

Abstract
Bluetongue virus (BTV), a member in the family Reoviridae, is a re-emerging animal disease infecting cattle and sheep. With its recent outbreaks in Europe, there is a pressing need for efficacious antivirals. We presented here the identification and characterization of a novel virostatic molecule against BTV, an aminothiophenecarboxylic acid derivative named compound 003 (C003). The virostatic efficacy of C003 could be improved via chemical modification, leading to a de novo synthesized compound 052 (C052). The 50% effective concentrations (EC50) of C003 and C052 were determined at 1.76±0.73 µM and 0.27±0.12 µM, respectively. The 50% cytotoxicity concentration (CC50) of C003 was over 100 µM and the CC50 of C052 was at 82.69 µM. Accordingly, the 50% selective index (SI50) of C003 and C052 against BTV was over 57 and 306, respectively. The inhibitory effect of C003/C052 on BTV-induced apoptosis was also confirmed via the inhibition of caspase-3/-7 activation post BTV infection. C003/C052 could inhibit BTV induced CPE even when added as late as 24 h.p.i., indicating that they might act at late stage of viral life-cycle. C003/C052 could reduce over two-logs of both the progeny virus production and the number of genomic viral RNA copies. Interestingly, both the activation of host autophagy and viral protein expression were inhibited post BTV infection when cells were treated with C003 and C052, suggesting that C003/C052 might act as virostatic agents via inhibiting host autophagy activation. Although further investigations might be needed to pin down the exact mechanism of C003/C052, our finding suggested that these compounds might be potent lead compounds with potential novel mechanism of action against BTV.
Introduction
Bluetongue virus (BTV), a double-strand RNA (dsRNA) virus, is the prototype virus in the genus Orbivirus within the family Reoviridae. The segmented BTV dsRNA genome encodes ten viral proteins (VP), including seven structural proteins (VP1-7) and three non-structural (NS) proteins, NS1, NS2 and NS3 [1], [2], [3]. As a member of arboviruses, BTV is transmitted by certain species of Culicoides biting midges, including C. variipennis and C. imocola [4], [5]. Bluetongue disease is a non-contagious viral disease affecting domestic animals including sheep and cattle primarily, as well as wild ruminants such as buffalo, antelope, deer, elk and camels. BTV disease is one of the most important diseases of domestic livestock, causing $3 billion per year loss worldwide [6], [7]. In sheep, the disease is acute, and mortality is accordingly high [8]. BTV is listed under the Office International des Epizooties (OIE) Terrestrial Animal Health Code –2009 [2]. Exotic BTV is also listed in the “USDA High Consequence Livestock Pathogens." Due to its economic significance, BTV has been the subject of extensive molecular virology and structural biology studies [9], [10], [11]. The recent development of the BTV reverse genetics system facilitates our understanding toward the BTV viral life-cycle, the structural and functional interrelationship among viral proteins [12], [13], [14]. Hence, BTV is now one of the well-characterized viruses [6], [9], [15].
The main prevention and control measures in endemic areas include active surveillance programs, animal quarantine and movement restrictions, vaccination and insect control measures [2], [16], [17], [18]. Vaccination is used as the most effective and practical measure to minimize losses related to BTV disease and to potentially interrupt the cycle from infected animal to vector [18], [19], [20]. However, due to the complexity of the virus, including the twenty-four different serotypes that have the ability of causing variable diseases, BTV is still endemic in many regions despite the high vaccine coverage in sheep and cattle [21], [22], [23], [24]. The 1998–2001 outbreak of BTV-8 in the Mediterranean Basin is the greatest epizootic of the disease on record, showing that BTV has extended its range northwards into areas of Europe that were never affected before, and has since persisted in many of these locations [6], [25], [26], [27]. In 2006, BTV-8 has reached three northern Europe countries, including the Netherlands, Belgium, and Germany [28], [29], and further spread to surrounding countries, reaching Switzerland by the end of October 2007 [6], [30], [31], [32].
Vaccination of individuals/animals during an outbreak can prove effective, however, protection of an individual/animal from the threat may not occur for two or more weeks after the initial vaccination [33]. Hence, only a drug can be offered as a therapeutic treatment of individuals/animals in an endemic area [33]. Surprisingly, there are no antiviral currently available against BTV disease. Recently, anti-BTV drug discovery has been implemented and potential antiviral lead compound(s) has been identified [34]. The development and validation of a cytopathic effect (CPE)-based assay led to the screening of the NIH Molecular Libraries Small Molecule Repository (MLSMR), with 194,950 small molecule compounds then. Further studies, using various primary, secondary and confirmatory assays, confirmed 185 structures that were grouped into six analog series corresponding to six scaffolds enriched within the active set compared to their distribution in the library [34]. Based on the results from the previous studies [34], we selected and evaluated the virostatic efficacy of a cluster of active compounds, in particular, the aminothiophenecarboxylic acid derivatives. Furthermore, aiming to understand their mechanism of action, various studies were carried out to determine which viral life stage(s) these compounds acted on, focusing on how these virostatic compounds protect cells from BTV-induced apoptosis.
Results
Thiophene Derivatives as Lead Virostatic Agents against BTV
Based on the previously identified six compound clusters [34], we selected 2–4 representative compounds from each cluster and assessed their virostatic efficacies against BTV, including the 50% effective concentration (EC50) and the 90% effective concentration (EC90). The EC50 of these lead compounds were determined between 0.7 µM and 20 µM, respectively (results not shown). Interestingly, the four thiophene derivative compounds from cluster I, i.e. compound 001 through 004 (C001, C002, C003 and C004), showed potent virostatic efficacy against BTV, especially the aminothiophenecarboxylic acid derivatives C001 and C003. Using the ten-concentration dose response assay, the EC50s of C001 and C003 were determined at 0.69±0.15 µM and 1.76±0.73 µM, respectively (Fig. 1A and B). The EC90s of C001 and C003 were at 6.20±1.39 µM and 15.86±6.56 µM, respectively (Fig. 1A and B). The above results indicated that both C001 and C003 could be potential novel virostatic agents against BTV. Additionally, the 50% cytotoxicity concentration (CC50) of both C001 and C003 were over 100 µM (Fig. 1A and B). Accordingly, the selective indexes (SI50), which equals to EC50/CC50, were above 145 and 57 for C001 and C003, respectively, indicating that both compounds were highly selective against BTV. Considering the compound solubility and availability, we selected C003 as the lead virostatic agent for further evaluation, including structure-activity relationship (SAR) analysis, chemical modification and mechanism of action studies.
Figure 1. The virostatic efficacy and cytotoxicity were showed in (A) C003 and (B) C001.
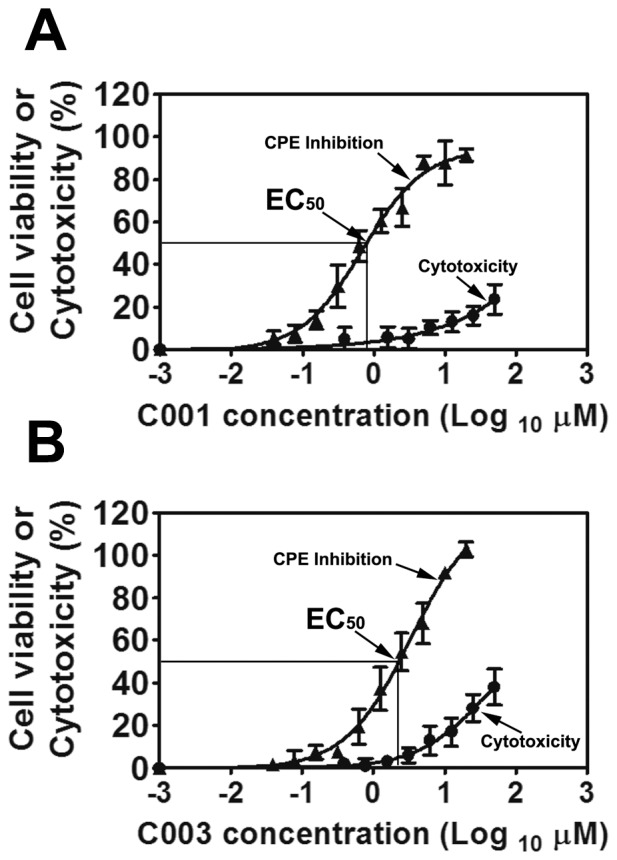
Cells were infected with BTV at MOI of 0.01 with or without C003 or C001 at various concentrations. At 72 h.p.i, cell viability (triangles) or cytotoxicity (circles) was measured using the CellTiter-Glo reagent. Each data point represents means and standard deviation (SD) from five replicates.
