

Abstract
Enteropathogenic Escherichia coli (EPEC) is a major cause of diarrheal disease in young children, yet symptoms and duration are highly variable for unknown reasons. Citrobacter rodentium, a murine model pathogen that shares important functional features with EPEC, colonizes mice in colon and cecum and causes inflammation, but typically little or no diarrhea. We conducted genome-wide microarray studies to define mechanisms of host defense and disease in C. rodentium infection. A significant fraction of the genes most highly induced in the colon by infection encoded CXC chemokines, particularly CXCL1/2/5 and CXCL9/10, which are ligands for the chemokine receptors CXCR2 and CXCR3, respectively. CD11b+ dendritic cells were the major producers of CXCL1, CXCL5, and CXCL9, while CXCL2 was mainly induced in macrophages. Infection of gene-targeted mice revealed that CXCR3 had a significant but modest role in defense against C. rodentium, whereas CXCR2 had a major and indispensable function. CXCR2 was required for normal mucosal influx of neutrophils, which act as direct antibacterial effectors. Moreover, CXCR2 loss led to severe diarrhea and failure to express critical components of normal ion and fluid transport, including ATPase β2-subunit, CFTR, and DRA. The antidiarrheal functions were unique to CXCR2, since other immune defects leading to increased bacterial load and inflammation did not cause diarrhea. Thus, CXCR2-dependent processes, particularly mucosal neutrophil influx, not only contribute to host defense against C. rodentium, but provide protection against infection-associated diarrhea.
Enteropathogenic Escherichia coli (EPEC)3 is an important cause of diarrheal disease and mortality worldwide, especially among infants and young children in the developing world (1). EPEC is distinguished from other diarrheagenic E. coli by its ability to form attaching/effacing (A/E) lesions characterized by intimate attachment to the epithelium and localized microvillus loss, combined with a lack of classical enterotoxins. The infection is accompanied by watery diarrhea, fever, vomiting, and abdominal pain, and it can lead to significant mortality. The mechanisms governing infection-associated diarrhea are not well understood, but they appear to be multifactorial (2). A decrease in the absorptive surface epithelium, disruption of tight junctions and epithelial barrier function, and release of secretagogues by inflammatory cells have been proposed to mediate diarrhea, although the relative importance of these and possibly other mechanisms remains unknown (2–4).
Progress in elucidating the pathogenesis of EPEC infection has been hampered by the dearth of suitable animal models. Instead, model pathogens with characteristics similar to EPEC have been employed widely, including rabbit EPEC and Citrobacter rodentium in mice. C. rodentium causes A/E lesions and expresses key virulence genes, including intimin, espA, espB, and tir, that are functionally exchangeable with the corresponding genes in EPEC. Infection of adult mice with C. rodentium leads to self-limiting epithelial hyperplasia and mucosal inflammation (5–8). Effective host defense against the pathogen requires CD4+ T cells, B cells, mast cells, and neutrophils (9–11). IgG Abs contribute to bacterial clearance, whereas secretory IgA or IgM plays a limited role in host defense (12, 13). Several immunoregulatory cytokines, including IFN-γ, TNF-α, IL-6, IL-12, IL-17, and IL-22, control innate or adaptive immune responses against the bacteria, since mice lacking the respective cytokines have a reduced ability to clear infection (14–19). However, the histologically apparent complexity of the host response that accompanies infection (5, 7) suggests that numerous other gene products are likely to be involved in orchestrating mucosal defense against the bacteria.
Expression analysis of large numbers of genes by microarrays has reached a technical maturity that now allows routine application to many biological systems. To date, such expression studies have mostly been conducted in cell culture models, as they allow tight control over experimental conditions. However, a number of in vivo applications have demonstrated the utility of microarrays in defining previously elusive biological processes in complex physiological settings (20–22). Based on such prior successes, we set out to examine the global gene expression profile associated with host defense against the model pathogen, C. rodentium. Our expression studies and the mechanistic pursuit of the findings unexpectedly revealed a novel role of CXC chemokines and neutrophils in controlling not only bacterial load but also the diarrheal response to infection, thus providing new insights into the importance of host components in determining diarrhea after infection with an A/E lesion-forming enteric pathogen.
