

Abstract
Marijuana cannabinoids such as Δ9-tetrahydrocannabinol (THC) have been shown in experimental systems to bias T helper immunity towards Th2 and away from Th1. This effect if broadly applicable to humans could have important implications in Th2-mediated diseases such as allergy. In the current study, we examined the effect of cannabinoids on serum immunoglobulin IgE levels in immunized mice and also examined the role of cannabinoid receptors in the response. The method involved pre-injecting mice with cannabinoid receptor agonists and antagonists followed 18–24 hours later with an immunizing injection with two different antigen/adjuvant combinations. This treatment was followed 2–3 weeks later with a booster injection of antigen and the subsequent bleeding of mice 1–2 weeks later for serum immunoglobulin analysis by ELISA. Our results showed that THC injection enhanced total IgE serum levels in response to antigen immunization even under conditions of deficient cannabinoid receptor 2 (CB2) and cannabinoid receptor 1 (CB1) activity and furthermore the increase in IgE was accompanied by a decrease in serum IgG2a. In addition, we observed that l-α-lysophosphatidyliniositol (LPI) increased serum IgE levels and that IgE levels were higher in CB2 deficient mice and suppressed by the CB2 agonist, Gp1a. These results suggest that in this IgE induction model in mice, non-selective cannabinoids such as THC increase IgE through receptors other than CB1 and CB2 but that CB2 receptors do play a suppressive role in the control of serum IgE levels.
Keywords: cannabinoid receptors, delta-9-tetrahydrocannabinol, immunoglobulin E (IgE), GPR55, CB2, allergy
Introduction
MJ cannabinoids have been shown to modulate various immune functions through mechanisms involving CB1 and CB2 cannabinoid receptors. However, several studies concluded that other receptors are in involved (Begg et al., 2005) because immune modulation by THC persists in CB1 and 2 deficient mice (Walter and Stella, 2004; Lu et al., 2006a; Springs et al., 2008). A third cannabinoid receptor, GPR55, has been reported and may account for some of the effects observed in the relative absence of CB1 and CB2. GPR55 is stimulated by THC as well as other cannabinoid ligands (Ryberg et al., 2007) with one of these ligands, lysophosphatidylinositol (LPI), speculated to be the natural endogenous ligand for this receptor (Oka et al., 2009).
Among the various immune mechanisms modulated by cannabinoids, T helper (Th) cell biasing has been reported with a suppression of Th1 and enhancement of Th2 immunity (Klein, 2005). This biasing effect of Th cells has also been observed with other neuroimmune agents such as morphine (Roy et al., 2001) and could partially explain the decrease in neuroinflammatory symptoms associated with Th1 activity (Maresz et al., 2007) or the increase in serum IgE levels (Th2 activity) observed in marijuana smokers (Rachelefsky et al., 1976). In the first report of cannabinoid-induced Th biasing, a suppression of cell-mediated immunity and splenocyte IFNg production was accompanied by increasing serum levels of IgG1 antibodies and splenocyte IL4 (Newton et al., 1994). Because the different subclasses of antibodies are regulated by Th cytokines with IL4 increasing the synthesis of IgG1 and IgE (Roper et al., 1990), these results suggested that THC might increase the production of the allergic antibody, IgE, in addition to IgG1.
In the current study, we wanted to see if THC increased the production of IgE in mice immunized with several standardized antigens and if so what is the role of cannabinoid receptors. Our results showed that THC injection enhanced total IgE serum levels in response to antigen immunization under CB2 and CB1 deficient conditions and the increase in IgE was accompanied by a decrease in serum IgG2a. In addition, we observed that the GPR55 agonist, LPI, increased serum IgE levels and that IgE levels were higher in CB2 deficient mice and were suppressed by the CB2 agonist, Gp1a. These results suggest that in this IgE induction model in mice, non-selective cannabinoids such as THC increase IgE through receptors other than CB1 and CB2 but that CB2 receptors doo play a suppressive role in the control of serum IgE levels.
