

Summary
Background
von Willebrand Factor (VWF) is a glycoprotein that plays an important role in primary hemostasis. VWF is synthesized and stored in endothelial cells (ECs) and megakaryocytes/platelets. Plasma VWF is primarily derived from ECs and is generally believed to be essential for hemostasis. VWF synthesized in megakaryocytes is stored in platelet α-granules from which it is released following platelet activation. The relative contribution of VWF stored in ECs or megakaryocytes/platelets, or present in plasma to hemostasis is not clear.
Objectives
We investigated whether EC-derived VWF plays the major role in hemostasis while the contribution of platelet-derived VWF is negligible, or if platelet-derived VWF also significantly contributes to hemostasis.
Methods and Results
Mice expressing VWF only in ECs (EC-VWF) or platelets (Plt-VWF) were created by reciprocal bone marrow transplantation between C57BL/6J (WT) and VWF knockout mice (VWF−/−). Plasma VWF levels in EC-VWF were similar to WT. Plt-VWF mice had a trace amount of VWF in their plasma while VWF levels in platelet lysate were comparable to WT. Tail bleeding time was normal in EC-VWF. Interestingly, Plt-VWF showed partially corrected bleeding time and significantly decreased blood loss volume compared to VWF−/−. Adhesion of platelets perfused over immobilized collagen under shear stress was significantly higher in both EC-VWF and Plt-VWF compared to VWF−/−.
Conclusion
VWF synthesized in ECs is sufficient to support hemostasis in VWF−/− mice, and VWF produced in megakaryocytes/platelets can also contribute to hemostasis in the absence of EC-derived VWF.
Keywords: bone marrow transplantation, endothelial cells, platelets, von Willebrand Disease, von Willebrand Factor
Introduction
von Willebrand Factor (VWF) is a large, multimeric glycoprotein that mediates platelet adhesion to subendothelium at sites of vascular injury. Deficiency or dysfunction of VWF causes a bleeding disorder known as von Willebrand disease (VWD) [1]. VWF synthesized in endothelial cells (ECs) is either constitutively secreted into plasma or stored in Weibel-Palade bodies (WPB) [2, 3]. In addition to EC production, approximately 15% of total VWF is produced and stored in α-granules of platelets and megakaryocytes, although very little is thought to be released into plasma until platelet activation [1]. Therefore, plasma VWF is primarily derived from endothelial synthesis, and is generally believed to play the major role in hemostasis. However, clinical observations have suggested that platelet VWF is also important to control bleeding symptoms of VWD patients. Type 1 VWD patients having normal levels of platelet VWF (“platelet normal” subgroup) are described to have almost normal bleeding times in contrast to the prolonged bleeding times found in patients with low platelet VWF [4–6], and this may include a group of patients having decreased VWF survival (VWD type 1C) [7]. Platelets derived from “platelet normal” type 1 VWD patients have also been shown to adhere normally to collagen ex vivo [8, 9]. Similarly type 3 VWD patients that have platelet VWF have been suggested to have milder clinical manifestations [10]. Moreover, in type 3 VWD patients, VWF replacement therapy sometimes does not normalize bleeding and platelet transfusion is beneficial, suggesting that platelet-derived VWF is important for hemostasis [11–13]. On the other hand, although normal dogs do not have VWF in platelets, they do not exhibit a bleeding diathesis [14]. This has added to the perception that the role of platelet VWF is less important than plasma VWF. Thus, a role for platelet-derived VWF in hemostasis has yet to be fully clarified. In this study, we explored the contributions of endothelial and platelet-derived VWF in hemostasis using reciprocal bone marrow transplantation (BMT) between VWF knockout (VWF−/−) and C57BL/6J (WT) mice [15].
Materials and Methods
Animal procedures
Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. All mice used in this study were on the C57BL/6 background, and isoflurane (Phoenix Pharmaceuticals, St. Joseph, MO, USA) or 2.5% tribromoethanol (Sigma-Aldrich, St Louis, MO, USA) were used for anesthesia when necessary. Bone marrow (BM) cells were collected from femurs and tibia of mice, and bone marrow mononuclear cells (BMMNCs) were isolated using ficoll as described previously [16]. Six- to 8-week-old VWF−/− or WT mice (recipients) were conditioned with a lethal dose of 1100 cGy total body irradiation using a Gammacell 40 Exactor cesium irradiator (Best Theratronics Ltd, Ottawa, Ontario, Canada). Twenty-four hours after irradiation, 1 × 107 BMMNCs were infused by retro-orbital vein injection. Recipients were analyzed beginning at 8 weeks after BMT.