Materials and Methods
Mice and infection protocol
All wild-type and gene-targeted mice of either gender were obtained from The Jackson Laboratory and were at least 8 wk old for experiments. For infections, C. rodentium (a gift from Dr. B. Vallance, British Columbia Children’s Hospital, Vancouver, Canada) was grown overnight in Luria-Bertani broth at 37°C, harvested by centrifugation, and resuspended in fresh broth at 2.5 × 109/ml. Mice were infected by oral gavage with 200 μl (5 × 108 bacteria) of the suspension. To determine bacterial numbers in the stool, fecal pellets were collected from individual mice, weighed, and homogenized in 5 ml of PBS. For enumerating bacteria in spleen and liver, each organ was homogenized in 2 ml of PBS. Serial dilutions of the homogenates were plated onto MacConkey agar, and the number of CFU was determined after overnight incubation at 37°C. The detection limit of the CFU assay was 103 CFU/g feces and <101 CFU per liver or spleen. The identity of representative colonies was verified by PCR analysis of the C. rodentium espB gene as described (12). All animal studies were reviewed and approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Histological analysis
Organs were removed and fixed in 10% phosphate-buffered formalin (Fisher Scientific) for 24 h. Colons were opened longitudinally, cleaned, and processed as “Swiss rolls” before fixation. Fixed tissues were embedded in paraffin, and 5-μm sections were prepared and stained with H&E. For immunohistological studies, tissues were frozen in OCT compound (Sakura Finetek), cut on a cryomicrotome, air-dried, fixed in acetone, and incubated with 0.3% H2O2 for 20 min at room temperature to inactivate endogenous peroxidase. After blocking with PBS containing 2% rabbit serum (Abcam), sections were incubated overnight at 4°C in the same buffer containing 1 μg/ml rabbit anti-mouse myeloperoxidase (MPO; Abcam), a 1/1000 dilution of rabbit anti-C. rodentium (from B. Vallance) (12), or 1 μg/ml normal rabbit IgG (Jackson ImmunoResearch Laboratories) as a control. Sections were washed, stained for 30 min at room temperature with 2 μg/ml HRP-conjugated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), developed with 3,3′-diaminobenzidine/H2O2 (Vector Laboratories), and counterstained with Gill’s hematoxylin (Fisher Scientific). Alternatively, sections were stained with 3 μg/ml Cy3-conjugated goat anti-rabbit IgG (R&D Systems) and mounted in media containing 4′,6′-diamidino-2-phenylindole (Vector Laboratories).
Microarray and real-time PCR analysis
RNA was isolated from whole colon tissue using TRIzol reagent (Invitrogen). RNA quality was assessed with an Agilent Technologies bioanalyzer. Total RNA was processed and hybridized to CodeLink Mouse Whole Genome Bioarrays (carrying 34,790 unique probes; GE Healthcare). Biotin-labeled first-strand cDNA from 2.5 to 5 μg of total RNA was used to hybridize arrays in 6× standard saline citrate phosphate/EDTA/30% formamide at 25°C for 18 h. Hybridized slides were washed and stained with streptavidin-Alexa 647 conjugate (Invitrogen/Molecular Probes). The processed slides were scanned at 10 μm resolution with an Axon 4000B Scanner (Molecular Devices). Positive and negative bacterial control genes were spotted on the CodeLink arrays, and specific bacterial mRNA was used to spike the cDNA synthesis reaction. Background correction was performed by the CodeLink software. For each array, RNA was pooled from four to six mice. Filtering was conducted by deleting probes that had low or ambiguous values. Numbers were normalized by dividing raw intensity values by medians for each chip. The complete microarray data sets are available on the National Center for Biotechnology Information Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) under accession no. GSE16847.
For PCR analysis, RNA was treated with Turbo DNase-free (Ambion) to remove contaminating DNA. Reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) and real-time PCR amplification using the Mesa Green 2× SYBR mix (Eurogentec). Amplification of the expected single products was confirmed on 1% agarose gels stained with ethidium bromide. Primers and PCR product sizes are listed in supplemental Table I.4 Relative changes in target mRNA levels were calculated by the 2ΔΔCt method, with GAPDH as the reference standard.
Isolation of epithelial and lamina propria cells
The colon was opened, incubated for 10 min at room temperature in PBS with 5% FCS and 1 mM DTT to remove mucus, and cut into 5-mm pieces. Tissue pieces were incubated in HBSS (without Ca and Mg) with 5 mM EDTA, 5% FCS, 10 mM HEPES (pH 7.3), and 1 mM DTT for 2 × 20 min at 37°C with constant shaking. Detached epithelial cells were passed through a nylon mesh strainer (100-μm pore size; Fisher Scientific) and collected by centrifugation. The remaining tissue pieces were washed and incubated twice at 37°C for 30 min in RPMI 1640 medium containing 1 mg/ml collagenase D (Roche Applied Science) and 100 μg/ml DNase I (Worthington Biochemical). After each incubation, mixtures were vortexed for 30 s, and suspensions were passed through a 40-μm cell strainer. Cells from both digestions were combined and collected by centrifugation.
Flow cytometry
Cell suspensions were stained with optimal concentrations of fluorochrome-conjugated Abs against mouse CD11b, CD11c, CD45, Gr-1 (all from eBioscience), and/or CXCR2 (R&D Systems) for 30 min on ice. Cells were washed and fixed with 2% paraformaldehyde in PBS before analysis on a BD Biosciences FACSCalibur flow cytometer. Cell sorting of stained but unfixed cells was performed on a MoFlo high-speed cell sorter (Beckman Coulter). Macrophages were identified as CD45+CD11b+CD11c− cells. Dendritic cells were identified as CD45+CD11c+ cells and further subdivided into CD11b+ and CD11b− subsets. All other leukocytes were identified as CD45+CD11b−CD11c− cells.
