

Abstract
Copper catalyzed azide-alkyne cycloaddition (CuAAC) chemistry is reported for the construction of previously unknown 5-(1H-1,2,3-triazol-1-yl)-4,5′-bithiazoles from 2-bromo-1-(thiazol-5-yl)ethanones. These novel triazolobithiazoles are shown to have cystic fibrosis (CF) corrector activity and, compared to the benchmark bithiazole CF corrector corr-4a, improved logP values (4.5 vs. 5.96).
Keywords: CuAAC, Triazolobithiazole, Cystic Fibrosis, CF corrector
1. Introduction
Cystic Fibrosis (CF) is a genetic disease affecting ~1 in 2,500 Caucasians1 which, in its most common form, is caused by the deletion of phenylalanine at position 508 in the CF transmembrane conductance regulator protein (ΔF508-CFTR).2,3 Our CF small molecule discovery program identified bithiazoles that partially rescue ΔF508-CFTR cellular misprocessing (CF ‘correctors’; Figure 1). In exploring this structural class, positions “a”, “b”, and “c” on the bithiazole scaffold were extensively modified.4 The work reported here follows from earlier studies targeting pyrazolothiazoles where we had shown that this compound class, which uniquely allows access to analogs exploring position “d” (not addressable with bithiazoles), was found to afford improved water solubility vis-à-vis bithazoles – albeit with lower CF corrector activity.5
Figure 1.

Bithiazole, pyrazolothiazole, and triazolobithiazole CF correctors.
This reduction in CF corrector activity in going from bithiazoles to pyrazolothiazoles caused us to consider exploring position “e” on the bithiazole scaffold, hypothesizing that placing a triazole moiety at C5 might give improved aqueous solubility while maintaining or perhaps improving corrector activity. Herein, we report the development of chemistry for the construction of novel 5-(1H-1,2,3-triazol-1-yl)-4,5′-bithiazoles, along with CF corrector activity and logP data. Retrosynthetic analysis suggested that triazole formation might be accomplished by copper catalyzed azide-alkyne cycloadditon (CuAAC) through a 2-bromo-1-(thiazol-5-yl)ethanone intermediate (Figure 1) and that a second bromination, now alpha to both the carbonyl and triazole moieties, might allow for α-bromoketone → thiazole formation.
2. Results and discussion
2.1. Chemistry
The requisite starting thiazoles (1a,b: R1 = t-Bu and Ph, respectively) were prepared as detailed in Scheme 1. Thiourea was condensed with 3-chloro-2,4-pentadione to afford 1-(2-amino-4-methylthiazol-5-yl)ethanone in nearly quantitative yield.6 The amino group in this 2-aminothiazole was then coupled with either pivalic (→ 1a; 85%) or benzoic (→ 1b; 79%) acid in CDI-mediated reactions to give the targeted amidothiazoles. After some experimentation, bromination alpha to the ketone carbonyl in 1 was effectively accomplished with pyridinium tribromide and 33 wt. % hydrobromic acid in acetic acid. Bromides 2a and 2b were thus obtained on ~1 gram scale in 75% and 63% overall yield, respectively, from 3-chloro-2,4-pentadione.
Scheme 1.
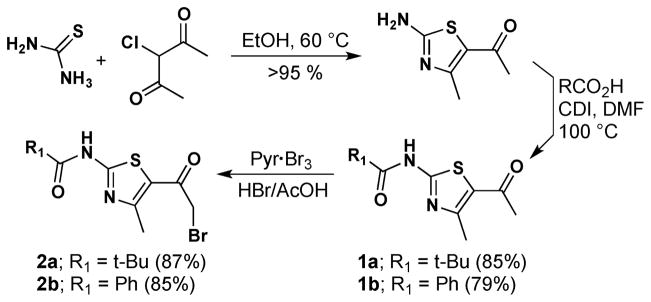
Synthesis of 2-bromo-1-(thiazol-5-yl)ethanones 2a,b.
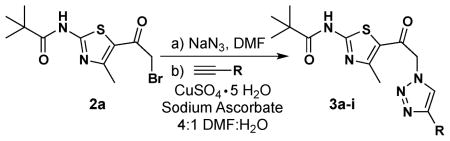
α-Bromoketone 2 was next stirred in DMF with sodium azide at room temperature to obtain, in situ, the corresponding azido compound. On completion of the displacement, as monitored by TLC (1:1 EtOAc:hexanes), the appropriate alkyne, copper catalyst, sodium ascorbate, and water (250 μL per mL of DMF) were added to the reaction flask and the contents were stirred for an additional 6 h. Workup, consisting of dilution with water and extraction with EtOAc, followed by flash column chromatographic purification, delivered the targeted 1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanone (3). Using this methodology, a small collection of analogs was synthesized in moderate to good yield from α-bromoketone 2a (Table 1).
Table 1.
One-pot synthesis of 1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanones 3a–i.
| |||
|---|---|---|---|
| entry | compound | R | yield |
| 1 | 3a | -Ph | 85% |
| 2 | 3b | -CH2OH | 76% |
| 3 | 3c | -CH2OCH3 | 73% |
| 4 | 3d | -CH2N(CH3)2 | 56% |
| 5 | 3e | -(CH2)3CN | 68% |
| 6 | 3f | -CH2N(CH2CH2)O* | 74% |
| 7 | 3g | -COOCH3 | 52% |
| 8 | 3h | -4-Cl-Ph | 95% |
| 9 | 3i | -CH2CH2OH | 70% |
morpholine
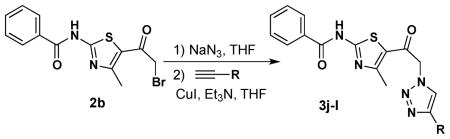
Surprisingly, it was discovered that employing α-bromoketone 2b as the starting material generally failed to give triazolothiazole products under the one-pot conditions outlined in Table 1; there was extensive product decomposition under the required prolonged reaction times. As a result, reactions employing 2b were modified – two-pot reaction with isolated azide and the use of copper(I) iodide as catalyst (see Table 2) – which allowed for shorter reaction times; with these modifications, three of the alkynes delineated in Table 1 led to product (see Table 2).