Mouse tail bleeding times were determined as previously described [17]. Briefly, a 3 mm portion of distal tail tip was removed using a sterile scalpel blade and the wounded tail was immersed in isotonic saline maintained at 37°C. The point at which complete cessation of visible blood flow occurred was defined as the bleeding time. When bleeding did not cease within 10 minutes, the tail was cauterized and bleeding time was recorded as 600 seconds.
For VWF release studies, mice were infused with epinephrine (American Regent, Shirley, NY, USA) by subcutaneous injection at a dose of 0.5 mg kg−1 body weight. In preliminary experiments, a significant increase of plasma VWF was observed in WT mice following epinephrine administration at doses of 0.5 mg kg−1 body weight and higher. Blood samples were collected before and 30 minutes after infusion.
To reconstitute a low-level of plasma VWF in VWF−/− mice, blood was collected from FVIII-deficient mice (F8−/−) or WT mice using D-Phe-Pro-Arg-chloromethylketone (PPACK) (Calbiochem, La Jolla, CA, USA) at a final concentration of 50 μM as an anticoagulant. VWF:Ag in pooled plasma was measured by ELISA and the amount required to reconstitute 100 mU mL−1 of plasma VWF (calculated by body weight, diluted in saline) was injected into VWF−/− mice. The final concentration of PPACK contained in 200 μl of plasma (the volume injected varied slightly depending on body weight) was 10μM (1 μg in total). Injection of 10 μg of PPACK had been confirmed to not prolong the bleeding time of WT mice. Fifteen minutes after retro-orbital plasma infusion, 50μl of blood was collected to measure plasma VWF levels and tail bleeding assays were performed.
Ex vivo perfusion assay
Platelet interaction with immobilized type I collagen (Chrono-Log, Havertown, PA, USA) was assessed using the VenaFlux Platform (Cellix, Dublin, Ireland). Type I collagen (50 μg mL−1) was coated on Vena8Fluor+Biochips overnight at 4°C in a humid chamber. The channels were blocked with 3% bovine serum albumin (BSA) in Hanks’ balanced salt solution (HBSS) for 1 hour at room temperature. Mouse blood was drawn from the vena cava using PPACK (50 μM final) and heparin (50 mU mL−1 final) as anticoagulants. Platelets were labeled in whole blood by addition of mepacrine to a final concentration of 40 μM (Quinacrine dihydrochloride) (Calbiochem, La Jolla, CA, USA). Perfusion assays were performed at a shear rate of 2000 s−1. Microfluidic chambers were observed using a Hamamatsu Orca R2 camera (Hamamatsu Photonics, Bridgewater, NJ, USA). The area occupied by platelet thrombi was calculated using Metamorph software (Molecular Devices, Sunnyvale, CA, USA).
VWF and FVIII analysis
VWF antigen (VWF:Ag) was measured by enzyme-linked immunosorbent assay (ELISA) using anti-mouse VWF monoclonal antibody (344.3, produced in our lab) as capture antibody and biotinylated anti-human VWF polyclonal antibody (DAKO, Carpinteria, CA, USA) as detection antibody. Normal pooled plasma from C57BL/6J WT mice was used as a reference and defined as 1 U mL−1. For analysis of VWF:Ag in platelets, washed platelets were prepared from citrated whole blood (0.38% final concentration). Whole blood was diluted 1:2 with modified Tyrode’s buffer (20mM HEPES pH7.4, 137mM NaCl, 2.5mM KCl, 5.5mM glucose, 0.25% BSA) containing 50ng mL−1 Prostaglandin E1 (PGE1) (Sigma-Aldrich), centrifuged at 150 × g for 5 minutes at room temperature (RT), and platelet-rich plasma (PRP) was collected. Platelets were washed by sedimentation at 800 × g for 5 minutes in modified Tyrode’s buffer containing PGE1 and lysed in phosphate-buffered saline (PBS) containing 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS). Plasma FVIII activity (FVIII:C) was quantified using the Coatest VIII:C/4 Kit (DiaPharma, Franklin, OH, USA) as previously described [18]. Plasma samples were diluted 1:40 in 1 X Coatest buffer before FVIII:C analysis.
Statistical analysis
Data are presented as mean ± SD. Groups were compared by the unpaired Student t test (Prism, GraphPad Software Inc, San Diego, CA, USA). Probability values <0.05 were considered statistically significant.