Immunoblotting
Tissues were lysed for 30 min on ice in a buffer containing 150 mM NaCl, 5 mM KCl, 10 mM HEPES, 0.5 mM EDTA, 0.2 mM EGTA, 1 mM sodium fluoride, 1 mM vanadate, 0.05% Nonidet P-40, 1 mM DTT, and a protease inhibitor cocktail (Roche Applied Science). After centrifugation to remove debris, equal protein amounts (20 μg) were boiled for 10 min in loading buffer (50 mM Tris (pH 6.8), 100 mM DTT, 2% SDS, 40% glycerol, and 0.2% bromophenol blue) and size-separated by electrophoresis on a 10% Tris-glycine polyacrylamide gel (Bio-Rad Laboratories). Proteins were electrotransferred to a polyvinylidene difluoride membrane, which was blocked and stained overnight at 4°C with rabbit anti-mouse DRA/SLC26A3 (Santa Cruz Biotechnology) in a buffer of 10 mM Tris (pH 7.6), 5% BSA, and 0.05% Tween 20, followed by incubation with HRP-conjugated goat anti-rabbit IgG and detection by ECL (GE Healthcare).
Neutrophil killing assay
A suspension of bone marrow cells, obtained from the femurs of C57BL/6 mice, was layered on top of two Histopaque layers (densities of 1.083 and 1.119 g/ml) (Sigma-Aldrich) and centrifuged at 1200 rpm for 30 min. Cells at the interface between the two Histopaque layers were collected and washed in HBSS. Neutrophil purity was >90% as determined by Giemsa staining. Cells (5 × 107/ml) were pretreated for 1 h with mouse TNF-α and/or IFN-γ (50 ng/ml each) (both from PeproTech), and C. rodentium was added at a multiplicity of infection of 1:1 in 100 μl of RPMI 1640 medium. After incubation at 37°C for up to 6 h with light agitation, cultures were diluted 1/10 in water to lyse the neutrophils, and serial dilutions were plated onto MacConkey agar.
Analysis of colonic cytokine production
Colon pieces (1 × 1 mm) were placed into 12-well plates in 500 μl of RPMI 1640 medium containing 50 μg/ml each streptomycin and penicillin (both from Sigma-Aldrich) and incubated for 6 h at 37°C. Supernatants were centrifuged and frozen at −80°C. In some experiments, colon tissues were homogenized in PBS with 0.1% Tween 20 and a cocktail of protease and phosphatase inbibitors (Roche Applied Science), centrifuged to remove debris, and supernatants were frozen at −80°C. Cytokine levels were determined by ELISA (R&D Systems) and were normalized to tissue weight.
ELISA
Blood was collected by tail vein bleeding, diluted 1/10 into PBS containing 2 mM EDTA, centrifuged, and stored at −80°C until use. C. rodentium, grown overnight in Luria-Bertani broth, were washed extensively in PBS, and 108 bacteria were added in 50 μl per well of a 96-well polystyrene plate. After overnight air drying, bacteria were fixed for 5 min at room temperature with 0.15% glutaraldehyde in 0.15 M phosphate buffer (pH 7.0), followed by blocking with 0.15 M glycine in 15 mM phosphate buffer (pH 7.0), and plates were incubated overnight at 4°C with PBS containing 5% nonfat dry milk and 0.5% Tween 20. After incubation with serum samples for 2 h, HRP-conjugated goat Abs against mouse IgG or IgM (SouthernBiotech) were applied to the plates for 1 h at room temperature. Bound peroxidase was visualized with tetramethylbenzidine-H2O2 in acetate buffer, and reactions were stopped with sulfuric acid and read at 450 nm.
MPO analysis
Colon pieces were snap-frozen in liquid nitrogen and stored at −80°C. Tissues were homogenized in hexadecyltrimethylammonium bromide buffer (Sigma-Aldrich), cleared by centrifugation, and the supernatant was added to a solution containing o-dianisidine (Sigma-Aldrich) and H2O2. Absorbance of the colorimetric reaction was measured at 600 nm. Purified MPO of known activity was used as a standard. MPO activity is expressed in units per gram of wet tissue.
Fecal water and ion transport studies
For fecal water analysis, stool was collected for 1 h from individual mice, weighed, dried for 24 h at 95°C, and weighed again. Percentage of fecal water was calculated by dividing the difference of dry and total weight by the total weight. For electrophysiological studies, a 3-cm segment of mid-distal colon was stripped of the muscle layer and cut into smaller sections that were then mounted on Ussing chamber inserts (Warner Instruments) with a window area of 0.07 cm2. Tissues were bathed in a modified oxygenated Ringer’s solution (140 mM Na+, 5.2 mM K+, 1.2 mM Ca+, 0.8 mM Mg+, 120 mM Cl−, 25 mM HCO3−, 2.4 mM H2PO4−, 0.4 mM HPO42−, and 10 mM glucose) at 37°C. The tissues were short-circuited by an automated voltage clamp, and the current (Isc) required to obtain zero potential difference across the tissues was monitored as an indication of net active ion transport. To determine transepithelial resistance, a defined voltage peak was transiently applied across the tissue, the resulting current was measured, and resistance was calculated by Ohm’s law.
