

Abstract
A gold-catalyzed regioselective homodimerization of aliphatic terminal alkynes is described. Bulky and less Lewis acidic tBuXPhosAuNTf2 is the preferred catalyst, and the additive, anhydrous NaOAc, substantially facilitates the reaction.
Keywords: gold, dimerization, regioselectivity, terminal alkynes, enynes
Dimerization of terminal alkynes provides a rapid and atom-economic access to synthetically versatile enynes1 and can be promoted by an amazingly broad range of metals including various transition metals such as Ni,2 Y,3 Ru,4 Rh,5 Pd,6 Ir,7 lanthalides,8 and actinides9 and main group metals.10 Three enyne isomers, Z-linear, E-linear and branched (gem) can be obtained with various levels of regio- and/or stereoselectivities following either a ‘head-to-head’ or a ‘head-to-tail’ union.
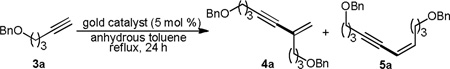
Albeit recent rapid development in homogeneous gold catalysis,11 there are still many well-established reactions, including dimerization of terminal alkynes, that have not succumbed to gold catalysis. As a part of our general interest in discovering new gold chemistry and probing potential unique reactivities of gold catalysts, we decided to study dimerization of terminal alkynes using soluble gold catalysts. Since gold complexes can readily activate alkynes toward nucleophilic attacks and alkynylgolds are known to be nucleophilic,12 we anticipated, as shown in Scheme 1, that the alkynylgold B, formed from the terminal alkyne-gold complex A, might act as the nucleophile to attack its precursor, thereby affording enynes 1 and 2 upon subsequent protodeauration. Interestingly, two different gold complexes would be involved in the key C-C bond formation step. Good ratios of 1/2 was anticipated as a Markovnikov-type anti addition to the terminal C-C triple bond should be preferred. Herein, we disclose our preliminary results.
scheme 1.

Proposed gold-catalyzed dimerization of terminal alkynes.
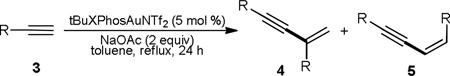
We began with screening different gold catalysts and reaction conditions for the dimerization of ethereal alkyne 3a. After some preliminary studies with different solvents at various reaction temperatures, it was found that the reaction was best conducted in refluxing toluene. Gold complexes that are prone to thermal decomposition such as Ph3PAuNTf2 (Table 1, entry 1) was totally ineffective due to gold precipitation. Bulky and less Lewis acidic gold catalysts are more stable upon heating and therefore capable of promoting the dimerization. These catalysts included IPrAuNTf213 (Table 1, entry 2), BrettPhosAuNTf214 (Table 1, entry 3) and tBuXPhosAuNTf2 (Table 1, entry 4). Among them, tBuXPhosAuNTf2 was the most effective. However, catalyst decomposition still occurred, and the overall yields were low. We reasoned that the addition of a base might make the gold catalyst more stable and moreover facilitate the formation of alkynylgold intermediates of type B. Consequently, various bases (2 equivalents) were tested. While stronger bases such as Na2CO3 (Table 1, entry 5) and Cs2CO3 (Table 1, entry 6) were proven deleterious, NaHCO3 (Table 1, entry 7) was surprisingly effectively in facilitating the reaction. Anhydrous NaOAc proved to be even better, and the isolated yield was 83% (Table 1, entry 8). Importantly, the branched ‘head-to-tail’ dimer 4a was formed with high selectivity over the linear Z-enyne 5a, formed via a ‘head-to-head’ dimerization. We were curious whether soluble acetate would perform the same feat. To our surprise, no reaction was observed with either 2 equivalents or 6 mol % of Me4N+OAc−. It is most likely that soluble OAc− reacted with the gold catalyst to form tBuXPhosAuOAc, which is not catalytically active. 2-bromopyridine (pKa of its conjugated acid: 0.7115), a soluble base but less basic than AcO− (pKa of HOAc: 4.76), could promote the reaction but to a less extent (Table 1, entry 11). 1,2-Dichloroethane was an inferior solvent for this reaction (Table 1, entry 12). Control experiments (Table 1, entries 13 and 14) established the indispensable role of the gold catalyst in this reaction as neither its absence nor substituting it with AgNTf2 led to any observable reaction.
Table 1.