Results
Establishment of murine models that produce VWF only in endothelial cells or platelets
To determine the relative roles of EC-derived and platelet-derived VWF, we established model mice producing VWF only in ECs or platelets. WT mouse bone marrow (BM) cells were transplanted into lethally irradiated VWF−/−, creating a “platelet-restricted” VWF model (Plt-VWF). Endogenous VWF−/− BM cells are ablated by lethal dose irradiation and transplanted WT BM cells produce platelets expressing VWF in these recipients. Conversely, VWF−/− BM was transplanted into WT mice, generating an EC-specific VWF model (EC-VWF). EC-VWF mice maintain VWF expression in ECs but BM cells are replaced with VWF−/− mouse BM cells so platelets do not produce VWF.
Plasma VWF levels in EC-VWF (1284 ± 369 mU mL−1, n = 16) were similar to WT (1013 ± 385 mU mL−1, n = 15) (Fig. 1A). EC-VWF platelets did not contain VWF, similar to VWF−/− mice (Fig. 1B). VWF in the platelet lysate of Plt-VWF mice (136 ± 14 mU/108 platelets, n = 7) was comparable to that of WT (139 ± 23 mU/108 platelets, n = 11) (Fig. 1B). Unexpectedly, Plt-VWF mice had a trace amount of VWF in their plasma (46 ± 15 mU mL−1, n = 16). VWF serves as a carrier protein for FVIII and FVIII activity (FVIII:C) is known to be diminished in type 3 VWD patients and in VWF−/− mice [15]. FVIII:C in EC-VWF (582 ± 231 mU mL−1, n = 9) was comparable to that of WT (629 ± 238 mU mL−1, n = 9) (Fig. 1C). FVIII:C in Plt-VWF (133 ± 81 mU mL−1, n = 23) was markedly lower than EC-VWF or WT mice, but was significantly higher than VWF−/− mice (38 ± 20 mU mL−1, n = 13), possibly due to the trace VWF level in the plasma of Plt-VWF mice.
Fig. 1.

Murine models of EC-VWF and Plt-VWF established by crossed BMT. (A) Plasma samples and (B) platelet lysates collected from WT, VWF−/−, EC-VWF, and Plt-VWF mice were analyzed by ELISAto measure VWF antigen levels. EC-VWF mice had WT levels of plasma VWF, but no platelet VWF. Plt-VWF mice had WT levels of platelet VWF and a trace amount of plasma VWF. (C) FVIII activity in mouse plasma was quantified by chromogenic assay. FVIII:C of EC-VWF mice were similar to WT mice. Plt-VWF mouse plasma presented low FVIII:C but the levels were significantly higher than VWF−/− mice. NS, not statistically significant, *P < 0.05
Platelet adhesion on collagen under flow
To determine if platelet VWF contributes to platelet adhesion and thrombus formation on collagen, whole blood samples drawn from WT, VWF−/−, EC-VWF and Plt-VWF were perfused over immobilized type I collagen at a shear rate of 2,000 s−1. Defective thrombus formation was observed with VWF−/−, demonstrating that platelet adhesion and thrombus formation are dependent upon VWF at this high shear rate (Figure 2A) [19]. As expected, the percent of surface area coverage observed with EC-VWF after 2 minutes of perfusion (11.6 ± 3.1%, n = 8) was similar to that of WT (13.3 ± 3.8%, n = 14) (Figure 2B). Interestingly, blood samples derived from Plt-VWF showed significantly improved platelet deposition and thrombus formation compared to VWF−/−. Surface coverage was significantly higher in Plt-VWF compared to VWF−/− (7.7 ± 3.5%, n = 9, vs 1.0 ± 0.4%, n = 5), indicating that platelet VWF plays a role in platelet adhesion to collagen. To demonstrate the contribution of low level plasma VWF, VWF−/− mouse blood samples supplemented with WT mouse plasma to a final VWF concentration of 50 mU ml−1 were perfused over collagen under flow. Surface coverage was slightly higher (3.7 ± 1.5%, n = 3) compared to VWF−/−. Most of the adherent platelets observed were single platelets or small aggregates, rather than the larger sized thrombi observed in Plt-VWF mouse blood. This result suggests that a low level of plasma VWF may mediate initial platelet recruitment, but platelet VWF released locally upon activation is required for subsequent thrombus formation.
Fig. 2.