Improving C003 Virostatic Efficacy via Chemical Modification
C003, or 2-{[3-(2-furyl)acryloyl]amino}-4-(4-methylphenyl)-3-thiophenecarboxylic acid, is an aminothiophenecarboxylic acid derivative with a structure showed in Figure 2. To improve the potential of C003 as an anti-BTV lead structural framework, it was divided into 5 zones for synthetic target goals. Figure 3 illustrates our results from a variation in zone 5 by replacing the furan of C003 with a thiophene (IC2a or C052) and pyridine (IC3a or C055).
Figure 2. The compound structure of C003 was showed.

The compound was divided into five zones as indicated.
Figure 3. The de novo synthesis of C052 (2a) and C055 (3a) were illustrated.

Each number here represents each intermediate compound (ic) as described in the text.
Our approach to C052 (IC2a) and C055 (IC3a) involved a convergent pathway whereby (1) the Gewald reaction was employed for the thiophene unit (IC4 of zone 1) and (2) a Horner-Wadsworth-Emmons reaction of 2-thiophenecarboxaldehyde (IC5) and 2-pyridinecarboxaldehyde (IC6) with malonic acid followed by acid chloride formation provided a means to IC7 and IC8, the zone 5 modifiers. Bringing these units (IC4 and IC7/IC8) together produced IC9 and IC 10. Saponification of IC9 and IC10 gave the sodium salts IC2b and IC3b, which, upon acidification, yielded the desired final products C052 (IC2a) and C055 (IC3a) (Fig. 3).
Because of their potential as prodrugs of C052 (IC2a) and C055 (IC3a), esters IC9 and IC10, were also evaluated for their virostatic efficacies. Interestingly, while other analogs including C055 did not show any improvement of its virostatic efficacy, the de novo synthesized analogs, C052, showed an improved virostatic efficacy with EC50 at 0.27±0.12 µM and EC90 of 2.5±1.04 µM, respectively (Fig. 4). Similarly, C052 showed very little toxicity with CC50 at 82.69 µM. Consequently, the SI50 of C052, were at 306. Comparing with the EC50 and SI50 of C003, the EC50 of C052 were 6.5 times lower, and the SI50 was 5.4 times higher, indicating that the virostatic efficacy of C003 could be further improved via SAR and chemical modifications, leading to a more potent and selective virostatic agent against BTV.
Figure 4. The virostatic efficacy and cytotoxicity of C052 were showed as cells were infected with BTV at MOI of 0.01 in the presence of various concentrations of C052.

At 72 h.p.i, cell viability (triangles) or cytotoxicity (circles) was determined using CellTiter-Glo reagent. Each data point represents means and SD from five replicates.
Both C003 and C052 Prevented BTV Induced CPE
The CellTiter-Glo cell viability assay determines the number of viable cells in culture based on quantitation of cellular ATP presented, which signals the presence of metabolically active living cells [34], [35], [36]. To further confirm that C003 or C052 could prevent BTV-induced CPE, cell morphology changes at different h.p.i. were examined post BTV infection at MOI of 0.01. Without C003 or C052 treatment, CPE was observed as early as 48 h.p.i. in infected cells. At 72 h.p.i., the majority of infected cells were dislodged from the plate indicating CPE and cell lysis (Fig. 5B). When C003 at 10 µM or C052 at 2.5 µM was added to BTV-infected cells at 0 h.p.i., CPE was not observed up till 72 h.p.i. There was no sign of cell dislodging and lysis (Fig. 5D and F). Similar to previous observation, uninfected cells treated with C003 at 10 µM or C052 at 2.5 µM (Fig. 5C and E) showed no overt toxicity, with regard to cell division, morphology changes, with comparable cell viability to that of the control cells (Fig. 5A). This result confirmed that BTV-induced CPE was prevented after cells were treated with C003 or C052.
Figure 5. Both C003 and C052 protects BSR cells from BTV induced CPE/apoptosis.

In mock infected BSR cells (A), cells formed a monolayer with no cells detached from the plates. At 72 h.p.i., BTV infected cells showed CPE/apoptosis and were detached from the plate (B). When C003 at 10 µM was added to the BSR cells without BTV infection, there were no obvious changes in term of cell morphology (C). There were also no morphology changes when cells were treated with C003 and with BTV infection (D). Similarly, when C052 at 2.5 µM was added to the BSR cells without BTV infection, there were no cytotoxicity (E), and C052 could also block BTV-induced CPE/apoptosis (F).
C003 and C052 Prevented BTV-induced Apoptosis
Previous studies have showed that BTV infection in mammalian cells triggers apoptosis leading to CPE/cell lysis in vitro and the virus-induced pathogenesis in vivo [1], [37]. In order to further confirmed that C003 and/or C052 prevented BTV-induced CPE via blocking apoptosis, we examined the activation of caspase-3/7 in infected cells treated with the virostatic compounds at different h.p.i. Our result showed that BTV-induced caspase-3/7 activation, the main characteristics of apoptosis, was inhibited in C003/C052 treated cells. When BTV-infected cells were treated with C003 at 1.8 µM and 10 µM, there were 2.1- and 3.3-fold reductions of caspase-3/7 activity at 48 h.p.i, respectively, comparing with the caspase-3/7 activity in cells infected with BTV but without C003 treatment (Fig. 6A and B). When cells were treated with C052 at 0.27 µM and 2.5 µM, the inhibitions of caspase-3/7 activity were similar to the inhibition of C003 at similar concentrations (1.8 µM and 10 µM), including 2.0- and 2.3-fold decreases of caspase-3/7 activity at 48 h.p.i. (Fig. 6A and B). The above results indicated that C003 and/or C052 could inhibit BTV-induced caspase-3/7 activation, which further prevented BTV-induced apoptosis, consequently CPE and cell lysis.
Figure 6. Effects of C003 and C052 on BTV-induced caspase-3/7 activation.
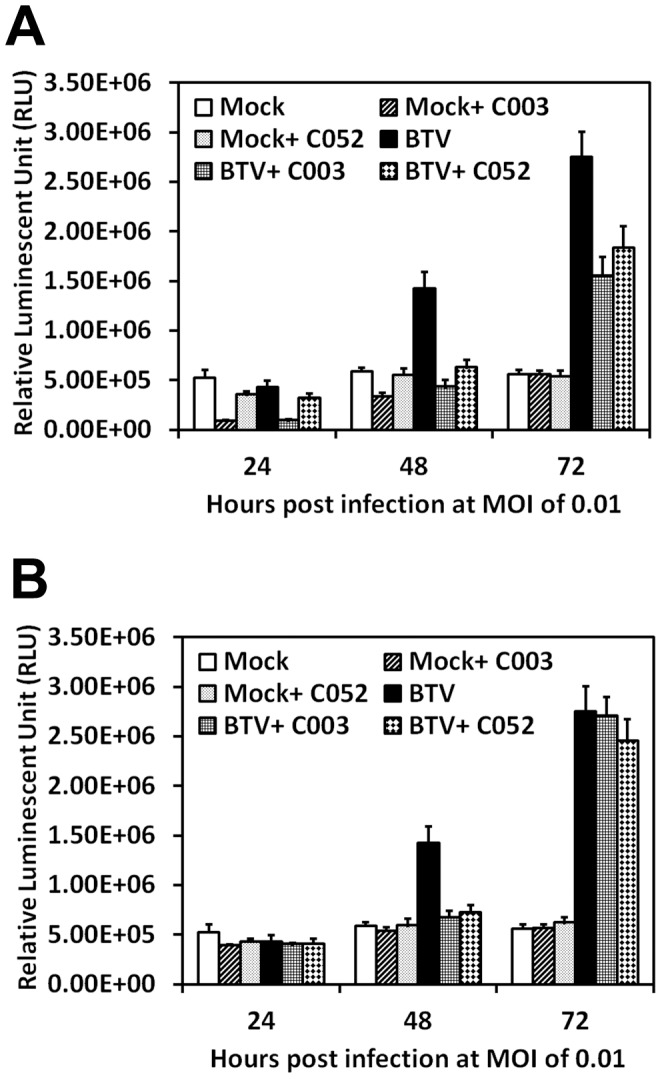
BSR cells were mock infected or infected with BTV at MOI of 0.01, and treated with C003 or C052 at their EC90 concentrations (A) or EC50 concentrations (B). At 24, 48 and 72 h.p.i, caspase-3/-7 activities were determined using the Caspase-Glo-3/7 reagent, and presented as relative fluorescent unit (RLU). Each data points represented mean values and SD from triplicates experiments.