Table 2.
Two-pot synthesis of 1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanones 3j–l.
| |||
|---|---|---|---|
| entry | compound | R | yield |
| 1 | 3j | -Ph | 69% |
| 2 | 3k | -CH2OCH3 | 75% |
| 3 | 3l | -CH2N(CH2CH2)O* | 55% |
morpholine
This unexpected click reaction problem with 2-bromo-1-(thiazolo-5-yl)ethanone 2b led us to investigate the cause of these poor yielding cycloadditions. Thinking the amide proton may be the problem (either its increased acidity or the fact that it is less sterically encumbered than in 2a), we decided to synthesize the N-methylated version of 2b (e.g., 6; Scheme 2). Heating a DMF solution of 1-(4-methyl-2-(methylamino)thiazol-5-yl)ethanone5 4 with benzoyl chloride delivered the acylated product 5, which was subsequently brominated in an analogous procedure to that used to prepare 2b. Bromide 6 was then subjected to a CuSO4/sodium ascorbate click reaction with propargyl alcohol and provided the targeted cycloadduct 7 in fair yield [45%; in contrast, the non-methyl analog of 6 (e.g., 2b) gave no cycloadduct with propargyl alcohol (result not shown)]. While other modifications such as the use of a Cu(I) ligand to stabilize the copper(I)-oxidation state might benefit these reactions,7 the results with close analogs 2a and 6 suggest that the benzamide moiety is the root cause of the failed cycloadditions with 2b.
Scheme 2.

Synthesis of N-methyl 1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanone 7.
With triazolothiazole 3 in hand, we addressed the two key questions central of the strategy alluded to in Figure 1: (i) would bromination of 3 result in the formation of a stable and useable 2-bromo-1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanone and (ii) would the 2-bromo-2-(1H-1,2,3-triazol-1-yl)ethanone substructure in this bis-heterocycle react with N-substituted thioureas to give 5-(1H-1,2,3-triazol-1-yl)-4,5′-bithiazoles? Given that a pivalamide moiety at C2′ proved to be slightly more efficacious in corrector activity than the corresponding benzamide analog,4,5 we addressed both questions using 1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanone 3a. After some experimentation, we found that bromination of 3a proceeded most effectively by treatment with elemental bromine in dioxane at 60 °C.8 Workup, consisting of quenching with 10% NaHSO3 and EtOAc extraction, delivered 2-bromo-1-(thiazol-5-yl)-2-(1H-1,2,3-triazol-1-yl)ethanone 8a in 45% yield (Scheme 3). While the yield for 3a → 8a was modest, product isolation was straightforward and 8a was stable to manipulation. The Knorr condensation9 of 8a with various thioureas was also straight- forward, yielding 5-(1H-1,2,3-triazol-1-yl)-4,5′-bithiazoles 9–14 obtained in 56–92% yield. Saponification of the ester moiety in 14 (aq. KOH, THF) delivered acid analog 15.
Scheme 3.

Synthesis and corrector data for 5-(1H-1,2,3-triazol-1-yl)-4,5′-bithiazoles 9–15.
2.2. ΔF508-CFTR corrector activity
The ΔF508-CFTR corrector activities of triazolobithiazoles 9–1510 were evaluated by measurement of I− influx in epithelial cells expressing ΔF508-CFTR and a fluorescent halide sensor as previously described.11 The resulting Vmax and EC50 data are tabulated in Scheme 3. While none of the triazolobithiazoles had ΔF508-CFTR corrector activity as good as benchmark bithiazole corr-4a, the results with analogs 13 and 14 are encouraging (Figure 2).
Figure 2.
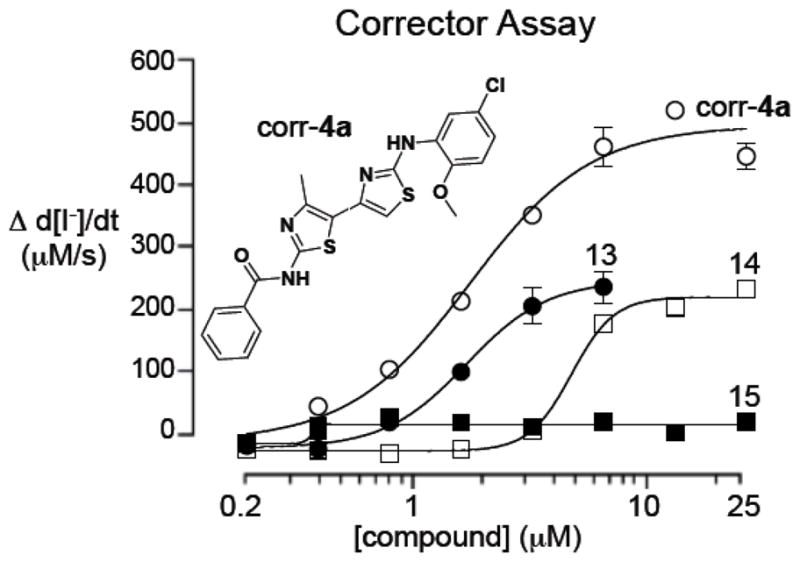
Dose-response showing I− influx in 3F508-CFTR cells treated for 24 h with triazolobithiazoles 13–15 or corr-4a and stimulated by a cAMP agonist (forskolin) and potentiator (genistein) (S.E., n=4).