Platelet adhesion on type I collagen under flow. (A) Platelets in mouse whole blood from WT, VWF−/−, EC-VWF, and Plt-VWF were labeled with mepacrine and perfused over Vena8Fluor+Biochips coated with type I collagen at a shear rate of 2,000 s−1. Images were taken after 120 seconds of perfusion. (B) Percent of surface area covered by platelets after 120 seconds of perfusion was calculated using Metamorph software and plotted for each group of mice. Platelet adhesion of EC-VWF mouse blood samples to type I collagen were similar to WT mice. Surface coverage of Plt-VWF mouse blood samples was significantly higher than VWF−/− mice. *P < 0.05, **P < 0.001
Tail bleeding assay
The hemostatic capacity of Plt-VWF and EC-VWF was also tested in vivo using a tail bleeding time assay. The bleeding time of WT mice (n = 7) was 2.7 ± 1.2 minutes while no VWF−/− mice stopped bleeding within 10 minutes (n = 8) (Fig. 3A) [15]. Bleeding time was normal in EC-VWF (2.3 ± 1.1 minutes, n = 11) and Plt-VWF had a partially corrected bleeding time (7.8 ± 3.3 minutes, n = 16) with bleeding stopped within 10 minutes for 6 out of 16 mice tested. Blood loss volume was significantly reduced in both EC-VWF and Plt-VWF (43.7 ± 34.2 μl, n = 11 and 102.1 ± 90.1 μl, n = 16) compared to VWF−/− (239.3 ± 177.4 μl, n = 8) (Fig. 3B). These results indicate that VWF produced by ECs alone is sufficient to support hemostasis. Platelet VWF can also contribute to local hemostasis in the absence of EC-derived VWF, albeit somewhat less effectively.
Fig. 3.

Tail bleeding assays. (A) WT, VWF−/−, EC-VWF, and Plt-VWF were analyzed by tail bleeding time assays 10 weeks after transplantation. (B) The amount of blood lost into collection tubes was determined by measuring hemoglobin content. Bleeding time and blood loss volume were normal in EC-VWF. Plt-VWF had a partially corrected bleeding time and significantly reduced blood loss volume compared to VWF−/−. *P < 0.05
Since Plt-VWF mice had normal VWF in platelets and very low but detectable VWF in plasma, we questioned whether improvement of bleeding phenotype in Plt-VWF mice is due to platelet-stored VWF or the low level of plasma VWF. To answer this question, plasma collected from FVIII−/− mice (as a VWF source) was intravenously injected into VWF−/− mice to reconstitute plasma VWF to a level similar to Plt-VWF (132 ± 35 mU mL−1, n=9). None of these injected mice stopped tail bleeding within 10 minutes. To investigate the contribution of the low levels of plasma FVIII observed in Plt-VWF mice, WT mouse plasma was infused into VWF−/− mice. Plasma FVIII (133 ± 53 mU mL−1, n=5) as well as VWF levels (83 ± 27 mU mL−1, n=5) were reconstituted to levels similar to or slightly higher than Plt-VWF mice. None of the tested animals stopped tail bleeding within 10 minutes. Together, these experiments suggest that platelet-stored VWF is the significant contributor to hemostasis in Plt-VWF mice.
Epinephrine releases VWF in EC-VWF but not in Plt-VWF
VWF is stored in EC WPB and platelet α-granules for regulated release [20]. Administration of desmopressin (DDAVP) releases VWF from ECs in humans but not in mice. To examine the releasable pool of VWF in mice, epinephrine was administered [21]. Blood samples were collected from WT, EC-VWF, and Plt-VWF mice before and 30 minutes after subcutaneous injection of epinephrine at a dose of 0.5mg kg−1. The levels of plasma VWF increased 1.8 ± 0.4-fold (n = 4) and 2.0 ± 0.8-fold (n = 4) in WT and EC-VWF mice, respectively, confirming an epinephrine-releasable pool of VWF in WPB of ECs (Figure 4). There was not a significant increase in the plasma VWF levels of Plt-VWF mice after infusion. These results show that EC-VWF mice have epinephrine-releasable VWF in ECs. Plt-VWF mice, on the other hand, do not have an endothelial storage pool of VWF and plasma VWF levels were not significantly changed after epinephrine administration.
Fig. 4.