The Effect of C003 and C052 on Virus Progeny Production
To understand whether C003 and C052 could disrupt the productive BTV viral life-cycle, we analyzed their effect on BTV progeny productions. BSR were infected with BTV at MOI of 0.01 and cells were treated with C003 or C052 at 10 µM or 2.5 µM, respectively. At different h.p.i, cells and supernatants were collected, and BTV titers in these samples were determined by the conventional plaque assay. At 24 h.p.i., there were one log differences of virus progeny production in cells treated with C003 or C052 at 10.0 µM and 2.5 µM, respectively, when compared with that in samples collected from BTV-infected only cells. There were three- or two-log reductions of virus progeny production in samples collected at 48 h.p.i. and 72 h.p.i. after treated with C003 or C052, respectively (Fig. 7A). Similarly, the reduction of progeny virus production was also noticeable in the supernatant samples after treated with C003 or C052, with three- or two-log reductions of virus progeny titer, respectively, comparing with that in samples collected from BTV-infected supernatant without virostatic compound treatment (Fig. 7B).
Figure 7. The effects of C003 and C052 on BTV progeny production.
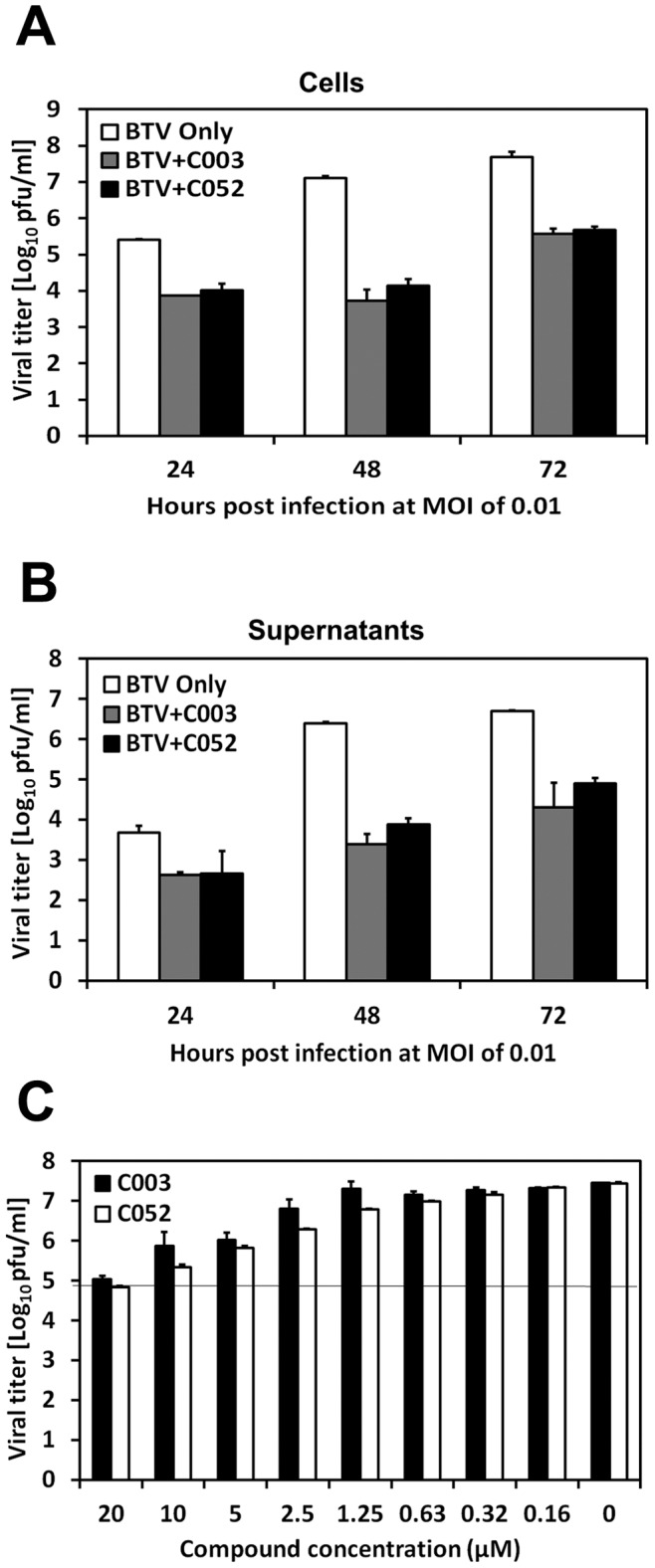
BTV infected cells were treated with C003 (10 µM) or C052 (2.5 µM). At different time post infection as indicated in figures, cell (A) and supernatant (B) samples were collected separately. BTV progeny productions were evaluated using the standard plaque assay. In a separate experiment, the plaque reduction assay (C) was carried by mixing C003 or C052 with the overlay and applied directly to BTV-infected cells. At 72 h.p.i, plaques were counted and analyzed. Each data points represented the average values and SD from of three independent replicates.
The inhibition of C003 and C052 to BTV virus progeny production was further confirmed using the plaque reduction assay. C003 or C052 at different concentration were mixed with the agrose overlay and applied to BTV infected cells (MOI of 0.01) directly. The inhibition of BTV plaque formation was then examined at 72 h.p.i. The effects of C003 and C052 on the BTV plaque reduction were in a dose-dependent fashion. At 72 h.p.i., a one-log reduction of plaque formation was observed, when C003 at 2.5 µM and C052 at 1.25 µM were applied to the BTV infected cells, respectively. When applied at higher concentrations, i.e. 20.0 µM of C003 or C052, to the infected cells, more than two-log of plaque reductions were observed (Fig. 7C). There was no difference in terms of the size or formation of the plaques (results not shown). This result showed that C003 and C052 could reduce the viral plaque formation directly.
Time-of-addition Assay
Since C003/C052 showed no overt toxicity in vitro, yet prevented BTV induced CPE/apoptosis/cell lysis and reduced the virus progeny production, we initiated the mechanism of action studies with the time-of-addition assay to determine the possible stage(s) of viral life-cycle that could be affected by C003 and C052. In order to observed both the enhancement and reduction of virostatic efficacy at different time of addition, we used two concentrations of virostatic compound for both C003 and C052, i.e., the C003 at 1.76 µM and 20 µM, and C052 at 0.27 µM and 2.5 µM, respectively. We first observed that adding virostatic compound C003 or C052 at 1 or 2 hours prior to BTV infection, i.e. −1 and −2 h.p.i., did not change their virostatic efficacies. This result indicated that both C003 and C052 might not act on the early stage of viral life-cycle, such as virus entry (Fig. 8A and B). Interestingly, when the compounds were added to BTV infected cells as later as 24 h.p.i., both C003 and C052 showed similar protection in term of the percentage of viable cells. When added at 32 h.p.i., the percentage of viable cells decreased in both C003 and C052 treatment samples, comparing with their protection in the initial 24 h.p.i., indicating that both C003 and C052 were less protective post certain stage(s) of viral life-cycle. When C003 and C052 were added at 48 h.p.i., both compounds showed no protection to BSR cells from BTV-induced CPE (Fig. 8A and B). Since the first cycle of BTV viral replication usually completed in infected cells within 24 h.p.i., our results suggested that C003 and C052 might not act at the early stage of BTV viral life-cycle, including virus entry and uncoating, but rather at the late stage of BTV viral life-cycle, such as virus replication, packaging, maturation and egress. Meanwhile, it is also possible that these compounds may act on host cellular machineries that were involved during late viral life-cycle.