2.3. Measured logP values
Encouraged by these results, we measured logP values (a measure of a compound’s hydrophilicity/hydrophobicity)12 of triazolobithiazoles 13–15. The reference for this study is the 5.96 logP of corr-4a, which Lipinski’s rules flag as a limitation.13 We determined the capacity factor from HPLC retention times to give correlated logP values. As shown in Figure 3, the logP values for compounds 13–15 were in the range 4.8–5.5.
Figure 3.
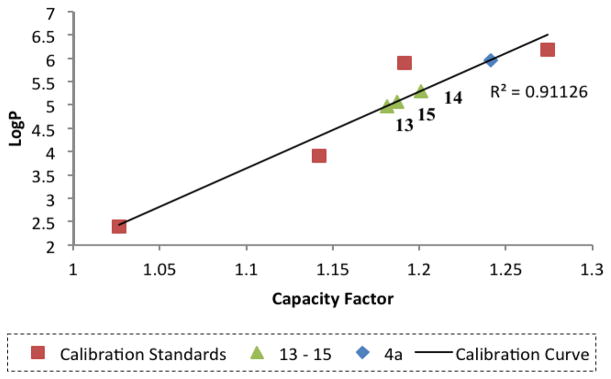
LogP measurement of triazolobithiazoles 13–15.
3. Conclusions
In summary, we have developed a practical synthesis of triazolobithiazoles, a previously unknown heterocyclic system. Several of the novel compounds reported here had ΔF508-CFTR corrector activity and improved logP compared to benchmark corrector corr-4a. These results will allow more extensive examination of triazolobithiazoles as potential development candidates for CF therapy.
4. Experimental section
4.1. Chemistry
General Experimental Procedures
All solvents and reagents were purchased from commercial suppliers and used without further purification. For reactions run in sealed microwave vials, oven-dried 5–10 mL or 10–20 mL vials containing a Teflon-coated stirrer bar and sealed with a Teflon-lined septum were used. Analytical thin layer chromatography was carried out on pre-coated plates (Silica gel 60 F254, 250 μm thickness) and visualized with UV light. Flash chromatography was performed with 60 Å, 35–70 μm silica gel. Concentration refers to rotary evaporation under reduced pressure. 1H NMR spectra were recorded on spectrometers operating at 300, 400, or 600 MHz at ambient temperature with DMSO-d6, MeOH-d4, CD3CN, acetone-d6 or CDCl3 as solvents. 13C NMR spectra were recorded on spectrometers operating at 75, 100, or 150 MHz at ambient temperature. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad), integration, coupling constant (Hz). Chemical shifts are reported in parts per million relative to DMSO-d6 (1H, δ 2.50; 13C, δ 39.52), CDCl3 (1H, δ 7.26; 13C, δ H, δ 0.00; 13C, δ 0.00). Infrared spectra were 77.16), or TMS (1 recorded on an ATI-FTIR spectrometer. The specifications of the LC/MS are as follows: electrospray (+) ionization, mass range 150–1500 Da, 20 V cone voltage, and C18 column (2.1 mm × 50 mm × 3.5 μm). LogP measurements were made as previously described.5
N-(5-acetyl-4-methylthiazol-2-yl)pivalamide (1a), N-(5-acetyl-4-methylthiazol-2-yl)benzamide (1b), N-(5-(2-bromoacetyl)-4-methylthiazol-2-yl)pivalamide (2a), and N-(5-(2-bromoacetyl)-4-methylthiazol-2-yl)benzamide (2b)
These compounds were prepared according to published methods and spectral data are in accord with established values.3
General procedure for copper catalyzed azide alkyne cycloaddition
A mixture of α-bromoketone (0.30 mmol) and sodium azide (22 mg, 0.34 mmol) were stirred in DMF (2 mL) at room temperature for 1 h. Alkyne (0.36 mmol) was added to the reaction followed by water (1 mL), CuSO4 (4 mg, 0.016 mmol), and sodium ascorbate (6 mg, 0.032 mmol). The solution was stirred for 6 h then diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organics were washed with water (45 mL) and brine (45 mL), dried over sodium sulfate, filtered, and concentrated. The resulting crude material was purified by flash chromatography.
N-(4-Methyl-5-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetyl)thiazol-2-yl)pivalamide (3a)
White solid (102 mg, 90%); mp 208–210 °C; IR (neat) vmax 3151, 3115, 2964, 2875, 1666, 1524, 1372, 1319, 1284, 1150, 1115, 968 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.13 (s, 1H), 7.94 (s, 1H), 7.86 (d, J = 7, 1H), 7.43 (t, J = 7, 2H), 7.34 (t, J = 7, 2H), 5.63 (s, 2H), 2.68 (s, 3H), 1.36 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 183.5, 177.0, 160.2, 159.0, 148.4, 130.8, 129.1, 128.5, 126.2, 121.8, 121.5, 57.4, 39.7, 27.4, 18.9; HRMS (ESI): calcd for [C19H21N5O2S + H]+ 384.1494, found 384.1486.
N-(5-(2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (3b)
White solid (77 mg, 76%); mp 185–186 °C; IR (neat) vmax 3324, 3235, 3138, 2977, 2933, 1688, 1670, 1493, 1324, 1137, 1039, 968 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.68 (s, 1H), 5.57 (s, 2H), 4.76 (s, 1H), 2.57 (s, 2H), 2.07 (s, 3H), 1.31 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 183.6, 178.2, 162.1, 157.6, 148.3, 124.2, 120.8, 57.4, 56.4, 39.8, 27.0, 18.1; HRMS (ESI): calcd for [C14H19N5O3S + H]+ 338.1287, found 338.1286.