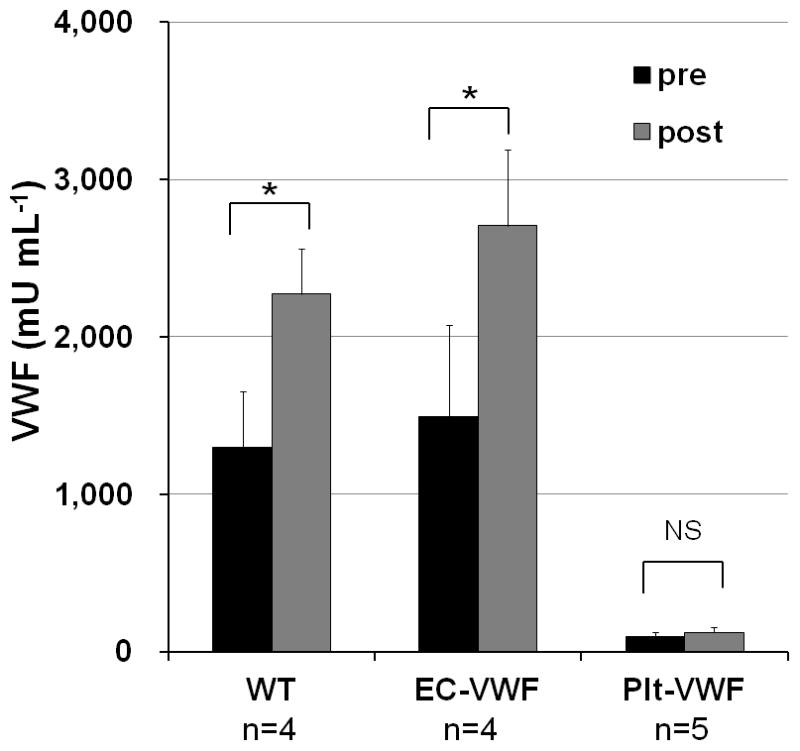
Epinephrine-stimulated release of VWF. Epinephrine was subcutaneously administered to animals. Plasma samples were collected before and 30 minutes after injection. VWF antigen levels were determined by ELISA. Plasma VWF levels were significantly increased by epinephrine infusion in EC-VWF mice and WT mice but not in Plt-VWF mice. NS, not significant, *P < 0.05
Discussion
In this study, we investigated the relative contribution of endothelial VWF and platelet VWF to hemostasis using crossed BMT between WT and VWF−/− mice. VWF synthesized in ECs was secreted into plasma and was also stored for regulated release which could be induced by epinephrine. The bleeding time of EC-VWF mice was normal. Interestingly, the bleeding time of Plt-VWF mice was partially corrected with bleeding stopping within 10 minutes in 38% of mice, and a significant decrease of blood loss volume compared to VWF−/− mice. Ex vivo perfusion assays with Plt-VWF blood also showed significantly enhanced thrombus formation on a type I collagen compared to VWF−/− mice. These results demonstrate that platelet VWF also contributes to platelet adhesion and plays a role in local hemostasis.
The only animal model in which platelet VWF has been studied is the VWD pig [22, 23]. Bowie EJ et al performed BM transplantation from a normal pig into a VWD pig, and showed an improvement of the bleeding diathesis and lower requirement of VWF replacement for correction of bleeding time [22]. However, this study was performed on a single recipient animal and a subsequent BMT study performed on multiple VWD pig recipients (n=4) did not reproduce improvement in their bleeding diathesis [23]. Thus, the role of platelet VWF studied in the pig model remained controversial. We show in the current study that platelet VWF contributes to hemostasis in mice. Since VWF is not synthesized in platelets in dogs, there may be a difference in the role of platelet VWF among species. As described in the introduction, clinical observations on VWD patients suggest that platelet VWF contributes to hemostasis in humans [4, 6, 11–13].
In our study, Plt-VWF mice had a trace amount of plasma VWF, the source of which is currently unknown. There are several potential explanations: (1) VWF may be artifactually released from activated platelets at the time of blood drawing, (2) WT donor-derived non-megakaryocytic/platelet lineage cells may produce and secrete VWF into plasma in recipients, (3) VWF synthesized in platelets is mostly stored in α-granules but a small portion is constitutively secreted into plasma. A low, but significantly higher level of FVIII activity in the plasma of Plt-VWF compared to VWF−/− suggests that VWF is present in plasma in vivo, thereby stabilizing FVIII in plasma, which would not be expected if VWF was released during sample processing (see Figure 1C). As to the second possibility, endothelial progenitor cells included in the BM cells could potentially be transplanted from WT, thereby producing and secreting VWF into the plasma of Plt-VWF mice [24]. If the residual amount of plasma VWF detected in Plt-VWF mice was from transplanted endothelial lineage cells, it should be releasable by epinephrine infusion. However, in our experiment, plasma VWF levels in Plt-VWF mice were not significantly increased after epinephrine infusion. Therefore, we saw no evidence supporting the hypothesis of endothelial lineage engraftment in Plt-VWF mice. With regard to the third hypothesis, our group has previously reported a transgenic hemophilia B mouse model with expression of human factor IX (FIX) under control of the platelet-specific integrin αIIb promoter. These transgenic mice demonstrated releasable storage of expressed FIX in α-granules with a small portion of FIX constitutively secreted into plasma, although the source of synthesis is not known [25]. Constitutive secretion of VWF from human megakaryocytes cultured in vitro has also been reported [26]. The majority of VWF produced in platelets is stored in α-granules but a small portion might be secreted into plasma. While the source of plasma VWF in Plt-VWF mice was not identified in the current study, our results indicate that it is unlikely that the plasma VWF observed in Plt-VWF mice was released artifactually from activated platelets during sample collection.