Statistical analysis
Data are expressed as means ± SEM. Differences were analyzed by t test, with p values of <0.05 considered significant. CFU counts were log10 transformed, and means and SEs of the mean were calculated from the log values. Samples without detectable C. rodentium colonies were assigned a log10 value equivalent to half of the detection limit of the CFU assay. Differences between CFU counts were evaluated by Mann-Whitney rank-sum test, with p values of <0.05 considered statistically significant. Results from males and females were combined, as no significant differences were detected between the genders in bacterial colonization or mucosal responses after infection.
Results
Global gene expression analysis of the colonic response to C. rodentium reveals prominent induction of chemokines
To broaden the understanding of the pathogenesis of enteric infections with A/E lesion-forming bacteria, we used a murine infection model with the prototypic pathogen, C. rodentium (8), and conducted a microarray analysis as an unbiased approach to obtain a genome-wide gene expression profile of the colonic response to infection. Intragastric administration of C. rodentium to normal C57BL/6 mice leads to peak infection levels by 1 wk, and bacterial clearance after 2–3 wk (12) (see Figs. 3A and 4A below). Accordingly, we extracted total RNA from the colon of infected mice at different times after infection (1–3 wk) and related expression levels to a parallel cohort of uninfected mice. The microarray studies revealed a wide spectrum of expression changes, whose exact characteristics were dependent on the selection criteria applied for determining significant and meaningful changes. To select suitable criteria, we first evaluated the reliability of the microarray data by assessing mRNA levels at different times after infection for 20 representative genes with real-time PCR as an independent, sensitive, and highly specific approach. Comparison of microarray and real-time PCR data showed a strong positive correlation, with greater changes in expression showing better reproducibility than lesser changes (Fig. 1A). Based on this observation, we subsequently focused on genes whose expression levels were >4-fold altered at any point after infection.
FIGURE 3.

C. rodentium infection of CXCR3-deficient mice. Cxcr3−/− (○) and wild-type mice (●) were orally infected with C. rodentium. A, Bacterial numbers in stool, liver, and spleen were determined by CFU assay. Data are means ± SEM (n = 20). *, p <0.05 by rank sum test compared with wild-type mice at the same time. B, Paraffin sections of the colon were prepared and stained with H&E. Photographs were taken with a ×20 lens and processed equally for inclusion in the figure. C, MPO activity in the colon of infected mice (2 wk) was determined by enzymatic assay, and was normalized against tissue weight. MPO activity in both groups of uninfected mice was <0.01 U/g. D, H&E-stained paraffin sections of the liver show that Cxcr3−/− mice, but not wild-type mice, had distinct necrotic areas (arrows). E, Serum was collected at the indicated times after infection, and IgM and IgG Abs against C. rodentium were determined by ELISA with whole bacteria as Ag. Data are means ± SEM (n ≥ 6 mice/group). In C and E, *, p < 0.05 by t test compared with wild-type mice.
FIGURE 4.

Importance of CXCR2 in host defense against C. rodentium. Cxcr2−/− mice (○) and wild-type mice (●) were infected with C. rodentium. A, Bacterial numbers in stool, liver, and spleen were determined by CFU assay. Data are means ± SEM (n = 16). *, p < 0.05 by rank sum test compared with wild-type mice at the same time. B and C, H&E-stained paraffin sections of colon and liver. D, Paraffin sections of the colon were prepared and stained by indirect immunofluorescence with Abs against C. rodentium (dark gray) and counterstained with a nuclear stain (light gray). Photographs in B–D were taken with a ×20 lens and processed equally for each panel. E, Serum was collected and tested by ELISA for IgM and IgG Abs against C. rodentium using whole bacteria as Ag. Data are means ± SEM (n ≥ 10/group). *, p < 0.05 by t test compared with wild-type mice.
FIGURE 1.