N-(5-(2-(4-(Methoxymethyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (3c)
White solid (76 mg, 73%); mp 154–156 °C; IR (neat) vmax 22977, 2933, 1679, 1537, 1493, 1368, 1315, 1226, 1146, 977 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 5.57 (s, 2H), 4.61 (s, 2H), 3.40 (s, 3H), 2.62 (s, 3H), 1.32 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 183.42, 177.20, 160.37, 158.84, 145.50, 124.65, 121.17, 66.08, 58.46, 57.27, 39.60, 27.21, 18.76; HRMS (ESI): calcd for [C15H21N5O3S + H]+ 352.1443, found 352.1437.
N-(5-(2-(4-((Dimethylamino)methyl)-1H-1,2,3-triazol-1-yl)-acetyl)-4-methylthiazol-2-yl)pivalamide (3d)
Beige solid(61 mg,56%); mp 70–71 °C (decomp); IR (neat) vmax 3138, 2960, 1679, 1528, 1368, 1310, 1230, 1141, 1035, 972 cm−1;1H NMR (600 MHz, CD3CN) δ 8.06 (s, 1H), 5.76 (s, 2H), 5.09 (s, 1H), 4.02 (s, 2H), 2.62 (s, 3H), 2.51 (s, 6H), 1.32 (s, 9H); 13C NMR (150 MHz, CD3CN) δ 185.0, 177.9, 161.3, 157.6, 140.4, 127.3, 122.2, 57.7, 52.8, 52.3, 42.7, 39.4, 26.3, 18.2; HRMS (ESI): calcd for [C16H24N6O2S + H]+365.1759, found 365.1753.
N-(5-(2-(4-(3-Cyanopropyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (3e)
White solid (76 mg, 68%); mp 72–73 °C (decomp); IR (neat) vmax 2969, 2853, 1679, 1537, 1493, 1368, 1315, 1226, 1137, 1039, 977 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.51 (s, 1H), 5.55 (s, 2H), 2.89 (t, J = 7, 2H), 2.60 (s, 3H), 2.42 (t, J = 7, 2H), 2.07 (p, J = 7, 2H), 1.32 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 183.7, 177.3, 160.5, 158.9, 146.0, 123.6, 121.1, 119.7, 57.3, 39.6, 27.2, 25.1, 24.4, 18.8, 16.7; HRMS (ESI): calcd for [C17H22N6O2S + H]+ 375.1603, found 375.1596.
N-(4-Methyl-5-(2-(4-(morpholinomethyl)-1H-1,2,3-triazol-1-yl)acetyl)thiazol-2-yl)pivalamide (3f)
White solid (91 mg, 75%); mp 183–184 °C; IR (neat) vmax 2964, 2929, 1675, 1533, 1497, 1426, 1372, 1319, 1146, 1110 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.51 (s, 1H), 7.62 (s, 1H), 5.56 (s, 2H), 3.67 (s, 6H), 2.59 (s, 3H), 2.49 (s, 4H), 1.31 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 183.5, 177.2, 160.3, 158.9, 144.6, 124.9, 121.2, 67.1, 57.3, 53.8, 53.5, 39.6, 27.2, 18.8; HRMS (ESI): calcd for [C18H26N6O3S + H]+ 407.1865, found 407.1861.
Methyl 1-(2-(4-methyl-2-pivalamidothiazol-5-yl)-2-oxoethyl)-1H-1,2,3-triazole-4-carboxylate (3g)
White solid (57 mg, 52%); mp 211–213 °C; IR (neat) vmax 3278, 3130, 2981, 1726, 1646, 1544, 1373, 1316, 1224, 1156, 996; 1H NMR (600 MHz, CDCl3) δ 9.40 (s, 1H), 8.27 (s, 1H), 5.64 (s, 2H), 3.96 (s, 3H), 2.66 (s, 3H), 1.36 (s, 9H); 13C NMR 13C NMR (150 MHz, CDCl3) δ 182.12, 176.86, 160.99, 160.25, 159.01, 140.20, 129.55, 120.73, 57.05, 52.24, 39.39, 27.01, 18.54; HRMS (ESI) calcd for [C15H19N5O4S + H]+ 366.1236, found 366.1233.
N-(5-(2-(4-(4-Chlorophenyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (3h)
White solid (124 mg, 95%); mp 255–256 °C; IR (neat) vmax 3120, 2969, 2933, 1679, 1661, 1528, 1457, 1368, 1315, 1235, 1137, 968; 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 8.55 (s, 1H), 7.90 (d, J = 8.5, 2H), 7.51 (d, J = 8.5, 2H), 6.01 (s, 2H), 2.67 (s, 3H), 1.25 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 185.74, 178.31, 162.24, 157.19, 145.79, 132.95, 130.28, 129.65, 127.51, 124.06, 122.68, 57.98, 39.71, 27.02, 19.12; HRMS (ESI): calcd for [C19H20ClN5O2S + H]+ 418.1104, found 418.1096.
N-(5-(2-(4-(3-Hydroxypropyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (3i)
White solid (76 mg, 70%); mp 193–196 °C; IR (neat) vmax 3144, 2930, 2872, 1685, 1529, 1489, 1374, 1315, 1140, 1043, 975 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 12.34 (s, 1H), 7.76 (s, 1H), 5.83 (s, 2H), 3.44 (t, J = 6.4, 2H), 2.66 (t, J = 7.6, 2H), 2.63 (s, 2H), 1.78 – 1.70 (m, 2H), 1.24 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 185.8, 178.0, 161.8, 156.6, 147.0, 123.9, 122.5, 60.5, 57.4, 39.5, 32.8, 26.8, 22.1, 18.8; HRMS (ESI): calcd for [C16H23N5O3S + H]+ 366.1592, found 366.1603.