Materials and Methods
Chemicals
The Research Technology Branch of the National Institute on Drug Abuse provided SR141716A (SR1; CB1 antagonist or inverse agonist), SR144528 (SR2; CB2 antagonist or inverse agonist), cannabidiol (CBD), and Δ-9-tetrahydrocannabinol (THC). L-α-lysophosphatidyliniositol (LPI), keyhole limpet hemocyanin (KLH), Sigma Adjuvant System® (RIBI), and ovalbumin (OVA) were obtained from Sigma (St Louis, MO). Imject® Alum (Pierce, Rockford, IL) and Gp1a (Tocris Bioscience, Ellisville, MO) were also used these studies.
Mice
Female BALB/c (National Cancer Institute-Harlan, Fredericksburg, MD were used at 8–10 wks of age. Cannabinoid CB2 receptor gene deficient mice (CB2−/−) on C57BL/6 background and C57B/6 mice littermates were bred by USF animal facility staff from stocks provided by Dr. Nancy Buckley (California State Polytechnic U). The mice were housed and cared for in the animal facility of University of South Florida Health Sciences Center, which is fully accredited by American Association for Accreditation of Laboratory Animal Care.
Treatments
The antagonists (SR1 or SR2), THC (Ki≤40nM CB1; Ki≤36 CB2) and Gp1a (Ki=0.037nM CB2; Ki=363nM CB1) were diluted first in DMSO (Sigma) to 50mg/ml and LPI was diluted in PBS followed diluting in mouse serum. When given, mice were injected intravenous (iv) with THC (8mg/kg; 200μg/mouse [ms]), Gp1a (2mg/kg; 50μg/ms), or LPI (3.2–4mg/kg;80–160μg/ms) 18 hrs prior to sensitization by intraperitoneal (ip) injection of each antigens (OVA or KLH). For C57BL/6 and CB2−/−, the treatments were performed with littermates. For the antagonist studies, the SR1 (4mg/kg; 100μg/ms) were injected iv 30 min prior to the THC treatment, SR2 (4mg/kg; 100μg/ms) 30 min prior to Gp1a or cannabidiol (CBD; 250μg/ms) or SR1 (100μg/ms) 30 min prior to LPI. For OVA sensitization, OVA (2mg/ml) added to equal amount Imject® Alum for 300μg of OVA in 300μl of ALUM that was injected intraperitoneal (ip) for 14–21 days and followed by a challenge of OVA/Alum (200μl/ms; ip). Mice were bled at 5–9 days after the challenge. Cardiac punctures of mice were done initially from 9 to 10 days before realizing that basically the same or higher levels of total IgE were obtainable from days 5 to 6. For KLH sensitization, KLH (1mg/ml) was combined with RIBI as per manufacturer's instruction for 100μg KLH in 100μl RIBI per mouse. This KLH/RIBI solution was injected ip (100μl/ms) for 14–21 days followed with challenge of KLH/RIBI (100μl; ip). Mice were bled at 5–6 days after boost.
ELISA for Serum Total IgE
Sera were diluted 1:5 or 1:10 and tested for Total IgE activity by sandwich ELISAs using a kit from BD-Pharmingen (San Diego, CA). Basically, the capture IgE antibody in 0.1 M carbonate buffer (pH9.5) was coated on medium-bind Costar EIA plates (Corning) followed by blocking buffer, standard and diluted serum, and detection biotinylated antibody plus streptavidin-horseradish peroxidase (HRP). The color was by adding 3,3',5,5'-Tetramethylbenzidine Liquid Substrate System (TMB; Sigma) for 5–30 min and the reaction stopped with 1N sulfuric acid. The plates were read at 450nm on Emax Microplate Reader (Molecular Devices; Mento Park, CA) Units were calculated from a standard curve run with each plate. The low-end sensitivity was 0.5 ng/ml.