Figure 8. The time-of-addition assay for C003 (A) and C052 (B).
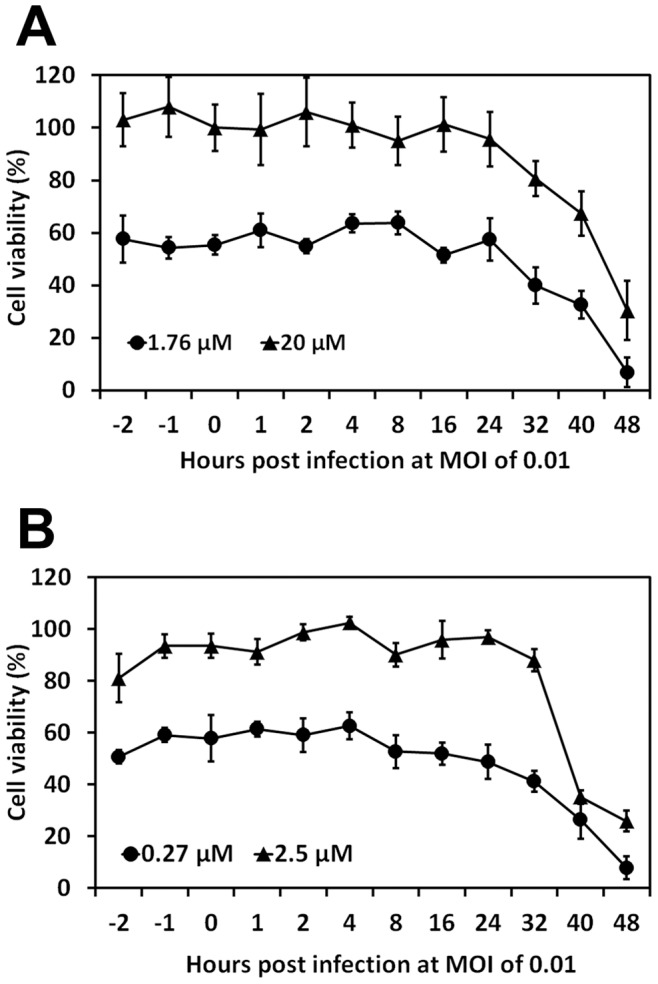
C003 at 20 and 1.76 µM, and C052 at 2.5 µM and 0.27 µM, respectively, were added to BTV infected cells at different h.p.i. as indicated, and the protection of C003 and C052 against BTV induced CPE, or cell viability, was measured using CellTiter-Glo reagent at 72 h.p.i. Each data points represented the average values and SD from of eight independent replicates.
Effect of C003 and C052 on BTV Viral Genomic RNA Replication
To determine whether C003/C052 acts on BTV viral life-cycle directly, we examined the effect of C003/C052 on viral genomic RNA replication in infected cells. BTV viral genomic RNA copies were determined in both cell and supernatant samples using the real time qRT-PCR (Fig. 9A and B). The correspondence of viral genomic RNA replication and virus progeny production in these samples were confirmed by evaluating the amount of infectious virus using standard plaque assay (Fig. 9C and D). Samples were collected post BTV infection and divided into cells and supernatants portion at different time in the initial 24 h.p.i., with and without treatments of C003 (10 µM) and C052 (2.5 µM), respectively. When infected at low MOI (0.01), the viral genomic RNA copies and progeny virus titers obtained from in cell samples represent the events occurred inside the cells for the initial viral-life cycle, including virus entry, replication and maturation within the cells. The viral genomic RNA copies and virus titers in the supernatant samples denotes the events post virus life-cycle inside the cells in the initial 24 h.p.i., including the amount of viruses exited from the cells. At early time points including 3 and 6 h.p.i., there were no differences in viral genomic RNA copies in samples collected from both supernatant and cells, whether treated with C003/C052 or not, indicating that early viral life cycle, such as the entry of BTV into the cells, were not interfered (Fig. 9 A and B). Meanwhile, there is no detectable progeny virus production in all samples (Fig. 9C and D). At 24 h.p.i., there were pronounced reductions, around two-log, of virus genomic RNA copies and virus titers in supernatant samples with C003/C052 treatment, when compared with these in the supernatant samples without C003/C052 treatment. In supernatant samples collected at 24 h.p.i from BTV infected but without C003/C052 treatment, there were 6.17×106 copies of BTV viral genomic RNA, while there were only 4.81×104 or 4.22×104 copies of BTV viral genomic RNA in samples treated with C003 or C052, respectively (Fig. 9B). This result indicated a pronounced effect of these virostatic agents at the late stage of viral life-cycle. However, C003/C052 might also inhibit virus replication directly, since there was a one-log difference in the viral genomic RNA copies and virus titers when comparing samples from infected cells with and without C003/C052 treatments (Fig. 9A).
Figure 9. The effects of C003 and C052 on BTV viral genomic RNA synthesized and progeny virus production at MOI of 0.01.

BTV infected cells were treated with C003 (10 µM) or C052 (2.5 µM), at different h.p.i. as indicated in figures. The viral genomic RNA was extracted and the copy number was determined using real-time qRT-PCR in cell (A) and supernatant (B) samples. The progeny virus titer in cells (C) and supernatants (D) were also analyzed using standard plaque assay.
Effect of C003/C052 on VP6 Expression
To further confirm the effect of C003/C052 on BTV viral life-cycle, we also analyzed viral protein synthesis in the above cell samples infected at MOIs of 0.01, with and without C003 or C052 treatments. VP6 of BTV possesses nucleoside triphosphatase, RNA binding, and helicase activities [9], [11], [38]. For a successful BTV life-cycle, VP6 is an integral part of a transcription complex essential for primary replication [14]. At MOI of 0.01, VP6 was not detectable at 24 h.p.i. in all samples, but was detected at 48 h.p.i. in BTV-infected cells without C003/C052 treatment (Fig. 10). When BTV-infected cells were treated with C003 at10 µM and C052 at 2.5 µM, respectively, there were either very low or no detectable VP6 expression in the cells at 48 h.p.i. (Fig. 10). This result supported our previous observations that C003/C052 reduced viral protein expression when infected at low MOI of 0.01.
Figure 10. Effects of C003 and C052 on BTV VP6 protein expression and LC3-I/LC3-II conversion were analyzed in sample collected at different h.p.i.
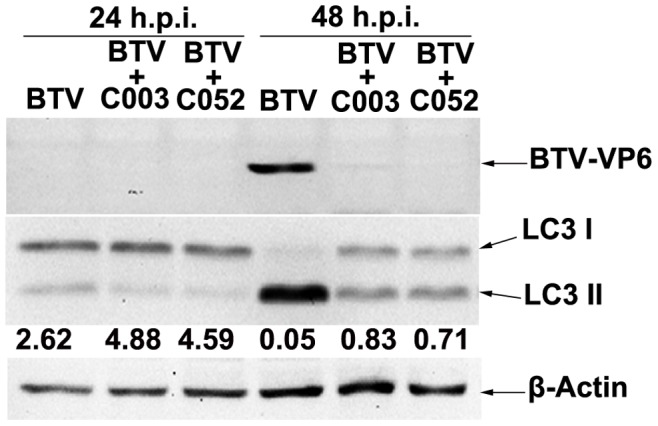
Cells were infected with BTV at MOI of 0.01, and treated with C003 (10 µM) or C052 (2.5 µM). VP6 expressions and LC3-I/LC3-II conversion in these samples were determined using the western blot.