N-(4-Methyl-5-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetyl)thiazol-2-yl)benzamide (3j)
α-Bromoketone 2b (50 mg, 0.147 mmol) was stirred at room temperature in DMF (4 mL) with sodium azide (10 mg, 0.161 mmol) for 2 h. The reaction was then diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organics were washed with brine (30 mL), dried over sodium sulfate, and filtered. The reaction was concentrated, dissolved in dry THF (2 mL) in a flask covered with tin foil, and triethylamine (77 μL, 0.558 mmol), copper(I) iodide (70 mg, 0.372 mmol), and phenylacetylene (16 mg, 0.161 mmol) were added. The reaction was stirred 12 h at room temperature and quenched by the addition of aq. NH4OH. The mixture was extracted with EtOAc (3 × 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, filtered and concentrated. The crude material was purified by recrystallization in EtOAc/hexanes to yield 3j as a white solid (38 mg, 69%); mp 222–224 °C; IR (neat) vmax 2910, 1682, 1533, 1481, 1192, 1030, 965cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.14 (d, J = 7.3, 2H), 7.89 (d, J = 7.2, 2H), 7.69 (t, J = 7.4, 1H), 7.58 (t, J = 7.6, 2H), 7.48 (t, J = 7.7, 2H), 7.36 (t, J = 7.4, 1H), 6.06 (s, 2H), 2.73 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 185.1, 184.9, 161.4, 146.0, 133.0, 131.2, 130.5, 128.7, 128.5, 128.2, 127.7, 125.0, 122.8, 122.1, 57.1, 26.1.; HRMS calcd for [C21H17N5O2S + H]+ 404.1181, found 404.1171.
N-(5-(2-(4-(Methoxymethyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)benzamide (3k)
White solid (42mg, 75%); mp 225–226 °C; IR (neat) vmax 3158, 2924, 1670, 1536, 1488, 1322, 1283, 1098, 965 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.91 (s, 1H), 7.97-7.95 (m, 2H), 7.72 (s, 1H), 7.70-7.68 (m, 1H), 7.59-7.55(m, 2H), 5.63 (s, 2H), 4.65 (s, 2H), 3.44 (s, 3H), 2.66 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 183.1, 164.9, 160.4, 133.7, 130.9, 129.2, 127.6, 124.4, 121.1, 65.9, 58.3, 57.1, 50.9, 30.4, 18.5; HRMS (ESI): calcd for [C17H17N5O3S + H] + 372.1130, found 372.1131.
N-(4-Methyl-5-(2-(4-(morpholinomethyl)-1H-1,2,3-triazol-1-yl)acetyl)thiazol-2-yl)benzamide (3l)
White solid (690 mg, 55%); mp 232–233 °C; IR (neat) vmax 2943, 2355, 2140, 2049, 1908, 1671, 1488, 1219, 1057, 965 cm−1;1H NMR (600 MHz, DMSO-d6) δ 8.18 (s, 1H), 8.00 (s, 1H), 7.72 (s, 1H), 7.62 (s, 1H), 5.95 (s, 1H), 3.66 (d, J = 28.1, 3H), 3.25 (s, 2H), 2.75 (s, 1H), 2.55 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 185.2, 166.0, 161.4, 156.2, 142.7, 132.9, 131.5, 128.6, 128.3, 125.4, 122.1, 66.0, 57.0, 52.7, 18.2; HRMS (ESI): calcd for [C20H22N6O3S + H]+ 427.1552, found 427.1543.
1-(4-Methyl-2-(methylamino)thiazol-5-yl)ethanone (4)
Prepared following a published method; spectral data in accordance with established values.5
N-(5-Acetyl-4-methylthiazol-2-yl)-N-methylbenzamide (5)
1-(4-Methyl-2-(methylamino)thiazol-5-yl)ethanone (1.27 g, 7.47 mmol) and N,N-diisopropylethylamine (1.55 mL, 8.96 mmol) were added to a flask charged with DMF (40 mL). The reaction flask was then heated to 100 °C and benzoyl chloride (1.29 mL, 11.2 mmol) was added. The reaction was stirred at 100 °C for 12 h and monitored by thin-layer chromatography. Upon completion, the reaction was poured into ice-cold water and a precipitate formed. The precipitate was collected by filtration and the beige solid (5) was used without further purification (1.82 g, 89%); mp 128–130 °C; IR (neat) vmax 2917, 1664, 1516, 1477, 1415, 1360, 1306, 1251, 1103, 1010, 946 cm−1;1H NMR (600 MHz, CDCl3) δ 7.58 – 7.52 (m, 3H), 7.52 – 7.47 (m, 2H), 3.66 (s, 3H), 2.69 (s, 3H), 2.53 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 191.2, 170.7, 160.3, 154.9, 133.8, 131.2, 128.7, 127.6, 126.0, 38.2, 30.6, 18.4; HRMS (ESI) calcd for [C14H14N2O2S + H]+ 275.0840, found 275.0855.