Anti-OVA IgG2a isotypes
ELISAs were done on the sera from above at 5–6 days. Basically OVA solution (10μg/ml) in 0.1 M carbonate buffer (pH9.5) was coated on medium-bind Costar EIA plates (Corning) and followed by blocking buffer. Sera were serially diluted with detection of the antibodies with anti-mouse IgG2a labeled with HRP. Color was developed with TMB (Sigma) and the reaction stopped with 1N sulfuric acid. The plates were read at 450nm on Emax Microplate Reader (Molecular Devices).
Statistical analysis
Data were analyzed by comparisons between groups using two-tailed Student's t-test.
Results
THC injection increases serum IgE antibodies
Our previous studies showed that THC injection prior to infection with Legionella pneumophila resulted in a selective increase in anti-Legionella IgG1 (Th2 antibody) and a decrease in IgG2a (Th1 antibody) (Newton et al., 1994). To see if another Th2 antibody class, IgE, was also increased by drug treatment, in vivo mouse immunization models with different antigens and adjuvants (OVA/ALUM and KLH/RIBI) were developed and serum levels of total IgE analyzed by ELISAs. BALB/c mice were pretreated with saline or THC (200μg/ms) 18 hrs prior to primary immunization with either OVA/ALUM (300μl/ms) or KLH/RIBI (100μl/ms) and then boosted with a second antigen injection 14–21 days later. Following the booster injections, the maximum serum IgE response times were selected and mice were bled either 9–10 days later (OVA/ALUM maximum) or 5–6 days (KLH/RIBI maximum) and serum IgE antibodies were determined by ELISA. As shown in Figure1, immunization with both antigens increased the serum level of IgE following the booster injections and furthermore, as seen previously measuring IgG1 (Newton et al., 1994), THC injection prior to the primary immunizations increased total IgE over normal serum and immune serum. Thus, remarkably, THC, injected at primary immunization with two different antigens enhanced IgE serum levels 4–5 weeks later following a booster injection.
Fig. 1.

THC injection increased serum IgE antibodies. Mice were immunized with OVA/ALUM (A) or KLH/RIBI (B) and boosted with the same antigen. Sera were collected at Day 9–10 (A) or 5 (B) and ELISAs performed as described in Methods. Data bars are sera from 4–10 mice/group +/− SEM. *=p≤0.05 vs normal; ** =p≤0.05 vs OVA/ALUM or KLH/RIBI
THC injection decreases serum IgG2a antibodies
In addition to examining drug effects on IgE antibodies, we also looked for effects on IgG2a antibodies. Sera from drug-treated and immunized BALB/c and C57BL/6 mice were examined for IgG2a antibodies specific for OVA antigen. Figure 2 shows that immunization led to a significant anti-OVA IgG2a serum titer when measured 5–6 days after boosting in either BALB/c (panel A) or C57BL/6 (panel B) mice. Importantly, the data also showed that THC injection significantly inhibited these antibodies levels confirming, along with the data in Figure 1, what we had seen before using Legionella antigens (Newton et al., 1994; Klein et al., 2000) that THC injection suppresses Th1 type antibodies while increasing Th2 type responses.
Fig. 2.

THC injection decreased serum lgG2a antibodies. The mice were immunized with OVA/ALUM followed by a boost of same antigen. Sera were collected from BALB/c (A) or C57BL/6 (B) mice 5–6 days following boosting. ELISAs were done as described in Methods. Data represented 4–6 mice per group ± SEM
CB2 receptor deficient mice show increased IgE serum levels
Recognizing that THC increased the level of the allergic antibody, IgE, we began an analysis of which cannabinoid receptors were involved. Initial studies were performed with mice of the C57BL/6 strain containing a targeted deletion of the CB2 receptor gene (Buckley et al., 2000). Wild type and CB2−/− mice were sensitized and challenged with OVA/ALUM (300μl/ms; 200μl/ms) and bled 9–10 days post-challenge. Surprisingly, the CB2 deficient mice showed increased rather than decreased serum IgE levels (Figure 3A) suggesting that CB2 receptors might have a negative regulatory effect on IgE production. Similar results were obtained sensitizing and challenging mice with KLH/RIBI in that the IgE response was higher in CB2−/− mice and THC was equipotent in wild type and CB2−/− mice and increased IgE in CB2−/− mice (Fig.3B).