Effect of C003 and C052 on BTV-induced Autophagy
Based on the data presented above, we hypothesized that C003/C052 might act on host machineries/pathways involved in the viral life-cycle, including the autophagic pathway. Autophagy is one of the major pathways for the degradation and turnover of long-lived proteins and organelles in cells during cell starvation or upon various stimuli, such as virus infection [39], [40], [41], [42]. Growing evidences suggest that autophagy actively participates in various pathogenic infections, including the formation of autophagosome for the replication of Influenza A virus [43], [44], [45], hepatitis C virus [46], [47] and Dengue virus [48]. The turnover of microtubule-associated protein light chain 3-I (LC3-I) to LC3-II, or the ratio of LC3-I and LC3-II, is the only biomarker that is reliably associated with the formation of autophagosome. This is also related to virus replication and the leading indicator of virus-induced apoptosis. Since C003 and C052 prevented BTV-induced apoptosis, we examined the activation and formation of autophagy in BTV-infected cells, with and without virostatic compound treatment. When infected at MOI of 0.01, autophagy was not activated at 24 h post BTV infection as the level of LC3-II showed no difference in sample with or without C003/C052 treatment. However, at 48 h.p.i., there was a turnover of LC3-I to LC3-II in BTV infected cells without C003/C052 treatment, with the LC3-I/LC3-II ratio at 0.05 which indicating most of LC3-I was cleaved into LC3-II (Fig. 10). The turnover of LC3-I to LC3-II was at a much lower level when C003 at 10µM or C052 at 2.5 µM was added to the BTV-infected cells, with the ratio of LC3-I/LC3-II at 0.83 and 0.71 respectively (Fig. 10). This result suggested that C003/C052 might act on interfering with BTV induced autophagy, consequently, inhibiting BTV-induced CPE.
Discussion
As a result of its economic significance and as a model system, BTV has been the subject of extensive molecular, genetic and structural studies, and now represents one of the most well characterized viruses [7], [10], [11], [15]. Much of the efforts were focused on developing vaccines against BTV, including the attenuated BTV and VLPs [18], [49], [50]. In contrast, it remains under-investigated to utilize the knowledge from the molecular and virological studies for antiviral drug discovery. Here, we reported the characterization of a novel class of compound as virostatic agents against BTV. These compounds, i.e. the thiophene derivatives, have been identified as active compounds in various assays, including as an enhancer of survival motor neuron protein splice variant expression; an activator for alpha-synuclein 5′UTR - 5′-UTR binding, and for STAT1 activation [51]. It is plausible that the aminothiophenecarboxylic acid derivative–C003 was identified as a novel compound against BTV, with potent virostatic efficacy, low cytotoxicity and high SI. More interestingly, the preliminary SAR analysis of C003 leads to an improved virostatic compound–C052, via de novo synthesis. Our studies suggest that using C003/C052 as lead compounds, it is possible to develop novel and potent antivirals against BTV via chemical modification, while maintain its low toxicity and high selectivity.
For the majority of RNA viruses, the induction of apoptosis, which facilitates the virus dissemination, is an essential step in the late stage of viral life-cycle. Interfering the host apoptotic machinery, especially inhibiting the virus induced apoptosis, will disrupt the successful viral life-cycle via blocking virus release [52], [53], [54]. Thus, host apoptotic response post virus infection is an attractive target for antiviral drug development because it is a common and indispensible late step for a productive viral life-cycle of many, if not all, viruses. Previous studies have shown that BTV infection triggers apoptosis via both intrinsic and extrinsic pathways [1], [37], [55]. The induction of apoptosis is involved in BTV-induced pathogenesis in vivo [56], [57]. Our mechanism of action studies indicated that C003 and C052 were active by protecting cells from BTV-induced apoptosis. The inhibition of caspsse-3/7 activation indicated the targets should be in the upstream of the cascade of caspase activation. However, when BSR cells were treated with pan-caspase inhibitor, including Z-VAD-FMK, cells were not protected from BTV-induced apoptosis. BTV-induced CPE was observed, and BTV replication was not interrupted (results not shown). This suggested that while apoptosis/CPE was inhibit by C003/C052, the mechanism of action of these virostatic compounds might not directly related to BTV-induced apoptotic pathways, rather as a results of inhibiting a late step in viral life-cycle or host machineries inducing upstream apoptotic signals.
Interestingly, our results showed that the host autophagy machinery/pathway was inhibited by C003 and/or C052, as LC3-II, the only reliable autophagy marker, was down-regulated when BTV-infected cells were treated with these compounds (Fig. 10). Autophagy is a highly conserved process in eukaryotes in which the cytoplasm, including excess or aberrant organelles, is sequestered into double-membrane vesicles and delivered to the degradative organelle, the lysosome/vacuole, for breakdown and eventual recycling of the resulting macromolecules [44], [47], [58]. Autophagy is activated as an adaptive response to a variety of extracellular and intracellular stimuli, including nutrient deprivation, hormonal or therapeutic treatment, aggregated and misfolded proteins, damaged organelles and microbial infection [58], [59], [60]. Although the autophagy pathway might be a host defense mechanism, different viruses have developed various strategies to counteract these antiviral mechanisms, and/or to utilize the autophagy machinery as proviral host factors favoring viral replication [46], [61]. Recently, a growing list of viruses has been shown to be targeted for autophagic degradation [62]. For instance, autophagy is actively involved in influenza A virus replication by triggering autophagosome formation, increasing the level of LC3-II and enhanced autophagic flux [45]. Dengue virus can also activate autophagic machinery that is favorable for viral replication [48], [63], and autophagosomes act as a site for translation and replication of dengue virus-2 and that its entry and replication are linked through an ongoing association with membranes of an endosomal–autophagosomal lineage [64], [65]. Similarly, induction of autophagy pathways can enhance the replication of poliovirus and coronavirus [66], [67]. During the infection of dsRNA virus rotavirus, nonstructural protein 4 (NSP4) colocalized with LC3 in cap-like structures associated with viroplasms, the site of nascent viral RNA replication, suggesting a possible new mechanism for the involvement of NSP4 in virus replication [68], [69]. Our finding showed that C003 and C052 interacted with autophagy machinery, which may lead to a new research avenue to develop novel antivirals intervene virus-autophagy interactions.
While virus entry and virus replication have been targets for extensive antiviral drug discovery and development, egress/dissemination of a newly synthesized progeny virus, equally important in relation to successful viral infection, receives much less attentions [70]. This is mainly due to the lack of information on the mechanism of egress/dissemination. Interestingly, further studies showed that autophagy may assist poliovirus egress. During Poliovirus and Coxsackievirus B3 infection, the co-localization of non-structural proteins with autophagy markers LC3-II, together with gene silencing and pharmacological experiments, indicate that autophagy proteins enhance viral replication and progeny virus yields. In BTV, the viral NS3 may act like the membrane protein of enveloped viruses and is responsible for intracellular trafficking and budding of virus particles [71]. Virostatics targeting virus egress would be predicted to prevent or slow down virus release and spread in an infected host, thus allowing more time for the individual’s immune system to respond and control the infection [72]. Further studies on the role of autophagy during BTV progeny virus egress/dissemination may help to understand the exact mechanism of action for C003/C052.
Our results extend our previous findings that showed cluster of lead compounds that prevent BTV induced CPE [34]. While further studies might be needed to define the exact mechanism of action for the virostatic agents C003/C052, our studies showed that these compounds may not interact with host apoptotic machinery, rather posses virostatic activity which block various signals to induce host apoptosis. The finding that host autophagy was involved in BTV life-cycle and C003/C052 might act on host autophagic machinery may open a new avenue to identify and develop novel small molecule antivirals that specifically interact with host autophagy machinery.
Materials and Methods
Cells, Virus Stock and Use of Compounds
BSR cells [73], a cloned derivative of baby hamster kidney (BHK) cells, were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA), supplemented with 5% fetal calf serum (FCS), 100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen). For all assays, unless otherwise noted, cells were plated in DMEM with optimized conditions as previously determined, including 1% FCS [34]. All cells were incubated and grown at 37°C, 5% CO2. The type 10 BTV (BTV-10) was plaque purified and propagated as described previously [1].
BTV-10 was titrated by standard plaque assay, stocked at 2.5×107 PFU/ml and further used for infection with the indicated multiplicity of infection (MOI). For virus replication, cell monolayers were washed with FCS-free growth medium and then incubated with viruses at the indicated MOI. Virus adsorptions were carried out for 2 h at 37°C, 5% CO2, followed by incubation in growth medium supplemented with 1% FCS. At various hours post infection (h.p.i.), cells and supernatants were collected. Cells were sonicated for 40 s and cell debris were removed by low-speed centrifugation (2,500 g). The virus titers in the resulting supernatants were determined on BSR cells at 37°C, 5% CO2, using a standard plaque assay as described previously [74].