N-(5-(2-Bromoacetyl)-4-methylthiazol-2-yl)-N-methylbenzamide (6)
N-(5-Acetyl-4-methylthiazol-2-yl)-N-methyl-benzamide (5; 100 mg, 0.36 mmol) and pyridinium tribromide (127 mg, 0.400 mmol) were added to a round bottom flask under a nitrogen atmosphere. The flask was charged with 1.5 mL of 33% HBr in acetic acid, then stirred at room temperature overnight. The reaction was quenched by the addition of water (2 mL) and a precipitate formed, which was collected by filtration. The filtrate was extracted with ethyl acetate (3 × 10 mL) and the precipitate was added to the combined organic extracts. These combined extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated. The crude material was purified by flash chromatography (2:1 EtOAc:hexanes) to afford 6 as an amorphous beige solid (108 mg, 85%); IR (neat) 2927, 1656, 1477, 1446, 1415, 1368, 1306, 1189, 1096, 1018 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.58 – 7.43 (m, 5H), 4.25 (s, 2H), 3.65 (s, 3H), 2.69 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 184.6, 170.8, 161.1, 157.8, 133.5, 131.4, 128.7, 127.6, 122.4, 38.4, 34.1, 18.6; HRMS calcd for [C14H13BrN2O2S + H]+ 352.9951 found 352.9927.
N-(5-(2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)-N-methylbenzamide (7)
N-(5-(2-Bromo-acetyl)-4-methylthiazol-2-yl)-N-methylben-zamide (6; 93.5 mg, 0.260 mmol) was dissolved in DMF (1.4 mL) and sodium azide (21.0 mg, 0.316 mmol) was added and stirred at room temperature for 30 min. The reaction was diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organics were washed with brine (30 mL), dried over sodium sulfate, filtered, and concentrated. This crude azide was reconstituted in a 4:1 DMF:H2O solution (1.5 mL) and copper(II) sulfate, sodium ascorbate, and propargyl alcohol were added. The reaction was stirred at room temperature for 2 h and monitored by thin layer chromatography. When complete, the reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL); the combined organics were washed with brine, dried over sodium sulfate, filtered, and concentrated. The resulting crude material was purified by flash chromatography (EtOAc) to yield 7 as a beige solid (44 mg, 45%); mp 161–163 °C; IR (neat) vmax 3321, 1684, 1655, 1475, 1410, 1299, 1016, 3064 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.68 (d, J= 6, 2H), 7.59 (t, J= 7.5, 1H), 7.54 (t, J= 7.5, 2H), 5.94 (s, 2H), 5.21 (t, J = 5.6, 1H), 4.56 (d, J= 5.6, 2H), 3.57 (s, 3H), 2.69 (s, 3H); 13C NMR (150 MHz DMSO-d6) δ 186.3, 171.0, 161.8, 155.7, 148.3, 134.1, 131.6, 129.0, 128.1, 124.8, 123.9, 57.6, 55.5, 38.6, 19.1; HRMS (ESI) calcd for [C17H17N5O3S + H]+ 372.1122 found 372.1097.
N-(5-(2-Bromo-2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetyl)-4-methylthiazol-2-yl)pivalamide (8a)
N-(4-Methyl-5-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetyl)thiazol-2-yl)pivalamide (3a; 50 mg, 0.14 mmol) was dissolved in dioxane (0.65 mL), the reaction flask was wrapped with tin foil, and the solution was heated to 60 °C. Elemental bromine (72 μL, 0.14 mmol) was added and the reaction was stirred for 5 h. The reaction was quenched by the addition of NaHSO3 (2 mL) and the resulting mixture was extracted with EtOAc (3 × 4 mL). The combined organics were washed with water and brine, dried over sodium sulfate, filtered, and concentrated. The crude material was purified by flash chromatography (2:1 EtOAc:hexanes) to afford 8a as a yellow amorphous solid (27 mg, 45%); IR (neat) vmax 2910, 2850, 1634, 1524, 1369, 1200, 1145, 1039, 970 cm−1 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 8.49 (s, 1H), 7.91 – 7.85 (m, 2H), 7.56 (s, 1H), 7.43 (t, J = 7.6, 2H), 7.35 (t, J = 7.3, 1H), 3.69 (s, 2H), 2.70 (s, 3H), 1.35 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 179.6, 176.9, 162.0, 161.0, 149.2, 130.1, 129.1, 128.8, 126.1, 121.5, 119.2, 55.6, 39.6, 27.4, 19.1; HRMS (ESI) calcd for [C19H20BrN5O2S + H]+ 462.0591, found 462.0589.
N-(5-(2-Bromo-2-(4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)-acetyl)-4-methylthiazol-2-yl)pivalamide (8b)
Yellow solid (16 mg, 35%); mp 120–121 °C; IR (neat) vmax 3170, 2964, 2930, 1686, 1527, 1367, 1321, 1139, 1036 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 11.46 (s, 1H), 8.76 (s, 1H), 8.33 (s, 1H), 5.18 (d, J = 5.8, 3H), 3.07 (s, 3H), 1.82 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 180.1, 178.0, 162.5, 161.5, 150.4, 124.1, 119.4, 57.9, 56.3, 39.9, 26.8, 18.8; HRMS (ESI) calcd for [C14H18BrN5O3S + H]+ 416.0392 found 416.0388.
Methyl 1-(1-bromo-2-(4-methyl-2-pivalamidothiazol-5-yl)-2-oxoethyl)-1H-1,2,3-triazole-4-carboxylate (8g)
Yellow solid (71 mg, 60%); mp 52–53 °C (decomp); IR (neat) vmax 3284, 2964, 1737, 1666, 1524, 1479, 1372, 1212, 1141, 1035, 972 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.42 (br s, 1H), 8.81 (s, 1H), 7.51 (s, 1H), 3.99 (s, 3H), 2.70 (s, 3H), 1.37 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 178.9, 177.2, 162.4, 161.5, 160.8, 141.0, 130.1, 118.7, 54.5, 52.6, 39.6, 27.2, 19.1; HRMS (ESI) calcd for [C15H18BrN5O4S + H]+ 444.0341 found 444.0340.