Fig. 3.
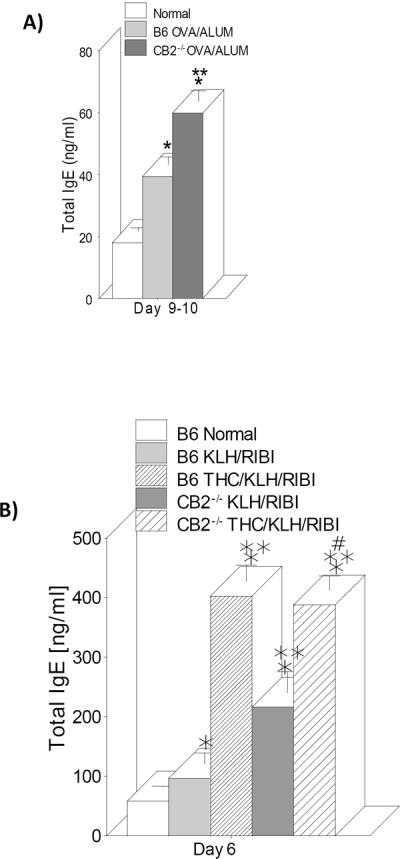
CB2 receptor deficient mice showed increased IgE serum levels. Wild-type and CB−/− mice were immunized and boosted with either OVA/ALUM (A) or KLH/RIBI (B) and IgE levels in serum determined 5–6 days after boosting. Some of the animals in (B) were pre-treated with THC. The IgE levels in non-treated wild-type (normal) and non-treated CB−/− mice were the same (data not shown). Data are expressed as mean+/−SEM with 4–6 mice per group. *=p<0.05 vs normal. **=p<0.05 vs OVA or KLH. #=p<0.05 vs CB2−/− KLH/RIBI.
THC injection increased IgE in the relative absence of CB1 and CB2
The above results suggested that CB2 was not involved in the THC-induced enhancement of IgE and we wanted to further examine this possibility and examine the role of CB1 in the THC effect. Wild type and CB2−/− C57BL/6 mice were immunized with OVA/ALUM and pretreated with either saline or THC alone or pretreated with the CB1 receptor antagonist, SR141716A, and THC prior to immunization. Figure 4 shows that serum IgE was increased by THC treatment to the same extent in wild-type mice, pretreated with SR141716A, and in CB2−/− mice, and that pre-treating CB2−/− mice with SR141716A further increased the THC effect. These results showed that the enhancing effect on IgE occurred in CB1 or CB2 blocked mice as well as in mice blocked for both receptors suggesting that neither CB1 nor CB2 was involved.
Fig. 4.