CPE-based Cell Viability Assay
The CPE-based cell viability assays, using the CellTiter-Glo® Luminescent Cell Viability kit (Promega Inc., Madison, WI), were carried out as described previously [34]. Cell viability was determined at indicated time points using CellTiter-Glo reagents following manufacturer’s instruction (Promega). Briefly, CellTiter-Glo buffer and the lyophilized CellTiter-Glo substrate (Promega) were thawed and equilibrated to room temperature prior to use. The homogeneous CellTiter-Glo reagent solution was reconstituted after mixing the lyophilized enzyme/substrate and the buffer reagent according to the manufacturer’s instructions. Meanwhile, the assay plates were also equilibrated to room temperature for 15 min. An equal volume (25 µl) of CellTiter-Glo reagents was added to each well by a MicroFlo select dispenser (BioTek, Winooski, VT). After incubated for 15 minutes at room temperature, luminescence signals were measured using Synergy-II multi-mode microplate reader (BioTek) with an integration time of 0.1 s.
The Ten-concentration Dose Response Assay and Cytotoxicity Assays
The virostatic efficacy assay for each compound was carried out under the optimized conditions as described previously [34]. Briefly, BSR cells were seeded into a 384-well black plate (Corning) using the MicroFlo select dispenser (BioTek). After 2 hours incubation at the 37°C, 5% CO2 and 80–95% humidity, ten different concentrations of each compound were serially diluted and added to each well. Cells were then infected with BTV at MOI of 0.01 via the Bio-Tek dispenser. Each compound treatment group, with BTV infection at denoted MOIs, includes eight replicates. Mock infection (cell only) and virus infection only controls were also included as positive and negative controls, respectively. Compound cytotoxicity assay were carried out in the same fashion, except without BTV infection. After incubated at 37°C, 5% CO2 and 80–95% humidity for 72 h, cell viability was determined using CellTiter-Glo reagents as described previously.
Time-of-Addition Assay
The Time-of-Addition assay was performed to further understand the virostatic activity of the previously identified compounds [34]. Briefly, BSR cells were seeded and incubated at 37°C, 5% CO2 and 80–95% humidity. Cells were then inoculated with BTV at MOI of 0.01, and incubated at the optimized conditions as described previously [34]. Selected virostatic compounds were diluted to the indicated concentrations and added to each well at different time pre- or post- BTV infection. The denoted −2 and −1 h.p.i. indicate that BSR cells were incubated with the selected virostatic compounds prior to BTV infection. For 0 h.p.i., the compound and BTV were added to the culture simultaneously. Treated cells were incubated at 37°C, 5% CO2 with 80–95% humidity. In parallel, cells without compounds and virus treatments were included as the mock infection control. Cells with compound treatment only were served as the control to monitor compound cytotoxicity. Cells with BTV infection but without compound treatment were also included as the virus infection only control. Each BTV and compound treatment group includes eight replicates. After incubated for 72 h, cell viability was determined using CellTiter-Glo reagent as described above. The ability of virostatic compound in inhibiting BTV-induced CPE were normalized using controls designated in the assay, where cell viability in the mock infected cells was designated as 100%, and cell viability in BTV infected cells as 0%.
The Caspase-Glo® 3/7 Apoptosis Assay
The Caspase-Glo® 3/7 apoptosis assay was accomplished using the Caspase-Glo® 3/7 assay kit (Promega) following the manufacturer’s instruction. Cell plating and incubation conditions, as well as the plate layout, were similar to that used in the CPE-based assay. BTV infection and compound addition were also in the similar fashion as described in the CPE-based virostatic efficacy assay. Caspase-Glo® 3/7 buffer and lyophilized Caspase-Glo® 3/7 substrate were equilibrated to room temperature before use. The Caspase-Glo® 3/7 buffer was transferred into the amber bottle containing Caspase-Glo® 3/7 substrate, and then mix by swirling or inverting the contents until the substrate is thoroughly dissolved to form the Caspase-Glo® 3/7 reagent. The premixed Caspase-Glo 3/7 reagents (Promega) were added at 1∶1 ratio (reagent : media) to the treated cells at 48 h.p.i., or otherwise noted in the text. After 40 minutes incubation in the dark at room temperature, luminescent signals were read with the Synergy-II multimode microplate reader (BioTek).
Real-time Quantitative Reverse Transcriptase PCR (qRT-PCR)
Total RNA was extracted from cells and supernatants as described for TRIzol LS Reagent (Invitrogen; Carlsbad, CA) according to the manufacturer’s instructions. Dried RNA pellets were re-suspended in 50 µl DEPC treated-water and stored at −80°C before RT- PCR amplification. Following the manufactures’ instruction, the real-time qRT-PCR was carried out using the “one step SRBY Ex Taq qRT-PCR" kit (Takara, Japan). The real-time qRT-PCR was performed in a single tube and amplified products were monitored in real-time using the 7500 Fast Real-Time PCR System (AB applied biosystems). Two BTV-specific primers were designated and used for the real-time qRT-PCR:
Forward primer: 5′-GATTGATGTTTACAGGGATGAGGT-3′
Reverse primer: 5′-TCTTCCTCTGCTTGCGTCCT- 3′
SDS-PAGE and Western Blot Analysis
Samples for SDS-PAGE and western blot analysis were prepared after boiled in protein dissociation buffer [10% (v/v) β-mercaptoethanol, 10% (wt/vol) SDS, 25% (v/v) glycerol, 10 mM Tris-HCl, pH 6.8), and 0.02% (wt/vol) bromophenol blue] for 10 min. Protein samples were loaded in 1× sample buffer and electrophoresis carried out on 12.5% SDS-polyacrylamide gels. Proteins were electroblotted on to Immobilon-P poly-vinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA) at 100 mA for 2 h by using a semidry electrotransfer apparatus. The PVDF membranes were immediately placed into 5% skimmed milk in TBS−0.1% Tween-20 (TBST) and incubated overnight. This step was followed by 1 h incubation in primary antibody diluted with 3% BSA in TBS. Mouse monoclonal anti-VP6 [75], mouse monoclonal anti-human LC3-II (Abgent, Inc., San Diego, CA) were used at the concentrations recommended by the manufacturers. Blots were washed three times for 10 min each in TBST and then incubated for 1 h in horseradish peroxidase-conjugated secondary antibody diluted in 3% BSA–TBST. Goat anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase (BD Biosciences, San Jose, CA) were used at 1∶5,000. Bound antibodies were detected by an enhanced chemiluminescence kit (Amersham, Piscataway, NJ, USA).
Plaque Reduction Assay
The plaque reduction assay was carried out in the 24-well plates by including compounds in the overlay agarose, which was applied directly to the BTV infected cell culture as described previously [76], [77]. Cells were initially inoculated with BTV at ten-fold series dilution with known initial BTV titer. After absorbing for 2 h at 37°C, 5% CO2 with 80–95% humidity, the inoculation was removed, washed with media and replaced by 1% agarose mixed with compound at the indicated concentration, ranging from 20.0 µM to 0.16 µM. Each concentration was triplicated. At 72 h.p.i., the number of plaques formed at different compound concentration was counted and the final virus titer was determined accordingly.
De novo Compound Synthesis
The structure and modification of C003 were illustrated in Figure 2 and 3. The preparation of intermediate compound 4 (IC4) followed an adaptation of the Gewald reaction [78] beginning with commercially available compound 4-methylacetophenone. IC7 and IC8 were achieved by modification of the Horner-Wadsworth-Emmons procedure [79] on commercially available 2-thiophenecarboxaldehyde and 2-pyridinecarboxaldehyde followed by a standard acid chloride formation with thionyl chloride. Combining IC4 with IC7 and IC8 was pursued as described in the literature [79], [80] for related systems. Standard saponification of the resultant IC9 and IC10 produced IC2b and IC3b that became the final products C052 (IC2a) and C055 (IC3a) upon routine acidification (Fig. 3). All compounds were characterized by high field NMR (Bruker AM 400) and high resolution mass spectral (Waters Q Premier API Quadrupole TOF Tandem mass spectrometer) analyses with purity confirmed by micro analytical elemental determinations.
Acknowledgments
We thank the technical assistances from Ms. Pulin Che and Mr. Hairui Su as assistant students during the course of the work.