N-(2-Amino-4′-methyl-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,5′-bithiazol-2′-yl)pivalamide (9)
N-(2-Amino-4′-methyl-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,5′-bithiazol-2′-yl)pivalamide (20 mg, 0.043 mmol) and thiourea (3 mg, 0.047mmol) were dissolved in ethanol (0.210 mL) and the mixture was stirred at 60 °C for 3 h. The reaction was then concentrated and the crude material dissolved in EtOAc (3 mL). The organics were washed with water (4 mL) and brine (4 mL), dried over sodium sulfate, filtered, and concentrated. The crude product was purified by flash chromatography (3:1 EtOAc:hexanes) to afford 9 (14 mg, 77%); mp 196–200 °C; IR (neat) vmax 2964, 2920, 1684, 1630, 1515, 1479, 1372, 1310, 1132, 970 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.0, 2H), 7.56 (s, 1H), 7.48 (t, J = 8.0, 2H), 7.43 (t, J = 8.0, 2H), 2.81 (s, 3H), 1.37 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 176.8, 171.0, 163.6, 157.2, 154.6, 129.1, 128.9, 126.7, 126.4, 125.3, 123.6, 39.4, 29.7, 27.1, 18.9; HRMS (ESI): calcd for [C20H21N7OS2 + H]+ 440.1327, found 440.1318.
N-(4′-Methyl-2-(methylamino)-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,5′-bithiazol-2′-yl)pivalamide (10)
Brown solid (18 mg, 92%); mp 136–137 °C; IR (neat) vmax 3231, 2964, 2929, 1675, 1533, 1400, 1301, 1221, 1150, 1026 cm−1;1H NMR (400 MHz, CDCl3) δ 7.74 – 7.66 (m, 3H), 7.35 (t, J = 7.5, 2H), 7.28 (d, J = 7.4, 1H), 6.68 (s, 1H), 2.91 (d, J = 4.7, 3H), 1.88 (s, 3H), 1.24 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 167.8, 158.4, 148.0, 146.2, 137.0, 129.8, 129.3, 129.1, 128.8, 126.1, 125.5, 121.9, 117.2, 39.4, 32.0, 27.4, 15.8; HRMS (ESI): calcd for [C21H23N7OS2 + H]+ 454.1483, found 454.1473.
N-(2-(Allylamino)-4′-methyl-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,5′-bithiazol-2′-yl)pivalamide (11)
White solid (11 mg, 56%); mp 204–206 °C; IR (neat) vmax 2969, 2924, 2720, 1679, 1617, 1519, 1484, 1368, 1324, 1146, 977 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 7.78 (d, J = 7.4, 2H), 7.73 (s, 1H), 7.41 (t, J = 7.4 3H), 7.33 (t, J = 7.4, 1H), 6.20 (s, 1H), 5.88 (ddd, J = 5.5, 10.2, 17.1, 1H), 5.32 (d, J = 17.1, 1H), 5.22 (d, J = 10.2, 1H), 3.92 (t, J = 5.5, 3H), 1.96 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 176.1, 166.2, 157.9, 148.0, 146.5, 136.6, 132.9, 130.0, 129.1, 128.7, 126.2, 121.7, 118.1, 117.5, 116.7, 48.0, 39.3, 27.4, 15.9; HRMS (ESI) calcd for [C23H25N7OS2 + H]+ 480.1632, found 480.1630.
N-(2-(5-Chloro-2-methoxyphenylamino)-4′-methyl-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,5′-bithiazol-2′-yl)pivalamide (12)
White solid (16 mg, 67%); mp 150–154 °C; IR (neat) vmax 2964, 1675, 1604, 1533, 1479, 1284, 1257, 1035 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.89 (s, 1H), 8.17 (d, J = 2.5, 1H), 7.82 – 7.77 (m, 3H), 7.41 (t, J = 7.6, 2H), 7.34 (t, J = 7.6, 1H), 6.98 (dd, J = 2.5, 8.6, 1H), 6.81 (d, J = 8.6, 1H), 3.90 (s, 3H), 2.11 (s, 3H), 1.29 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 176.1, 159.9, 158.1, 148.3, 147.2, 146.3, 136.8, 130.0, 129.9, 129.1, 128.8, 126.5, 126.3, 122.6, 121.7, 118.0, 117.5, 117.0, 111.2, 56.3, 39.3, 27.4, 16.2; HRMS (ESI): calcd for [C27H26ClN7O2S2 + H]+ 580.1356, found 580.1351.
N-(2-(5-Chloro-2-methoxyphenylamino)-5-(4-(hydroxyl-methyl)-1H-1,2,3-triazol-1-yl)-4′-methyl-4,5′-bithiazol-2′-yl)pivalamide (13)
Yellow solid (15 mg, 64%); mp 172–173 °C; IR (neat) vmax 2977, 1679, 1599, 1528, 1484, 1412, 1297, 1252, 1172, 1030 cm−1; 1H NMR (400 MHz, acetone-d6) δ 10.37 (s, 1H), 9.44 (s, 1H), 8.69 (d, J = 2.3, 1H), 7.93 (d, J = 6.2, 1H), 7.02 – 6.88 (m, 2H), 4.67 (s, 2H), 3.83 (s, 3H), 2.85 (s, 1H), 2.06 (s, 3H), 1.28 (s, 9H); 13C NMR (100 MHz, acetone-d6) δ 176.5, 160.1, 158.1, 149.2, 146.9, 137.4, 131.0, 125.4, 125.1, 121.7, 117.9, 116.6, 111.7, 111.0, 109.0, 78.5, 55.9, 39.1, 26.5, 15.9; HRMS (ESI): calcd for [C22H24ClN7O3S2 + H]+ 534.1149, found 534.1139.
