279:17079–17084 [DOI] [PubMed] [Google Scholar]
- 27. Guillot L, et al. 2002. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 168:5989–5992 [DOI] [PubMed] [Google Scholar]
- 28. Yu M, et al. 2006. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26:174–179 [DOI] [PubMed] [Google Scholar]
- 29. Kurt-Jones EA, et al. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401 [DOI] [PubMed] [Google Scholar]
- 30. Haynes LM, et al. 2001. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75:10730–10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsuzuki H, Tani T, Ueyama H, Kodama M. 2001. Lipopolysaccharide: neutralization by polymyxin B shuts down the signaling pathway of nuclear factor kappaB in peripheral blood mononuclear cells, even during activation. J. Surg. Res. 100:127–134 [DOI] [PubMed] [Google Scholar]
- 32. Rallabhandi P, et al. 2006. Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J. Immunol. 177:322–332 [DOI] [PubMed] [Google Scholar]
- 33. Visintin A, Iliev DB, Monks BG, Halmen KA, Golenbock DT. 2006. Md-2. Immunobiology 211:437–447 [DOI] [PubMed] [Google Scholar]
- 34. Visintin A, et al. 2006. MD-2 expression is not required for cell surface targeting of toll-like receptor 4 (TLR4). J. Leukoc. Biol. 80:1584–1592 [DOI] [PubMed] [Google Scholar]
- 35. Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. 2005. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J. Immunol. 175:6465–6472 [DOI] [PubMed] [Google Scholar]
- 36. Zuckerman SH, Qureshi N. 1992. In vivo inhibition of lipopolysaccharide-induced lethality and tumor necrosis factor synthesis by Rhodobacter sphaeroides diphosphoryl lipid A is dependent on corticosterone induction. Infect. Immun. 60:2581–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rossignol DP, Lynn M. 2002. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J. Endotoxin Res. 8:483–488 [DOI] [PubMed] [Google Scholar]
- 38. Kim HM, et al. 2007. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906–917 [DOI] [PubMed] [Google Scholar]
- 39. Park BS, et al. 2009. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458:1191–1195 [DOI] [PubMed] [Google Scholar]
- 40. Bukreyev A, Whitehead SS, Murphy BR, Collins PL. 1997. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J. Virol. 71:8973–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teng MN, Collins PL. 1998. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J. Virol. 72:5707–5716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson S, et al. 1997. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 176:1215–1224 [DOI] [PubMed] [Google Scholar]
- 43. Fulginiti VA, et al. 1969. Respiratory virus immunization. I. A field trial of two inactivated respiratory virus vaccines; an aqueous trivalent parainfluenza virus vaccine and an alum-precipitated respiratory syncytial virus vaccine. Am. J. Epidemiol. 89:435–448 [DOI] [PubMed] [Google Scholar]
- 44. Murphy BR, Sotnikov AV, Lawrence LA, Banks SM, Prince GA. 1990. Enhanced pulmonary histopathology is observed in cotton rats immunized with formalin-inactivated respiratory syncytial virus (RSV) or purified F glycoprotein and challenged with RSV 3-6 months after immunization. Vaccine 8:497–502 [DOI] [PubMed] [Google Scholar]
- 45. Prince GA, et al. 2000. Efficacy and safety studies of a recombinant chimeric respiratory syncytial virus FG glycoprotein vaccine in cotton rats. J. Virol. 74:10287–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moghaddam A, et al. 2006. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines. Nat. Med. 12:905–907 [DOI] [PubMed] [Google Scholar]
- 47. Delgado MF, et al. 2009. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 15:34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boukhvalova MS, et al. 2006. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 24:5027–5035 [DOI] [PubMed] [Google Scholar]
- 49. Prince GA, et al. 2001. Monophosphoryl lipid A adjuvant reverses a principal histologic parameter of formalin-inactivated respiratory syncytial virus vaccine-induced disease. Vaccine 19:2048–2054 [DOI] [PubMed] [Google Scholar]
- 50. Wright SD. 1991. Multiple receptors for endotoxin. Curr. Opin. Immunol. 3:83–90 [DOI] [PubMed] [Google Scholar]
- 51. McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY. 1967. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry 6:2363–2372 [DOI] [PubMed] [Google Scholar]
- 52. Roder C, Krusat T, Reimers K, Werchau H. 2000. Purification of respiratory syncytial virus F and G proteins. J. Chromatogr. B Biomed. Sci. Appl. 737:97–106 [DOI] [PubMed] [Google Scholar]
- 53. Cole LE, et al. 2006. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 176:6888–6899 [DOI] [PubMed] [Google Scholar]
- 54. Rallabhandi P, et al. 2008. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J. Biol. Chem. 283:24314–24325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shirey KA, Cole LE, Keegan AD, Vogel SN. 2008. Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J. Immunol. 181:4159–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rallabhandi P, et al. 2008. Differential activation of human TLR4 by Escherichia coli and Shigella flexneri 2a lipopolysaccharide: combined effects of lipid A acylation state and TLR4 polymorphisms on signaling. J. Immunol. 180:1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prohinar P, et al. 2007. Specific high affinity interactions of monomeric endotoxin.protein complexes with Toll-like receptor 4 ectodomain. J. Biol. Chem. 282:1010–1017 [DOI] [PubMed] [Google Scholar]