Funding Statement
This project was supported by grant 1R03MH08127-01 and 7R03MH08127-02 from NIH to Q. Li, and by the IMPACT funds from Department of Medicine at University of Alabama to Q. Li. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Li Q, Li H, Blitvich BJ, Zhang J (2007) The Aedes albopictus inhibitor of apoptosis 1 gene protects vertebrate cells from bluetongue virus-induced apoptosis. Insect Mol Biol 16: 93–105. [DOI] [PubMed] [Google Scholar]
- 2. Schwartz-Cornil I, Mertens PP, Contreras V, Hemati B, Pascale F, et al. (2008) Bluetongue virus: virology, pathogenesis and immunity. Vet Res 39: 46. [DOI] [PubMed] [Google Scholar]
- 3. Forzan M, Marsh M, Roy P (2007) Bluetongue virus entry into cells. J Virol 81: 4819–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mellor PS, Wittmann EJ (2002) Bluetongue virus in the Mediterranean Basin 1998–2001. Vet J 164: 20–37. [DOI] [PubMed] [Google Scholar]
- 5. Paweska JT, Venter GJ, Mellor PS (2002) Vector competence of South African Culicoides species for bluetongue virus serotype 1 (BTV-1) with special reference to the effect of temperature on the rate of virus replication in C. imicola and C. bolitinos. Med Vet Entomol 16: 10–21. [DOI] [PubMed] [Google Scholar]
- 6. Mellor PS, Carpenter S, Harrup L, Baylis M, Mertens PP (2008) Bluetongue in Europe and the Mediterranean Basin: history of occurrence prior to 2006. Prev Vet Med 87: 4–20. [DOI] [PubMed] [Google Scholar]
- 7. Mertens PP, Diprose J, Maan S, Singh KP, Attoui H, et al. (2004) Bluetongue virus replication, molecular and structural biology. Vet Ital 40: 426–437. [PubMed] [Google Scholar]
- 8. MacLachlan NJ, Conley AJ, Kennedy PC (2000) Bluetongue and equine viral arteritis viruses as models of virus-induced fetal injury and abortion. Anim Reprod Sci 60–61: 643–651. [DOI] [PubMed] [Google Scholar]
- 9. Roy P (2005) Bluetongue virus proteins and particles and their role in virus entry, assembly, and release. Adv Virus Res 64: 69–123. [DOI] [PubMed] [Google Scholar]
- 10. Roy P (2008) Functional mapping of bluetongue virus proteins and their interactions with host proteins during virus replication. Cell Biochem Biophys 50: 143–157. [DOI] [PubMed] [Google Scholar]
- 11. Roy P (2008) Bluetongue virus: dissection of the polymerase complex. J Gen Virol 89: 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyce M, Celma CC, Roy P (2008) Development of reverse genetics systems for bluetongue virus: recovery of infectious virus from synthetic RNA transcripts. J Virol 82: 8339–8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyce M, Roy P (2007) Recovery of infectious bluetongue virus from RNA. J Virol 81: 2179–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsuo E, Roy P (2009) Bluetongue virus VP6 acts early in the replication cycle and can form the basis of chimeric virus formation. J Virol 83: 8842–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maclachlan NJ, Drew CP, Darpel KE, Worwa G (2009) The pathology and pathogenesis of bluetongue. J Comp Pathol 141: 1–16. [DOI] [PubMed] [Google Scholar]
- 16. Szmaragd C, Wilson AJ, Carpenter S, Wood JL, Mellor PS, et al. (2010) The spread of bluetongue virus serotype 8 in Great Britain and its control by vaccination. PLoS One 5: e9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De A, Batabyal S, Biswas SK, Chand K, Singh RK, et al. (2009) Surveillance of bluetongue virus antibody in goats using a recombinant VP7-based indirect ELISA in the coastal saline area of West Bengal, India. Vet Ital 45: 339–346. [PubMed] [Google Scholar]
- 18. Bhanuprakash V, Indrani BK, Hosamani M, Balamurugan V, Singh RK (2009) Bluetongue vaccines: the past, present and future. Expert Rev Vaccines 8: 191–204. [DOI] [PubMed] [Google Scholar]
- 19.Wackerlin R, Eschbaumer M, Konig P, Hoffmann B, Beer M (2010) Evaluation of humoral response and protective efficacy of three inactivated vaccines against bluetongue virus serotype 8 one year after vaccination of sheep and cattle. Vaccine. [DOI] [PubMed]
- 20. Dungu B, Potgieter C, Von Teichman B, Smit T (2004) Vaccination in the control of bluetongue in endemic regions: the South African experience. Dev Biol (Basel) 119: 463–472. [PubMed] [Google Scholar]
- 21. Santman-Berends IM, Hage JJ, van Rijn PA, Stegeman JA, van Schaik G (2010) Bluetongue virus serotype 8 (BTV-8) infection reduces fertility of Dutch dairy cattle and is vertically transmitted to offspring. Theriogenology 74: 1377–1384. [DOI] [PubMed] [Google Scholar]
- 22. Saegerman C, Mellor P, Uyttenhoef A, Hanon JB, Kirschvink N, et al. (2010) The most likely time and place of introduction of BTV8 into Belgian ruminants. PLoS One 5: e9405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson AJ, Mellor PS (2009) Bluetongue in Europe: past, present and future. Philos Trans R Soc Lond B Biol Sci 364: 2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vandenbussche F, De Leeuw I, Vandemeulebroucke E, De Clercq K (2009) Emergence of bluetongue serotypes in Europe, part 1: description and validation of four real-time RT-PCR assays for the serotyping of bluetongue viruses BTV-1, BTV-6, BTV-8 and BTV-11. Transbound Emerg Dis 56: 346–354. [DOI] [PubMed] [Google Scholar]
- 25. Mehlhorn H, Walldorf V, Klimpel S, Schaub G, Kiel E, et al. (2009) Bluetongue disease in Germany (2007–2008): monitoring of entomological aspects. Parasitol Res 105: 313–319. [DOI] [PubMed] [Google Scholar]
- 26. Mehlhorn H, Walldorf V, Klimpel S, Schmahl G (2008) Outbreak of bluetongue disease (BTD) in Germany and the danger for Europe. Parasitol Res 103 Suppl 1 S79–86. [DOI] [PubMed] [Google Scholar]
- 27. Meiswinkel R, Goffredo M, Dijkstra EG, van der Ven IJ, Baldet T, et al. (2008) Endophily in Culicoides associated with BTV-infected cattle in the province of Limburg, south-eastern Netherlands, 2006. Prev Vet Med 87: 182–195. [DOI] [PubMed] [Google Scholar]
- 28. Toussaint JF, Sailleau C, Breard E, Zientara S, De Clercq K (2007) Bluetongue virus detection by two real-time RT-qPCRs targeting two different genomic segments. J Virol Methods 140: 115–123. [DOI] [PubMed] [Google Scholar]
- 29. Enserink M (2006) Emerging infectious diseases. During a hot summer, bluetongue virus invades northern Europe. Science 313: 1218–1219. [DOI] [PubMed] [Google Scholar]
- 30. Meroc E, Herr C, Verheyden B, Hooyberghs J, Houdart P, et al. (2009) Bluetongue in Belgium: episode II. Transbound Emerg Dis 56: 39–48. [DOI] [PubMed] [Google Scholar]
- 31.Worwa G, Hilbe M, Chaignat V, Hofmann MA, Griot C, et al.. (2010) Virological and pathological findings in Bluetongue virus serotype 8 infected sheep. Vet Microbiol. [DOI] [PubMed]
- 32. Worwa G, Thur B, Griot C, Hofmann M, MacLachlan JN, et al. (2008) [Bluetongue disease in Swiss sheep breeds: clinical signs after experimental infection with bluetongue virus serotype 8]. Schweiz Arch Tierheilkd 150: 491–498. [DOI] [PubMed] [Google Scholar]
- 33.Jonsson CB, White EL (2007) Launching an HTS campaign to discover new antivirals. European Pharmaceutical Review: 4.