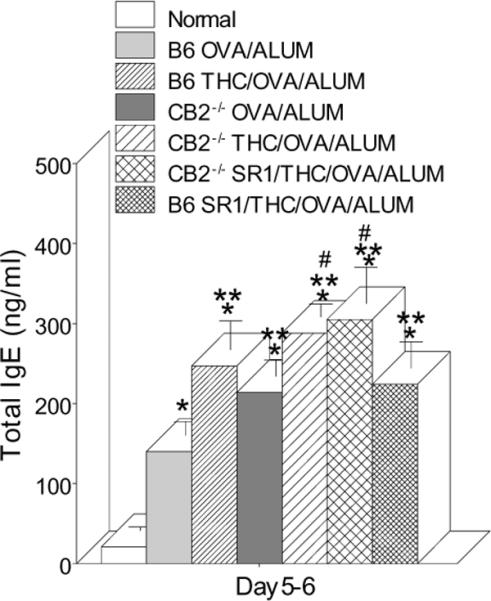
THC injection increased serum IgE in the absence of CB1 and CB2. CB−/− or wild type C57BL/6 mice were pretreated or not 18 hrs prior to immunization with OVA/ALUM. The mice were boosted and sera collected 5–6 days later for ELISA analysis. The IgE levels in non-treated wild-type (normal) and non-treated CB−/− mice were the same and SR1 treatment only had no effect (data not shown). Data bars are sera from 4–10 mice/group +/− SEM. *=p≤0.05 vs Normal; ** =p≤0.05 vs OVA/ALUM ; #=p≤0.05 vs CB2−/−OVA/ALUM
LPI injection increases serum IgE
The previous results suggested that neither CB1 nor CB2 were involved in the THC effect on IgE. This left open the possibility that the third cannabinoid receptor, GPR55, might be involved. To test this, mice were treated with a GPR55 agonist, l-α-lysophosphatidyliniositol (LPI) (Oka et al., 2009) (80–160μg/ms) followed by immunization with OVA/ALUM and boosting. Figure 5 shows LPI treatment elevated IgE in the serum; however, pretreatment with either cannabidiol (CBD; 250μg/ms) or SR141716A (SR1; 100μg/ms), both of which have been reported to have both antagonist and agonist activity (Sharir and Abood, 2010), did not attenuate the LPI effect nor increase the IgE response (data not shown).
Fig. 5.

LPI injection increased serum IgE. Lysophosphatidylinositol (LPI), a ligand for GPR55, pretreatment of BALB/c mice enhanced the serum levels of total IgE. The mice were pre-treated with either cannabidiol (CBD) or SR141716A (SR1) prior to LPI followed by sensitization of OVA/ALUM following a boost of same antigen. Treatment with either CBD or SR1 only had no effect on IgE levels (data not shown). The sera were collected at Day 6 after the boost and ELISAs were performed. Data bars represent serum from 4–14 mice/group +/− SEM. *=p≤0.05 vs Normal; **=p≤0.05 vs OVA/ALUM
Activation of CB2 receptor attenuated serum levels of IgE
The above results showed that CB2 is not involved in the IgE-enhancing effect of THC and in fact might lead to a suppression of the response. To test this directly, BALB/c mice were pretreated with the strong agonist for CB2 receptor (Ki of 0.037nM), Gp1a (50μg/ms), and then sensitized and challenged with OVA/ALUM. Figure 6 shows injection of Gp1a attenuated by about one third the serum IgE response to OVA/ALUM and this attenuation effect was inhibited by pretreatment with SR144528 (SR2; 100μg/ms), a CB2 antagonist/inverse agonist. Therefore, activation of CB2 with Gp1a has a negative effect on serum IgE levels and may be involved in down-regulating this immune response.
Fig. 6.

Activation of CB2 receptor attenuated serum levels of IgE. BALB/c mice were pretreated with Gp1a, a CB2 agonist (Ki=0.037nM) or Gp1a plus SR2 the CB2 antagonist, 18 hrs later the mice were sensitized with OVA/ALUM and boosted with same antigen. Sera were collected at Day 6 after the boosting and ELISAs performed. Treatment with SR2 only had no effect on IgE levels (data not shown). Data bars are serum values from 4–8 mice/group +/− SEM. *=p≤0.05 vs Normal; **=p≤0.05 vs OVA/ALUM and SR2/Gp1a/OVA/ALUM; #=p≤0.05 from OVA/ALUM
Discussion
Cannabinoids such as THC have been shown to modulate immune function and a large body of evidence suggests that both CB1 and CB2 receptors are involved (Klein and Cabral, 2006). However, not all immune modulation can be attributed to these two receptors leaving open the involvement of other receptors or molecular mechanisms (Begg et al., 2005). Some time ago it was observed that THC in animal models of immune activation biased Th immunity away from Th1 and toward Th2 (Newton et al., 1994; Zhu et al., 2000). Other neuroimmune agents were subsequently shown to bias T helper responses and the mechanism in the case of cannabinoids was partially due to effects on the helper biasing cytokines, IL-12, IFNγ, and IL-4, through mechanisms involving CB1 and CB2 receptors (Newton et al., 2009).