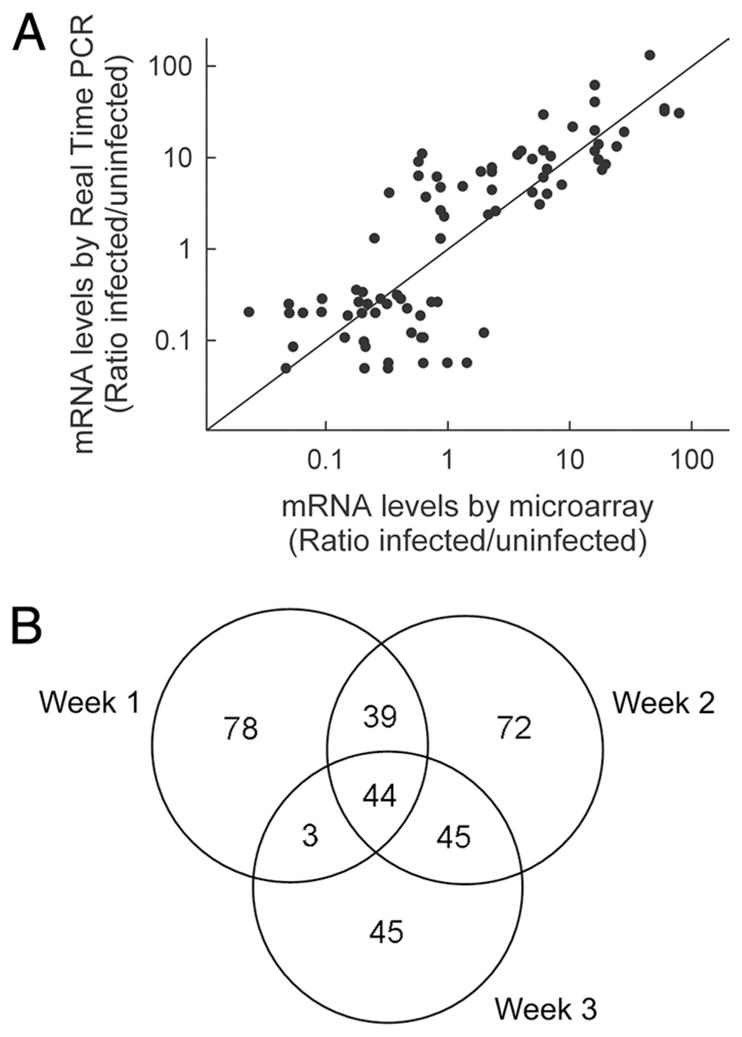
Microarray analysis of colonic gene expression after C. rodentium infection. Wild-type (C57BL/6J) mice were infected orally with 5 × 108 C. rodentium, or left uninfected. Total colon RNA was extracted after 1, 2, and 3 wk and subjected to microarray analysis. In parallel, mRNA levels of representative genes were assayed by real-time PCR. mRNA levels are expressed as ratio of the levels in infected over uninfected mice. A, Correlation of microarray data and real-time PCR results. Each datum point represents one gene at one time point. B, Venn diagram of all genes with a >4-fold increase in expression at any time after infection.
The microarrays employed in our studies carried 34,790 probes covering the entire murine genome. Of these, 577 probes (1.7%), representing the same number of unique genes, revealed a >4-fold increase (326 genes) or decrease (251 genes) in expression at any time after C. rodentium infection (Fig. 1B, supplemental Table II, and data not shown). Most changes were observed at weeks 1 and 2, while relatively fewer changes were seen after 3 wk. Increases dominated over decreases by ratios ranging from 2.2:1 after 1 wk to 1.5:1 after 3 wk. Furthermore, increased expression of most of the genes was confined to a limited period after infection, with only few genes showing significant increases throughout the 3-wk period and practically no genes exhibiting a biphasic expression pattern (Fig. 1B and supplemental Table II). Taken together, these results suggest that the initial host response to infection is characterized by induction of an array of “early response” genes, while the later clearance and healing phase is dominated by induction of a different set of genes.
Because we sought to elucidate the events important in the initiation of host defense against the bacteria, we focused on those genes for which the microarrays had revealed the earliest and greatest relative increase in expression (Table I and supplemental Table II). Of the top 20 genes in this category, 4 were CXC chemokines. Most of the other highly induced genes encode products with diverse functions, ranging from acute phase reactants (leucinerich α2-glycoprotein 1 and serum amyloid A3) and calcium-binding proteins with diverse intracellular and extracellular functions (S100A8 and S100A9) (23) to ion pumps (Na+/K+ ATPase, γ-subunit) and lectins with antimicrobial activity (Reg3α, Reg3β, and Reg3γ) (24).
Table I.
Induction of colonic gene expression after C. rodentium infectiona
| No. | Fold Change | Accession No. | Gene | Protein |
|---|---|---|---|---|
| 1 | 183 | NM_009114 | S100a9 | S100 calcium-binding protein A9 (calgranulin B) |
| 2 | 136 | NM_013650 | S100a8 | S100 calcium-binding protein A8 (calgranulin A) |
| 3 | 80 | NM_029796 | Lrg1 | Leucine-rich α2-glycoprotein 1 |
| 4 | 35 | NM_009141 | Cxcl5 | Chemokine (C-X-C motif) ligand 5 (LIX) |
| 5 | 35 | NM_011315 | Saa3 | Serum amyloid A3 |
| 6 | 28 | NM_008392 | Irg1 | Immunoresponsive gene 1 |
| 7 | 22 | NM_009140 | Cxcl2 | Chemokine (C-X-C motif) ligand 2 (MIP-2) |
| 8 | 21 | NM_011259 | Reg3a | Regenerating islet-derived 3α |
| 9 | 18 | NM_008361 | Il1b | IL-1β |
| 10 | 17 | NM_011260 | Reg3g | Regenerating islet-derived 3γ |
| 11 | 17 | NM_010819 | Clec4d | C-type lectin domain family 4, member d |
| 12 | 17 | NM_013415 | Atp1b2 | Na+/K+-ATPase β2-subunit |
| 13 | 16 | NM_001033199 | AI747448 | Calcium-activated chloride channel |
| 14 | 15 | NM_001146073 | Hexdc | Hexosaminidase (glycosyl hydrolase family 20) |
| 15 | 14 | NM_008176 | Cxcl1 | Chemokine (C-X-C motif) ligand 1 (KC) |
| 16 | 13 | NM_011036 | Reg3b | Regenerating islet-derived 3β |
| 17 | 12 | NM_009896 | Socs1 | Suppressor of cytokine signaling 1 |
| 18 | 12 | NM_008599 | Cxcl9 | Chemokine (C-X-C motif) ligand 9 (MIG) |
| 19 | 11 | NM_008489 | Lbp | LPS-binding protein |
| 20 | 11 | NM_175449 | Fam26f | Hypothetical protein LOC215900 |
Total RNA was extracted from the colon of C. rodentium-infected (week 1) and uninfected C57BL/6J mice and subjected to microarray analysis. Levels of mRNA are expressed as ratio of infected over uninfected tissues (fold change) and are sorted in decreasing order of induction for the top 20 genes.