- 34. Li Q, Maddox C, Rasmussen L, Hobrath JV, White LE (2009) Assay development and high-throughput antiviral drug screening against Bluetongue virus. Antiviral Res 83: 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phillips T, Jenkinson L, McCrae C, Thong B, Unitt J (2011) Development of a high-throughput human rhinovirus infectivity cell-based assay for identifying antiviral compounds. J Virol Methods 173: 182–188. [DOI] [PubMed] [Google Scholar]
- 36. Noah JW, Severson W, Noah DL, Rasmussen L, White EL, et al. (2007) A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res 73: 50–59. [DOI] [PubMed] [Google Scholar]
- 37. Mortola E, Larsen A (2009) [Bluetongue virus infection: signaling pathway activated during apoptosis]. Rev Argent Microbiol 41: 134–140. [PubMed] [Google Scholar]
- 38. Stauber N, Martinez-Costas J, Sutton G, Monastyrskaya K, Roy P (1997) Bluetongue virus VP6 protein binds ATP and exhibits an RNA-dependent ATPase function and a helicase activity that catalyze the unwinding of double-stranded RNA substrates. J Virol 71: 7220–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cherra SJ, 3rd, Dagda RK, Chu CT (2010) Review: autophagy and neurodegeneration: survival at a cost? Neuropathol Appl Neurobiol 36: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singletary K, Milner J (2008) Diet, autophagy, and cancer: a review. Cancer Epidemiol Biomarkers Prev 17: 1596–1610. [DOI] [PubMed] [Google Scholar]
- 41. Larsen KE, Sulzer D (2002) Autophagy in neurons: a review. Histol Histopathol 17: 897–908. [DOI] [PubMed] [Google Scholar]
- 42. Wileman T (2006) Aggresomes and autophagy generate sites for virus replication. Science 312: 875–878. [DOI] [PubMed] [Google Scholar]
- 43. Law AH, Lee DC, Yuen KY, Peiris M, Lau AS (2010) Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell Mol Immunol 7: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossman JS, Lamb RA (2009) Autophagy, apoptosis, and the influenza virus M2 protein. Cell Host Microbe 6: 299–300. [DOI] [PubMed] [Google Scholar]
- 45. Zhou Z, Jiang X, Liu D, Fan Z, Hu X, et al. (2009) Autophagy is involved in influenza A virus replication. Autophagy 5: 321–328. [DOI] [PubMed] [Google Scholar]
- 46. Dreux M, Chisari FV (2009) Autophagy proteins promote hepatitis C virus replication. Autophagy 5: 1224–1225. [DOI] [PubMed] [Google Scholar]
- 47. Dreux M, Gastaminza P, Wieland SF, Chisari FV (2009) The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 106: 14046–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heaton NS, Randall G (2010) Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8: 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartram DJ, Heasman L, Batten CA, Oura CA, Plana-Duran J, et al.. (2011) Neutralising antibody responses in cattle and sheep following booster vaccination with two commercial inactivated bluetongue virus serotype 8 vaccines. Vet J. [DOI] [PubMed]
- 50. Hamers C, Galleau S, Chery R, Blanchet M, Besancon L, et al. (2009) Use of inactivated bluetongue virus serotype 8 vaccine against virulent challenge in sheep and cattle. Vet Rec 165: 369–373. [DOI] [PubMed] [Google Scholar]
- 51.Pubchem Bioassay. MLS000673879 - Substance Summary (SID 24798053). http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?sid=24798053&loc=es_rss.
- 52. Clarke TE, Clem RJ (2003) Insect defenses against virus infection: the role of apoptosis. Int Rev Immunol 22: 401–424. [DOI] [PubMed] [Google Scholar]
- 53. Everett H, McFadden G (1999) Apoptosis: an innate immune response to virus infection. Trends Microbiol 7: 160–165. [DOI] [PubMed] [Google Scholar]
- 54. Koyama AH, Irie H, Fukumori T, Hata S, Iida S, et al. (1998) Role of virus-induced apoptosis in a host defense mechanism against virus infection. J Med Invest 45: 37–45. [PubMed] [Google Scholar]
- 55. Stewart ME, Roy P (2010) Role of cellular caspases, nuclear factor-kappa B and interferon regulatory factors in Bluetongue virus infection and cell fate. Virol J 7: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mortola E, Noad R, Roy P (2004) Bluetongue virus outer capsid proteins are sufficient to trigger apoptosis in mammalian cells. J Virol 78: 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nagaleekar VK, Tiwari AK, Kataria RS, Bais MV, Ravindra PV, et al. (2007) Bluetongue virus induces apoptosis in cultured mammalian cells by both caspase-dependent extrinsic and intrinsic apoptotic pathways. Arch Virol 152: 1751–1756. [DOI] [PubMed] [Google Scholar]
- 58. Yorimitsu T, Klionsky DJ (2005) Autophagy: molecular machinery for self-eating. Cell Death Differ 12 Suppl 2 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fimia GM, Piacentini M (2009) Toward the understanding of autophagy regulation and its interplay with cell death pathways. Cell Death Differ 16: 933–934. [DOI] [PubMed] [Google Scholar]
- 60. Gougeon ML, Piacentini M (2009) New insights on the role of apoptosis and autophagy in HIV pathogenesis. Apoptosis 14: 501–508. [DOI] [PubMed] [Google Scholar]
- 61. Dreux M, Chisari FV Viruses and the autophagy machinery. Cell Cycle 9: 1295–1307. [DOI] [PubMed] [Google Scholar]
- 62. Orvedahl A, Levine B (2009) Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 16: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, et al. (2008) Autophagic machinery activated by dengue virus enhances virus replication. Virology 374: 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khakpoor A, Panyasrivanit M, Wikan N, Smith DR (2009) A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J Gen Virol 90: 1093–1103. [DOI] [PubMed] [Google Scholar]
- 65. Panyasrivanit M, Khakpoor A, Wikan N, Smith DR (2009) Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J Gen Virol 90: 448–456. [DOI] [PubMed] [Google Scholar]
- 66. Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, et al. (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3: e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR (2004) Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 279: 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hyser JM, Utama B, Crawford SE, Estes MK Genetic divergence of rotavirus nonstructural protein 4 results in distinct serogroup-specific viroporin activity and intracellular punctate structure morphologies. J Virol 86: 4921–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, et al. (2006) Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol 80: 6061–6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roy P, Noad R (2006) Bluetongue virus assembly and morphogenesis. Curr Top Microbiol Immunol 309: 87–116. [DOI] [PubMed] [Google Scholar]
- 71. Celma CC, Roy P (2009) A viral nonstructural protein regulates bluetongue virus trafficking and release. J Virol 83: 6806–6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Harty RN (2009) No exit: targeting the budding process to inhibit filovirus replication. Antiviral Res 81: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Buchholz UJ, Finke S, Conzelmann KK (1999) Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mertens PP, Burroughs JN, Anderson J (1987) Purification and properties of virus particles, infectious subviral particles, and cores of bluetongue virus serotypes 1 and 4. Virology 157: 375–386. [DOI] [PubMed] [Google Scholar]
- 75. Kar AK, Roy P (2003) Defining the structure-function relationships of bluetongue virus helicase protein VP6. J Virol 77: 11347–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Landry ML, Stanat S, Biron K, Brambilla D, Britt W, et al. (2000) A standardized plaque reduction assay for determination of drug susceptibilities of cytomegalovirus clinical isolates. Antimicrob Agents Chemother 44: 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hayden FG, Cote KM, Douglas RG Jr (1980) Plaque inhibition assay for drug susceptibility testing of influenza viruses. Antimicrob Agents Chemother 17: 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sabnis RW, Rangnekar DW, Sonawane ND (1999) 2-aminothiophenes by the Gewald reaction. Journal of Heterocyclic Chemistry 36: 333–345. [Google Scholar]
- 79. El-Batta A, Jiang CC, Zhao W, Anness R, Cooksy AL, et al. (2007) Wittig reactions in water media employing stabilized ylides with aldehydes. Synthesis of alpha,beta-unsaturated esters from mixing aldehydes, alpha-bromoesters, and Ph3P in aqueous NaHCO3. Journal of Organic Chemistry 72: 5244–5259. [DOI] [PubMed] [Google Scholar]
- 80. Touchard FP (2004) New and efficient conditions for the Z-selective synthesis of unsaturated esters by the Horner-Wadsworth-Emmons olefination. Tetrahedron Letters 45: 5519–5523. [Google Scholar]