Reaction optimizationa
 | ||||
|---|---|---|---|---|
| entry | catalyst | additiveb | yieldc | 4a/5ad |
| 1 | Ph3PAuNTf2 | - | - | - |
| 2 | IPrAuNTf2 | - | 20% | 4.5/1 |
| 3 | BrettPhosAuNTf2 | - | 22% | 14/1 |
| 4 | tBuXPhosAuNTf2 | - | 28% | 10/1 |
| 5 | tBuXPhosAuNTf2 | Na2CO3 | 15% | 19/1 |
| 6 | tBuXPhosAuNTf2 | Cs2CO3 | - | - |
| 7 | tBuXPhosAuNTf2 | NaHCO3 | 68% | 5/1 |
| 8 | tBuXPhosAuNTf2 | NaOAc | 83%e | 14/1 |
| 9 | tBuXPhosAuNTf2 | Me4N+OAc− | - | - |
| 10 | tBuXPhosAuNTf2 | Me4N+OAc−f | - | - |
| 11 | tBuXPhosAuNTf2 | 2-BrPy | 39% | >20/1 |
| 12g | tBuXPhosAuNTf2 | NaOAc | 22% | 18/1 |
| 13 | - | NaOAc | - | - |
| 14 | AgNTf2 | NaOAc | - | - |
Reaction run in Schlenk tubes under nitrogen; [alkyne] = 0.1 M.
2 equivalent.
NMR yield using diethyl phthalate as the internal reference.
Ratio determined by crude 1H NMR.
Isolated yield.
6 mol % instead.
DCE as the reaction solvent.

The optimized reaction conditions were then applied to the reaction scope study. As shown in Table 2, aliphatic terminal alkynes such as 1-dodecyne (entry 1), cyclohexylacetylene (entry 2) and cyclopentylacetylene (entry 3) were all amenable to the dimerization, yielding the branched isomer (i.e., 4) in mostly good yields. Several functional groups such as sulfide (entry 4), phenyl (entry 5), imide (entry 6) and ester (entry 7) could be readily incorporated into aliphatic terminal alkynes without compromising much of the reaction efficiency. To our surprise, phenylacetylene was a poor substrate, and only 8% of the dimer product was formed after 24 h; moreover, 2-methylbut-3-yn-1-ene was not a suitable substrate (entry 9). Extension of this chemistry to heterodimerization led to mixtures of four branched enyne products with low overall yields.
Table 2.
Reaction scope of homodimerizationa
 | ||||
|---|---|---|---|---|
| entry | R | 4 | yield (4)b | 4/5c |
| 1 | n-decyl | 4b | 72% | 14/1 |
| 2 | cyclohexyl | 4c | 69% | 13/1 |
| 3d | cyclopentyl | 4d | 85% | 25/1 |
| 4d | 4e | 61% | 12/1 | |
| 5 | PhCH2CH2 | 4f | 61% | 11/1 |
| 6 | 4g | 78% | 10/1 | |
| 7 | 4h | 81% | 11/1 | |
| 8 | Ph | 4i | 8% | 12/1 |
| 9 | 2-propenyl | 4j | - | - |
Reaction run in Schlenk tubes under nitrogen; [alkyne] = 0.1 M.
Isolated yield and containing small amount of inseparable 5.
Determined by crude 1H NMR.
Reaction time: 48 h; catalyst loading: 10 mol %.
The reaction mechanism is likely following what we originally conceived in Scheme 1. The role of NaOAc is worth further commenting. It should facilitate the formation of B by deprotonation of complex A or trapping the released proton; HOAc thus formed should be capable of protodeauration under the elevated temperature in the final step. The gold complex thus generated is tBuXPhosAuOAc instead of tBuXPhosAuNTf2. However, it is likely that this inactive complex can be returned to its active form by switching counter anion with the in-situ generated NaNTf2, driven by the formation of crystalline and barely soluble NaOAc. On the contrary, when Me4N+OAc− was used, such a driving force was missing, and all the gold catalyst was in its inactive tBuXPhosAuOAc form.
In conclusion, a gold-catalyzed homodimerization of aliphatic terminal alkynes has been developed. The branched ‘head-to-tail’ enyne isomers are formed in mostly good yields and with excellent selectivities over the linear isomers. NaOAc was found to be effective as an additive in facilitating the reaction. The likely reaction mechanism entails a key C-C bond formation step involving two different gold species.
Supplementary Material
Acknowledgment
The authors are grateful for the generous financial support by NIH (R01 GM084254) and UCSB. LZ is a Sloan Fellow.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synlett.