The immune modulating ability of cannabinoids suggests that these drugs might be useful as therapeutics in immune diseases and indeed their efficacy has been tested in several disease models (Mackie, 2006). On the other hand, the modulating effect might contribute to disease and again a number of reports have addressed this issue (Roth et al., 2002). Increased IgE and allergic diseases would be expected in an animal being driven toward Th2 and indeed significantly elevated serum IgE was reported in 4 of 8 chronic marijuana-smoking adults (Rachelefsky et al., 1976) without allergic symptoms. This finding coupled with our observation that THC injection in mice increased IgG1 suggested to us that the immunocannabinoid system might be involved in the regulation of IgE and therefore be involved in the development of allergic disease. Initially, we observed that THC treatment of purified mouse B cells in culture promoted antibody class-switching from IgM to IgE and that CB2 receptors were involved in the response (Agudelo et al., 2008). To extend these cell culture findings to the whole animal, we wanted to see in the current study if THC injection could increase IgE in the blood of immunized mice.
We started the current studies with two different antigen/adjuvant combinations by duplicating the experimental paradigm used previously with Legionella antigens (Newton et al., 1994). The method involved pre-injecting mice with THC followed 18–24 hours later with an immunizing injection of antigen. This treatment was followed 2–3 weeks later with a booster injection of antigen and the subsequent bleeding of mice 1–2 weeks later for serum immunoglobulin analysis by ELISA. As with our previous studies analyzing IgG1 levels to Legionella, THC treatment resulted in increased serum IgE (Fig.1) showing that a single injection of THC prior to primary immunization quantitatively changed the IgE response to a secondary immune challenge weeks later. Also in accord with our previous studies examining IgG2a, THC treatment decreased IgG2a antibodies (Fig. 2). This effect on Ig levels was not detected after primary immunization (data not shown) but only after boosting suggesting the drug was affecting early immune cell mechanisms responsible for biasing secondary humoral responses. One of these mechanisms might be the T-helper cell polarizing function of dendritic cells as reported previously (Lu et al., 2006b) or an effect on B cells which have been shown to shift in culture from IgM to IgE producers following treatment with cannabinoids (Agudelo et al., 2008).
The effect of THC on IgE was intriguing to us because of the implications to allergic disease and we therefore wanted to pursue the role of cannabinoid receptors in the response. Initial studies using CB2−/− mice were quite surprising because these mice had not only a heightened IgE response to both antigens (Fig 3) but THC increased IgE equally in treated wild type and CB2−/− mice (Fig 3B) suggesting CB2 may actually be negatively regulating IgE immunity. Our previous work showed that CB2 was involved in increasing IgE in response to THC in cultured B cells and when combined with the current in vivo results suggested that THC injection is increasing IgE by affecting immune cells other than B cells and receptors other than CB2. Not having access to CB1/CB2 double knockout mice, we devised experiments using CB2−/− mice treated with the CB1 antagonist, SR141716A (SR1). Figure 4 shows THC treatment increased rather than decreased IgE levels not only in CB2−/− mice and SR1 treated mice but also in CB2−/− mice co-treated with SR1 suggesting that neither receptor was involved in the effect. That we observed a THC-induced increase in serum IgE is at variance with a previous report showing a drug-induced decrease (Jan et al., 2003); however, in the current study we injected much less drug which may account t for the difference. Furthermore, it is important to note that the THC effect on IgE we observed was quantitatively as robust in CB1 antagonized mice as in non-antagonized mice and CB2−/− mice suggesting that neither receptor was involved in THC enhancing IgE. This is at variance with other reports showing that CB1 and CB2 are sometimes only partially involved in drug effects such as suppression of lymphocyte and dendritic cell functions (Walter and Stella, 2004; Lu et al., 2006a; Springs et al., 2008).