Currently, options for severe VWD treatment are limited. Since VWD is a monogenic disease, it is a good candidate for gene therapy [27–29]. The large size of VWF cDNA has been thought to be a barrier for efficient gene transfer but several studies have shown ectopic expression of VWF in liver induced by hydrodynamic injection of VWF expression vectors in mice [27, 29]. One of the remaining problems to be solved is that it is not known whether cells other than ECs and platelets are able to produce fully active VWF. Recently recombinant VWF (rVWF) expressed in CHO cells has been developed as a new drug candidate for treating VWD patients [30]. rVWF co-expressed with FVIII in CHO cells still contained VWF propeptide and had to be exposed to recombinant furin to remove pro-peptide and achieve efficient binding with FVIII [31]. Thus, the cell type in which VWF is expressed is potentially problematic because the biosynthesis of VWF is a complicated process that requires extensive posttranslational processing including furin cleavage, glycosylation and multimerization. Susceptibility of ectopically expressed VWF to ADAMTS13 cleavage is also not known. Excessive expression of VWF or impaired ADAMTS13 cleavage may result in thrombotic complications. Our study suggests that the platelet could be a potential target for expression of VWF by gene transfer for the treatment of VWD. Platelets are one of the endogenous sources of VWF, and the bulk of VWF produced in platelets is stored in α-granules ready to be released upon platelet activation, thereby providing a high local concentration at sites of vascular injury. It is known that posttranslational modification of VWF synthesized in megakaryocytes differs in glycosylation from that of EC synthesis, which may result in differences in VWF function [32–34]. However, observations of VWD patients and normal individuals suggest the functionality of platelet VWF in hemostasis in humans [4–6, 10–13, 35, 36]. Thus, gene therapy using either transfected or transduced autologous hematopoietic stem cells to express VWF in platelets might be beneficial for long-term disease control in VWD patients refractory or unresponsive to DDAVP treatment. Still the hemostatic effect of platelet VWF shown in this study may not be adequate to correct the bleeding diathesis of human VWD patients, so genetic manipulation to increase expression and/or stability of VWF in platelets might be required to provide a basis for therapeutic application.
In this study, we showed that VWF produced by ECs was sufficient to support normal hemostasis. VWF produced in megakaryocytes/platelets was also shown to contribute to hemostasis in the absence of EC-derived VWF although bleeding time and platelet adhesion were not completely normalized. Taken together these data indicate that plasma VWF is a major determinant of hemostasis but platelet VWF also contributes to control bleeding, and the presence of both might be optimal for normal hemostasis.
Acknowledgments
This work was supported by National Institutes of Health grants HL-081588 (RRM), HL-33721 (RRM), HL-44612 (RRM), and American Heart Association Postdoctoral Fellowship (10POST261016) (SK).