Since chemokines were highly overrepresented among the most highly induced genes (i.e., they made up 20% of the top 20 genes, but only represent ~0.15% of all predicted murine genes) and since they are critical chemoattractants and activators of leukocyte subsets, we focused our further analysis on the entire group of chemokines represented on the microarrays. A distinct chemokine expression pattern was observed, with the highest early induction (>10-fold on week 1) observed for CXCL1, CXCL2, CXCL5, and CXCL9 (Table I), and modest early induction for CXCL3 (5.7-fold), CXCL10 (7.6-fold), and CXCL11 (4.8-fold). Limited or no early induction (<4-fold) was observed for the other CXC chemokines or for most CC chemokines, with the partial exception of CCL7 (6.2-fold) and CCL19 (4.4-fold). These data show that rapid induction of a distinct subset of CXC chemokines is a prominent feature of the colonic host response to C. rodentium.
Increased colonic CXCL expression after C. rodentium infection
To further investigate chemokine functions in host defense against C. rodentium, we focused on the two most highly induced groups of CXC chemokines, CXCL1/2/5 and CXCL9/10/11. These chemokines are cognate ligands for only two CXC chemokine receptors, CXCR2 and CXCR3, respectively, suggesting that the ligand interactions with these two receptors may be important in antibacterial host defense. Time course studies revealed a rapid induction of at least five of the six chemokines in the colon within 3 days after infection and maximal expression by 7–14 days (Fig. 2A). Marked, albeit lower, increases were also observed in jejunum, mesenteric lymph nodes, liver, and spleen, indicating that the chemokine responses were not limited to the site of infection (Fig. 2B). Furthermore, protein production followed mRNA changes, as demonstrated by ELISA in supernatants of short-term colon explant cultures for CXCL5 and CXCL9 as representatives of each chemokine group (Fig. 2C).
FIGURE 2.

Chemokine expression in response to C. rodentium infection. C57BL/6J mice were infected with C. rodentium or left uninfected (week 0), and mRNA expression was analyzed by real-time PCR for the indicated chemokines in the colon (A) or the indicated organs (B). Expression levels are shown relative to uninfected controls. Data are means ± SEM (n ≥ 4). C, Explants of whole colon tissue were cultured for 6 h, and levels of CXCL5 and CXCL9 in the supernatants were examined by ELISA. Data are means ± SEM (n ≥ 4). D, The indicated cell types were isolated from the colon of infected (2 wk) and uninfected mice by differential detachment (epithelial cells) or FACS (leukocyte subsets) and examined for CXC chemokine expression by real-time PCR. Data are shown as ratio of mRNA levels in cells from infected over uninfected mice. *, p < 0.05 by t test compared with the levels in uninfected mice.
We next examined the cellular source of the chemokines after infection. Epithelial cells and subsets of lamina propria cells were isolated and analyzed by real-time PCR for chemokine expression. CXCL1, CXCL5, and CXCL9 were most highly induced in CD11b+ dendritic cells, while CXCL2 was mainly induced in macrophages (Fig. 2D). CXCL5 was also modestly induced in epithelial cells and macrophages, and CXCL9 in epithelial cells. In contrast, CD11b− dendritic cells or any other CD45+ leukocytes were not a significant source of the four chemokines after infection (Fig. 2D). Thus, CD11b+ dendritic cells and macrophages were the major chemokine responders to C. rodentium.
CXCR3 in host defense against C. rodentium
As a next step toward elucidating the role of specific CXC chemokines in defense against C. rodentium, we determined the impact of the loss of ligand/receptor interactions on the pathogenesis of infection. For this, we employed mice with targeted deletions of the CXC chemokine receptors, CXCR2 and CXCR3, as several of their respective cognate ligands were induced after infection. Mice lacking CXCR3 are fertile and healthy under standard husbandry conditions (25). Upon oral C. rodentium challenge, CXCR3-deficient mice had significantly increased fecal CFU at 14 and 17 days after infection (Fig. 3A), times that represent the middle to late bacterial clearance phase. No significant differences in bacterial load were observed at 7 days or 21 days (Fig. 3A), indicating that CXCR3 was dispensable in the early innate control and ultimate eradication of the bacteria. Furthermore, CXCR3-deficient mice had significantly higher bacterial numbers in liver and spleen at 2 wk, but surprisingly also after 1 wk, at which time fecal CFU did not differ between knockout and wild-type mice (Fig. 3A).