Methyl 1-(2-(5-chloro-2-methoxyphenylamino)-4′-methyl-2′-pivalamido-4,5′-bithiazol-5-yl)-1H-1,2,3-triazole-4-carboxylate (14)
White solid (19 mg, 80%); mp 198–200 °C; IR (neat) vmax 3169, 2955, 1737, 1693, 1675, 1541, 1497, 1435, 1372, 1292, 1221, 1043 cm−1; 1H NMR (600 MHz, acetone-d6) δ 10.43 (s, 1H), 9.59 (s, 1H), 8.77 (s, 1H), 8.75 (d, J = 2.4, 1H), 7.03 (dt, J = 5.5, 8.7, 2H), 3.91 (s, 3H), 3.87 (s, 3H), 2.90 (s, 3H), 1.33 (s, 9H); 13C NMR (150 MHz, acetone-d6) δ 176.5, 160.8, 160.5, 158.1, 147.4, 147.0, 139.9, 139.2, 131.9, 130.8, 125.4, 122.0, 118.0, 116.0, 115.9, 111.8, 55.9, 51.6, 39.1, 26.5, 16.2. HRMS (ESI): calcd for [C23H24ClN7O4S2 + H]+ 562.1098, found 562.1092.
1-(2-(5-Chloro-2-methoxyphenylamino)-4′-methyl-2′-pivalamido-4,5′-bithiazol-5-yl)-1H-1,2,3-triazole-4-carboxylic acid (15)
Beige solid (14 mg, 62%); mp 206–208 °C; IR (neat) vmax 3169, 2955, 1737, 1693, 1675, 1541, 1497, 1435, 1372, 1292, 1221, 1043 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.57 – 13.19 (m, 1H), 11.92 (s, 1H), 10.32 (s, 1H), 9.07 (d, J = 0.9, 1H), 8.66 (s, 1H), 7.13 – 7.02 (m, 2H), 3.90 (s, 3H), 2.29 (s, 3H), 1.20 (s, 9H); 13C NMR (100 MHz, acetone-d6) δ 187.2, 161.4, 161.2, 158.8, 148.1, 140.7, 140.0, 132.6, 131.6, 126.1, 122.7, 118.8, 116.8, 116.4, 112.6, 56.6, 52.4, 39.8, 27.2, 17.0. HRMS (ESI): calcd for [C22H22ClN7O4S2 + H]+ 548.0941, found 548.0933.
Supplementary Material
Acknowledgments
The authors thank the Tara K. Telford Fund for Cystic Fibrosis Research at the University of California, Davis, the National Institutes of Health (Grants DK072517, GM089153 and HL073856), and the National Science Foundation [CHE-0910870; grants CHE-0910870, CHE-0443516, CHE-0449845, and CHE-9808183 supporting NMR spectrometers].
Footnotes
Copies of 1H-NMR, 13C-NMR, and HRMS spectra for all new compounds can be found in the supporting information file. This material is available free of charge via the Internet at http://pubs.acs.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bobadilla JL, Macek M, Fine JP, Farrell PM. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 2.Sharma M, Benharouga M, Hu W, Lukacs GL. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 3.As reported by the Cystic Fibrosis Foundation (http://www.cff.org/treatments/Therapies/Kalydeco/), “Kalydeco™ (generic name, ivacaftor; previously known as VX-770) is a new oral medication for the treatment of cystic fibrosis, approved by the U.S. Food and Drug Administration (FDA) in January 2012. The FDA approved Kalydeco for people ages 6 and older with the G551D mutation of CF.”
- 4.Yu G, Yoo CL, Yang B, Lodewyk MW, Meng L, El-Idreesy TT, Fettinger JC, Tantillo DJ, Verkman AS, Kurth MJ. J Med Chem. 2008;51:6044–6054. doi: 10.1021/jm800533c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye L, Knapp JM, Sangwung P, Fettinger JC, Verkman AS, Kurth MJ. J Med Chem. 2010;53:3772–3781. doi: 10.1021/jm100235h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Meades C, Wood G, Osnowski A, Anderson S, Yuill R, Thomas M, Mezna M, Jackson W, Midgley C, Griffiths G, Fleming I, Green S, McNae I, Wu S, McInnes C, Zheleva D, Walkinshaw MD, Fischer PM. J Med Chem. 2004;47:1662–1675. doi: 10.1021/jm0309957. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly PS, Zanatta SD, Zammit SC, White JM, Williams SJ. Chem Commun. 2008;2459–2461 doi: 10.1039/b719724a. [DOI] [PubMed] [Google Scholar]
- 8.Katritzky AR, Wu J, Wrobel L, Rachwal S, Steel PJ. Acta Chem Scand. 1993;47:167–175. [Google Scholar]
- 9.Hantzsch A. Justus Liebigs Ann Chem. 1889;250:257–273. [Google Scholar]
-
10.(a) In our previously reported SAR study of 148 methylbithiazole analogues focused on the peripheral amide and aniline substructures (e.g., circled regions “a” and “c”, respectively, of the bithiazole depicted in Figure 1), we established that piv-4a (see right) is a more effective corrector than corr-4a.10b This, coupled with the click chemistry problems associated with the benzamide series, suggested to us that proceeding with only the pivalamide series (e.g., triazolobithiazoles 9–15) was appropriate Yoo CL, Yu GJ, Yang B, Robins LI, Verkman AS, Kurth MJ. Bioorg Med Chem Lett. 2008;18:2610–2614. doi: 10.1016/j.bmcl.2008.03.037.
- 11.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJV, Verkman AS. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 12.Malik I, Sedlarova MA, Csollei CS, Andrianainty P, Kurfurst P, Vanco J. Chem Pap. 2006;60:42–47. [Google Scholar]
- 13.Ghose AK, Viswanadhan VN, Wendoloski JJ. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.