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
References
- 1.Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, Rick ME, Sadler JE, Weinstein M, Yawn BP. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA) Haemophilia. 2008;14:171–232. doi: 10.1111/j.1365-2516.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- 2.Weibel ER, Palade GE. New Cytoplasmic Components in Arterial Endothelia. J Cell Biol. 1964;23:101–12. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–60. doi: 10.1083/jcb.95.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannucci PM, Lombardi R, Bader R, Vianello L, Federici AB, Solinas S, Mazzucconi MG, Mariani G. Heterogeneity of type I von Willebrand disease: evidence for a subgroup with an abnormal von Willebrand factor. Blood. 1985;66:796–802. [PubMed] [Google Scholar]
- 5.Ruggeri ZM, Mannucci PM, Bader R, Barbui T. Factor VIII-related properties in platelets from patients with von Willebrand’s disease. J Lab Clin Med. 1978;91:132–40. [PubMed] [Google Scholar]
- 6.Gralnick HR, Rick ME, McKeown LP, Williams SB, Parker RI, Maisonneuve P, Jenneau C, Sultan Y. Platelet von Willebrand factor: an important determinant of the bleeding time in type I von Willebrand’s disease. Blood. 1986;68:58–61. [PubMed] [Google Scholar]
- 7.Haberichter SL, Balistreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, Manco-Johnson MJ, Gill JC, Montgomery RR. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108:3344–51. doi: 10.1182/blood-2006-04-015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fressinaud E, Federici AB, Castaman G, Rothschild C, Rodeghiero F, Baumgartner HR, Mannucci PM, Meyer D. The role of platelet von Willebrand factor in platelet adhesion and thrombus formation: a study of 34 patients with various subtypes of type I von Willebrand disease. Br J Haematol. 1994;86:327–32. doi: 10.1111/j.1365-2141.1994.tb04734.x. [DOI] [PubMed] [Google Scholar]
- 9.d’Alessio P, Zwaginga JJ, de Boer HC, Federici AB, Rodeghiero F, Castaman G, Mariani G, Mannucci PM, de Groot PG, Sixma JJ. Platelet adhesion to collagen in subtypes of type I von Willebrand’s disease is dependent on platelet von Willebrand factor. Thromb Haemost. 1990;64:227–31. [PubMed] [Google Scholar]
- 10.Kahr WHA, Pluthero FG, Blanchette VS, Robinson KS, Lillicrap D, James PD. Type 3 Von Willebrand Disease: Plasma Versus Platelets. Blood (ASH Annual Meeting Abstracts) 2009;114:3059. [Google Scholar]
- 11.Castillo R, Escolar G, Monteagudo J, Ordinas A, Garrido M, Moia M, Federici AB, Mannucci PM. Role for platelet von Willebrand factor in supporting platelet-vessel wall interactions in von Willebrand disease. Am J Hematol. 1989;31:153–8. doi: 10.1002/ajh.2830310303. [DOI] [PubMed] [Google Scholar]
- 12.Castillo R, Monteagudo J, Escolar G, Ordinas A, Magallon M, Martin Villar J. Hemostatic effect of normal platelet transfusion in severe von Willebrand disease patients. Blood. 1991;77:1901–5. [PubMed] [Google Scholar]
- 13.Castillo R, Escolar G, Monteagudo J, Aznar-Salatti J, Reverter JC, Ordinas A. Hemostasis in patients with severe von Willebrand disease improves after normal platelet transfusion and normalizes with further correction of the plasma defect. Transfusion. 1997;37:785–90. doi: 10.1046/j.1537-2995.1997.37897424399.x. [DOI] [PubMed] [Google Scholar]
- 14.Nichols TC, Bellinger DA, Reddick RL, Smith SV, Koch GG, Davis K, Sigman J, Brinkhous KM, Griggs TR, Read MS. The roles of von Willebrand factor and factor VIII in arterial thrombosis: studies in canine von Willebrand disease and hemophilia A. Blood. 1993;81:2644–51. [PubMed] [Google Scholar]
- 15.Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilcox DA, Shi Q, Nurden P, Haberichter SL, Rosenberg JB, Johnson BD, Nurden AT, White GC, 2nd, Montgomery RR. Induction of megakaryocytes to synthesize and store a releasable pool of human factor VIII. J Thromb Haemost. 2003;1:2477–89. doi: 10.1111/j.1538-7836.2003.00534.x. [DOI] [PubMed] [Google Scholar]
- 17.Ware J, Russell S, Ruggeri ZM. Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. Proc Natl Acad Sci U S A. 2000;97:2803–8. doi: 10.1073/pnas.050582097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, Cooley BC, Desai D, Morateck PA, Gorski J, Montgomery RR. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116:1974–82. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 20.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–46. doi: 10.1146/annurev.cb.06.110190.001245. [DOI] [PubMed] [Google Scholar]