Histological analysis of the colon revealed that CXCR3-deficient mice had modestly increased submucosal and mucosal infiltration with inflammatory cells, and greater epithelial ulceration after 2 wk (Fig. 3B), which was accompanied by elevated levels of the neutrophil marker MPO in colon homogenates (Fig. 3C). Moreover, the liver showed disseminated foci of necrotic tissue in CXCR3-deficient but not wild-type mice after infection (Fig. 3D). These and the CFU data suggest that CXCR3-dependent processes not only help to control bacterial clearance in the colon, but have additional functions in containing systemic infection and tissue damage.
Analysis of Ab titers against C. rodentium demonstrated that CXCR3 deficiency significantly delayed induction of specific IgG Abs, whereas specific IgM production was not affected (Fig. 3E). Delayed antibacterial IgG responses may partly explain the delayed bacterial clearance in the absence of CXCR3, since IgG contributes to immune defense against the bacteria (12, 13). Taken together, these studies show that CXCR3 has a significant but limited role in host defense against C. rodentium.
Loss of CXCR2 severely compromises C. rodentium clearance but not development of specific Ab responses
Upon realization that CXCR3 was of limited importance in antibacterial defense, we turned to CXCR2, the other chemokine receptor whose cognate ligands were strongly and rapidly induced after infection. Mice lacking CXCR2 are fertile and generally healthy, although they display disturbances in constitutive neutrophil trafficking (26). After oral C. rodentium infection, CXCR2-deficient mice had bacterial numbers comparable to wild-type controls after 1 wk, but subsequently displayed a marked defect in controlling and eradicating the infection. They continued to have detectable fecal CFU counts for at least 4 wk, whereas wild-type mice had cleared the bacteria by 3 wk (Fig. 4A). In parallel, increased bacterial numbers were found in liver and spleen of CXCR2-deficient mice (Fig. 4A).
Histologically, CXCR2-deficient mice exhibited increased signs of colonic inflammation and ulceration after 2 wk, while infection-associated epithelial hyperplasia normally found in wild-type mice was attenuated in the knockout mice (Fig. 4B). No pathological changes were observed in the liver of either CXCR2-deficient or wild-type mice (Fig. 4C), suggesting that CXCR2, unlike CXCR3, had no unique role in preventing infection-associated liver damage. Immunohistological staining demonstrated that CXCR2-deficient mice had greater bacterial colonization of the colon surface and deeper infiltration of C. rodentium into colonic crypts (Fig. 4D). In some crypts of knockout mice, bacteria were found throughout the entire crypt including the base, whereas bacterial colonization was always limited to the colon surface in wild-type mice (Fig. 4D).
In contrast to CXCR3 knockout mice, CXCR2-deficient mice were not impaired in their ability to mount specific Ab responses against the bacteria, since they had elevated (rather than decreased) levels of antibacterial IgM and IgG relative to wild-type mice after 2 wk and no significant differences after 3 wk (Fig. 4E). Taken together, these results indicate that CXCR2 plays an important and indispensable role in mucosal defense against C. rodentium.
CXCR2 deficiency abolishes infection-associated influx of neutrophils as critical effector cells
Given the key role of CXCR2 in host defense against C. rodentium, we began to explore the mechanism by which the receptor mediates its physiologic functions. Flow cytometric analysis showed that the number of CXCR2+ cells in the colon increased by ~9-fold 1 wk after infection of wild-type mice (Fig. 5A). The vast majority (95%) of these cells were positive for the neutrophil marker Gr-1. Conversely, a large proportion (84%) of Gr-1+ cells were also CXCR2+ (Fig. 5A). Consistent with the flow cytometrically detected influx of neutrophils, levels of the neutrophil-specific marker MPO increased 8-fold in the colon of infected mice (Fig. 5B).
FIGURE 5.

Neutrophil involvement in host defense against C. rodentium. A, C57BL/6 mice were infected with C. rodentium or left uninfected (week 0). Lamina propria cells were isolated from the colon and examined by flow cytometry for CXCR2 and Gr-1 expression. Numbers in the quadrants indicate the percentage of each cell population relative to total lamina propria cells. B, Time course analysis of Gr-1+ cells (left panel) and MPO activity (right panel) in the colon of wild-type (●) and Cxcr2−/− mice (○). Data are means ± SEM (n ≥ 4/group). *, p < 0.05 by t test compared with wild-type mice. C, Frozen sections of the colon were prepared and stained by an indirect immunoperoxidase technique with an Ab against MPO (dark gray), and counterstained with hematoxylin (light gray). Enlargements of the mid-crypt regions are shown in the insets. Images were taken with a ×20x lens and processed equally for either the low- or high-magnification panels. D, Neutrophils were prepared from the bone marrow of wild-type mice and were preincubated for 1 h with TNF-α, IFN-γ, or TNF-α plus IFN-γ, or left unstimulated. C. rodentium was added at a multiplicity of infection of 1:1, and total bacterial numbers in the cultures after hypotonic cell lysis were determined by CFU assay at the indicated times. Data are means ± SEM (n = 3/group) of a representative experiment (*, p < 0.05 by t test relative to untreated neutrophils). Two additional experiments yielded similar results.