A third cannabinoid receptor, GPR55, has been described that responds to THC and other natural and synthetic agonists (Sharir and Abood, 2010; Begg et al., 2005; Lauckner et al., 2008; Ross, 2009) and also responds to endogenous lipids such as anandamide and LPI. Regarding the function of this receptor, a significant amount of work has been done using GPR55 transfected cells while many of the whole animal in vivo experiments have used GPR55−/− mice (Sharir and Abood, 2010). The literature suggests that, in addition to THC, LPI can act as an agonist and that CBD and SR1 can be both agonistic and antagonistic depending upon the system studied (Sharir and Abood, 2010). To look for a role of GPR55 in our model, we injected mice with combinations of LPI, SR1, and CBD. Figure 5 shows that LPI can significantly increase IgE when combined with OVA/ALUM immunization and although both CBD and SR1 had shown some antagonistic effect in transfected cells, they had no agonistic (not shown) or antagonistic effect in our animal injection model. Our findings are among the few reports showing that LPI has an immune effect when administered to animals. It would be of interest to reexamine our immunization model with LPI in GPR55−/− mice looking for a link between the expression of this receptor and the up-regulation of IgE. These mice have been shown to have altered cytokine responses following an inflammatory challenge (Staton et al., 2008).
Finally, from the above results, it appears that THC injection causes a change during the primary immune response leading to augmented IgE production and that receptors other than CB1 or CB2 are involved. However, in addition to increasing IgE, the endocannabinoid system might also be involved in suppressing the response. This was suggested by our results in Figure 3 wherein IgE was higher in treated CB2−/− mice. Thus, to test the suppressive effect we injected mice with the CB2 agonist, Gp1a, and measured IgE. Figure 6 shows that indeed Gp1a can suppress the IgE response and that this effect is mediated by CB2 because it is attenuated by the CB2 antagonist.
In summary, our findings suggest that cannabinoid receptors are involved in the immune regulation of IgE and that overall serum levels of reaginic antibodies depend on a balance between enhancing receptors (possibly GPR55) and suppressing receptors (CB2). Thus, CB2 may be a negative regulator of IgE and defects in the expression or function of this receptor may account for diseases such as allergy wherein too much IgE is produced. We are planning experiments to examine CB2 expression in allergic patients to see if a defect in the gene or protein is associated with disease severity.
Acknowledgements
This work supported in part by National Institutes of Health grant No. RO1 DA019824 from the National Institute on Drug Abuse. We would like to thank Dr. Nancy E. Buckley (California State Polytechnic University) for providing the CB2−/− mice and helpful discussions.
Footnotes
Conflict of Interest. The authors declare no conflict of interest.
References
- Agudelo M, Newton C, Widen R, Sherwood T, Nong L, Friedman H, Klein T. Cannabinoid Receptor 2 (CB2) Mediates Immunoglobulin Class Switching from IgM to IgE in Cultures of Murine-Purified B Lymphocytes. Journal of NeuroImmune Pharmacology. 2008;3:35–42. doi: 10.1007/s11481-007-9088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, Liu J, Kunos G. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- Jan TR, Farraj AK, Harkema JR, Kaminski NE. Attenuation of the ovalbumin-induced allergic airway response by cannabinoid treatment in A/J mice. Toxicol Appl Pharmacol. 2003;188:24–35. doi: 10.1016/s0041-008x(03)00010-3. [DOI] [PubMed] [Google Scholar]
- Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- Klein TW, Cabral GA. Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol. 2006;1:50–64. doi: 10.1007/s11481-005-9007-x. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton CA, Nakachi N, Friedman H. Delta 9-tetrahydrocannabinol treatment suppresses immunity and early IFN-gamma, IL-12, and IL-12 receptor beta 2 responses to Legionella pneumophila infection. J Immunol. 2000;164:6461–6466. doi: 10.4049/jimmunol.164.12.6461. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Newton C, Perkins I, Friedman H, Klein TW. Role of cannabinoid receptors in Delta-9-tetrahydrocannabinol suppression of IL-12p40 in mouse bone marrow-derived dendritic cells infected with Legionella pneumophila. Eur J Pharmacol. 2006a;532:170–177. doi: 10.1016/j.ejphar.2005.12.040. [DOI] [PubMed] [Google Scholar]
- Lu T, Newton C, Perkins I, Friedman H, Klein TW. Cannabinoid treatment suppresses the T-helper cell-polarizing function of mouse dendritic cells stimulated with Legionella pneumophila infection. J Pharmacol Exp Ther. 2006b;319:269–276. doi: 10.1124/jpet.106.108381. [DOI] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med. 2007;13:492–497. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- Newton CA, Klein TW, Friedman H. Secondary immunity to Legionella pneumophila and Th1 activity are suppressed by delta- 9-tetrahydrocannabinol injection. Infect Immun. 1994;62:4015–4020. doi: 10.1128/iai.62.9.4015-4020.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton CA, Chou PJ, Perkins I, Klein TW. CB(1) and CB(2) cannabinoid receptors mediate different aspects of delta-9-tetrahydrocannabinol (THC)-induced T helper cell shift following immune activation by Legionella pneumophila infection. J Neuroimmune Pharmacol. 2009;4:92–102. doi: 10.1007/s11481-008-9126-2. [DOI] [PubMed] [Google Scholar]
- Oka S, Toshida T, Maruyama K, Nakajima K, Yamashita A, Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: a possible natural ligand for GPR55. J Biochem. 2009;145:13–20. doi: 10.1093/jb/mvn136. [DOI] [PubMed] [Google Scholar]
- Rachelefsky GS, Opelz G, Mickey MR, Lessin P, Kiuchi M, Silverstein MJ, Stiehm ER. Intact humoral and cell-mediated immunity in chronic marijuana smoking. J Allergy Clin Immunol. 1976;58:483–490. doi: 10.1016/0091-6749(76)90192-5. [DOI] [PubMed] [Google Scholar]
- Roper RL, Conrad DH, Brown DM, Warner GL, Phipps RP. Prostaglandin E2 promotes IL-4-induced IgE and IgG1 synthesis. J Immunol. 1990;145:2644–2651. [PubMed] [Google Scholar]
- Ross RA. The enigmatic pharmacology of GPR55. Trends Pharmacol Sci. 2009;30:156–163. doi: 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Roth MD, Baldwin GC, Tashkin DP. Effects of delta-9-tetrahydrocannabinol on human immune function and host defense. Chem Phys Lipids. 2002;121:229–239. doi: 10.1016/s0009-3084(02)00159-7. [DOI] [PubMed] [Google Scholar]
- Roy S, Balasubramanian S, Sumandeep S, Charboneau R, Wang J, Melnyk D, Beilman GJ, Vatassery R, Barke RA. Morphine directs T cells toward T(H2) differentiation. Surgery. 2001;130:304–309. doi: 10.1067/msy.2001.116033. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther. 2010;126:301–313. doi: 10.1016/j.pharmthera.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springs AE, Karmaus PW, Crawford RB, Kaplan BL, Kaminski NE. Effects of targeted deletion of cannabinoid receptors CB1 and CB2 on immune competence and sensitivity to immune modulation by Delta9-tetrahydrocannabinol. J Leukoc Biol. 2008;84:1574–1584. doi: 10.1189/jlb.0508282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, Chong E, Mander PK, Green PJ, Billinton A, Fulleylove M, Lancaster HC, Smith JC, Bailey LT, Wise A, Brown AJ, Richardson JC, Chessell IP. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LX, Sharma S, Stolina M, Gardner B, Roth MD, Tashkin DP, Dubinett SM. Delta-9-tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J Immunol. 2000;165:373–380. doi: 10.4049/jimmunol.165.1.373. [DOI] [PubMed] [Google Scholar]