- 21.Shi Q, Fahs SA, Kuether EL, Cooley BC, Weiler H, Montgomery RR. Targeting FVIII expression to endothelial cells regenerates a releasable pool of FVIII and restores hemostasis in a mouse model of hemophilia A. Blood. 2010;116:3049–57. doi: 10.1182/blood-2010-03-272419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowie EJ, Solberg LA, Jr, Fass DN, Johnson CM, Knutson GJ, Stewart ML, Zoecklein LJ. Transplantation of normal bone marrow into a pig with severe von Willebrand’s disease. J Clin Invest. 1986;78:26–30. doi: 10.1172/JCI112560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols TC, Samama CM, Bellinger DA, Roussi J, Reddick RL, Bonneau M, Read MS, Bailliart O, Koch GG, Vaiman M, et al. Function of von Willebrand factor after crossed bone marrow transplantation between normal and von Willebrand disease pigs: effect on arterial thrombosis in chimeras. Proc Natl Acad Sci U S A. 1995;92:2455–9. doi: 10.1073/pnas.92.7.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey AS, Fleming WH. Converging roads: evidence for an adult hemangioblast. Exp Hematol. 2003;31:987–93. doi: 10.1016/j.exphem.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Zhang G, Shi Q, Fahs SA, Kuether EL, Walsh CE, Montgomery RR. Factor IX ectopically expressed in platelets can be stored in alpha-granules and corrects the phenotype of hemophilia B mice. Blood. 116:1235–43. doi: 10.1182/blood-2009-11-255612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sporn LA, Chavin SI, Marder VJ, Wagner DD. Biosynthesis of von Willebrand protein by human megakaryocytes. J Clin Invest. 1985;76:1102–6. doi: 10.1172/JCI112064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Meyer SF, Vandeputte N, Pareyn I, Petrus I, Lenting PJ, Chuah MK, VandenDriessche T, Deckmyn H, Vanhoorelbeke K. Restoration of plasma von Willebrand factor deficiency is sufficient to correct thrombus formation after gene therapy for severe von Willebrand disease. Arterioscler Thromb Vasc Biol. 2008;28:1621–6. doi: 10.1161/ATVBAHA.108.168369. [DOI] [PubMed] [Google Scholar]
- 28.De Meyer SF, Vanhoorelbeke K, Chuah MK, Pareyn I, Gillijns V, Hebbel RP, Collen D, Deckmyn H, VandenDriessche T. Phenotypic correction of von Willebrand disease type 3 blood-derived endothelial cells with lentiviral vectors expressing von Willebrand factor. Blood. 2006;107:4728–36. doi: 10.1182/blood-2005-09-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marx I, Lenting PJ, Adler T, Pendu R, Christophe OD, Denis CV. Correction of bleeding symptoms in von Willebrand factor-deficient mice by liver-expressed von Willebrand factor mutants. Arterioscler Thromb Vasc Biol. 2008;28:419–24. doi: 10.1161/ATVBAHA.107.159442. [DOI] [PubMed] [Google Scholar]
- 30.Turecek PL, Schrenk G, Rottensteiner H, Varadi K, Bevers E, Lenting P, Ilk N, Sleytr UB, Ehrlich HJ, Schwarz HP. Structure and function of a recombinant von Willebrand factor drug candidate. Semin Thromb Hemost. 2010;36:510–21. doi: 10.1055/s-0030-1255445. [DOI] [PubMed] [Google Scholar]
- 31.Turecek PL, Mitterer A, Matthiessen HP, Gritsch H, Varadi K, Siekmann J, Schnecker K, Plaimauer B, Kaliwoda M, Purtscher M, Woehrer W, Mundt W, Muchitsch EM, Suiter T, Ewenstein B, Ehrlich HJ, Schwarz HP. Development of a plasma- and albumin-free recombinant von Willebrand factor. Hamostaseologie. 2009;29 (Suppl 1):S32–8. [PubMed] [Google Scholar]
- 32.Matsui T, Shimoyama T, Matsumoto M, Fujimura Y, Takemoto Y, Sako M, Hamako J, Titani K. ABO blood group antigens on human plasma von Willebrand factor after ABO-mismatched bone marrow transplantation. Blood. 1999;94:2895–900. [PubMed] [Google Scholar]
- 33.Williams SB, McKeown LP, Krutzsch H, Hansmann K, Gralnick HR. Purification and characterization of human platelet von Willebrand factor. Br J Haematol. 1994;88:582–91. doi: 10.1111/j.1365-2141.1994.tb05077.x. [DOI] [PubMed] [Google Scholar]
- 34.Brown SA, Collins PW, Bowen DJ. Heterogeneous detection of A-antigen on von Willebrand factor derived from platelets, endothelial cells and plasma. Thromb Haemost. 2002;87:990–6. [PubMed] [Google Scholar]
- 35.McGrath RT, McRae E, Smith OP, O’Donnell JS. Platelet von Willebrand factor--structure, function and biological importance. Br J Haematol. 2010;148:834–43. doi: 10.1111/j.1365-2141.2009.08052.x. [DOI] [PubMed] [Google Scholar]
- 36.Rodeghiero F, Castaman G, Ruggeri M, Tosetto A. The bleeding time in normal subjects is mainly determined by platelet von Willebrand factor and is independent from blood group. Thromb Res. 1992;65:605–15. doi: 10.1016/0049-3848(92)90210-2. [DOI] [PubMed] [Google Scholar]