Primary Data for this article are available online at http://www.thieme-connect.com/ejournals/toc/synlett and can be cited using the following DOI: (number will be inserted prior to online publication).
References
- 1.For a review, see: Bustelo E, Dixneuf PH. In: Handbook of C-H Transformations : Applications in Organic Synthesis. Dyker G, editor. Vol. 1. Wiley-VCH: Weinheim; 2005. pp. 62–72.
- 2.For an example, see: Ogoshi S, Ueta M, Oka M-a, Kurosawa H. Chem. Commun. 2004:2732–2733. doi: 10.1039/b409851j.
- 3.For an example, see: Komeyama K, Kawabata T, Takehira K, Takaki K. J. Org. Chem. 2005;70:7260–7266. doi: 10.1021/jo0509206.
- 4.For an example, see: Chen X, Xue P, Sung HHY, Williams ID, Peruzzini M, Bianchini C, Jia G. Organometallics. 2005;24:4330–4332.
- 5.(a) Kovalev IP, Yevdakov KV, Strelenko YA, Vinogradov MG, Nikishin GI. J. Organomet. Chem. 1990;386:139–146. [Google Scholar]; (b) Boese WT, Goldman AS. Organometallics. 1991;10:782–786. [Google Scholar]; (c) Lee C-C, Lin Y-C, Liu Y-H, Wang Y. Organometallics. 2004;24:136–143. [Google Scholar]; (d) Weng W, Guo C, Celenligil-Cetin R, Foxman BM, Ozerov OV. Chem. Commun. 2006:197–199. doi: 10.1039/b511148j. [DOI] [PubMed] [Google Scholar]
- 6.For earlier examples, see: Trost BM, Chan C, Ruhter G. J. Am. Chem. Soc. 1987;109:3486–3487. Luo FT, Fwu SL, Huang WS. Tetrahedron Lett. 1992;33:6839–6840.
- 7.(a) Ohmura T, Yorozuya S-i, Yamamoto Y, Miyaura N. Organometallics. 2000;19:365–367. [Google Scholar]; (b) Ghosh R, Zhang X, Achord P, Emge TJ, Krogh-Jespersen K, Goldman AS. J. Am. Chem. Soc. 2007;129:853–866. doi: 10.1021/ja0647194. [DOI] [PubMed] [Google Scholar]; (c) Ez-Zoubir M, d'Herouville FLB, Brown JA, Ratovelomanana-Vidal V, Michelet V. Chem. Commun. 2010;46:6332–6334. doi: 10.1039/c0cc00721h. [DOI] [PubMed] [Google Scholar]
- 8.For examples, see: Heeres HJ, Teuben JH. Organometallics. 1991;10:1980–1986. Yoshida M, Jordan RF. Organometallics. 1997;16:4508–4510.
- 9.For examples, see: Dash AK, Wang JX, Berthet JC, Ephritikhine M, Eisen MS. J. Organomet. Chem. 2000;604:83–98. Dash AK, Gourevich I, Wang JQ, Wang J, Kapon M, Eisen MS. Organometallics. 2001;20:5084–5104.
- 10.For an example, see: Dash AK, Eisen MS. Org. Lett. 2000;2:737–740. doi: 10.1021/ol9903584.
- 11.For selected reviews, see: Fürstner A, Davis PW. Angew. Chem. Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. Corma A, Leyva-Pérez A, Sabater MJ. Chem. Rev. 2011;111:1657–1712. doi: 10.1021/cr100414u. Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m. Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. Wang S, Zhang G, Zhang L. Synlett. 2010:692–706.
- 12.(a) Li C-J. Acc. Chem. Res. 2010;43:581–590. doi: 10.1021/ar9002587. [DOI] [PubMed] [Google Scholar]; (b) Skouta R, Li C-J. Tetrahedron. 2008;64:4917–4938. [Google Scholar]; (c) Lo VK-Y, Liu Y, Wong M-K, Che C-M. Org. Lett. 2006;8:1529–1532. doi: 10.1021/ol0528641. [DOI] [PubMed] [Google Scholar]
- 13.For earlier studies using this catalyst, see: Li G, Zhang L. Angew. Chem. Int. Ed. 2007;46:5156–5159. doi: 10.1002/anie.200701449. Ricard L, Gagosz F. Organometallics. 2007;26:4704–4707.
- 14.For the initial report on BrettPhos ligand, see: Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358. For the first study using BrettPhosAu+, see: Ye L, He W, Zhang L. Angew. Chem. Int. Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624.
- 15.Linnell R. J. Org. Chem. 1960;25:290–290. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.