In contrast to wild-type mice, neutrophil numbers did not increase in the colon of infected CXCR2-deficient mice, as determined by flow cytometry for Gr-1+ cells and MPO assay (Fig. 5B). Immunohistological staining confirmed that CXCR2-deficient mice were severely impaired in colonic neutrophil influx, whereas wild-type mice exhibited marked mucosal infiltration in the sub-epithelial surface and pericryptal regions (Fig. 5C). These data demonstrate that CXCR2 is critical for neutrophil influx into the colon in response to C. rodentium infection.
Because CXCR2 deficiency led to a loss of normal mucosal neutrophil recruitment, we asked whether these cells can act as direct effectors against the bacteria. Neutrophils were isolated from the bone marrow and tested for their ability to kill C. rodentium. Resting neutrophils could not kill the bacteria over a 6-h incubation period (Fig. 5D). However, stimulation with TNF-α induced significant killing capacity in the neutrophils, which was further enhanced by costimulation with IFN-γ (Fig. 5D). Both cytokines are known to be important in host defense against C. rodentium (14, 15). These results indicate that activated neutrophils kill C. rodentium, which can explain their role in antibacterial defense (11).
Critical role of CXCR2 in protection against infection-associated diarrhea
In addition to impaired bacterial clearance, we observed that infection of CXCR2-deficient mice, but not wild-type controls, was accompanied by marked diarrhea. Thus, analysis of fecal water content showed that diarrhea began within 7 days after infection and persisted for 2–3 wk, whereas no evidence of increased fecal water content was observed in wild-type mice at any time after infection (Fig. 6A). The underlying mechanisms of diarrhea were local in nature, since electrophysiological studies of explanted colonic mucosa in Ussing chambers demonstrated reversal of the normally positive baseline Isc in wild-type mice to a negative baseline Isc in infected CXCR2-deficient mice (Fig. 6B). These results are consistent with a loss of normal sodium and/or chloride uptake mechanisms in the colon, which can contribute to diarrhea by removing the driving force for passive water absorption from the lumen. In contrast, trans-mucosal resistance was not significantly different between knockout or wild-type mice before or after infection (data not shown), indicating that the epithelium was not differentially compromised by infection.
FIGURE 6.

Severe diarrhea in C. rodentium-infected Cxcr2−/− mice. Cxcr2−/− mice (open symbols and bars) and wild-type mice (closed symbols and bars) were infected with C. rodentium. A, Fecal water content was determined at the indicated times after infection. Data are means ± SEM (n = 6/group). *, p < 0.05 by t test relative to wild-type mice. B, Electrophysiological analysis of net ion transport (baseline short-circuit current, Isc) in Ussing chamber-mounted, muscle-stripped colon segments from infected and uninfected (week 0) mice. Data are means ± SEM (n = 6/group). *, p < 0.05 by t test. C, Expression levels of the indicated ion transporters and pumps in the colon of infected (week 2) and uninfected mice were determined by real-time PCR and are shown as ratios of infected over uninfected mice. Data are means ± SEM (n = 3/group). *, p < 0.05 by t test. D, Immunoblot analysis of DRA and GAPDH in infected and uninfected (week 0) mice. For each lane, colon extracts from four to six mice were pooled before analysis.
To define the molecular basis of the apparent defects in ion transport in CXCR2-deficient mice, we analyzed the expression of key ion transporters in the colon. Wild-type mice showed marked increases in mRNA expression for ATPase (β2-subunit), CFTR, and DRA, and modest increases for NHE3 and PAT1 upon infection (Fig. 6C). In contrast, CXCR2-deficient mice were severely impaired in the induction of these ion transporters. Immunoblots confirmed that DRA was strongly induced in wild-type mice, but only weakly in their CXCR2-deficient littermates in response to infection (Fig. 6D). Expression levels of the epithelium-specific product villin were not different between the groups (data not shown), indicating that differential loss of epithelial cells was not responsible for the observed differences in ion transporter expression. Because deficiency of DRA and NHE3 can cause diarrhea due to a failure to absorb chloride or sodium ions from the lumen, respectively (27, 28), these data strongly suggest that the failure to normally induce expression of these ion transporters is responsible for the infection-induced diarrhea in the absence of CXCR2.
Neutrophil influx rather than bacterial load determines diarrhea phenotype
The electrophysiological alterations were not simply related to the increased bacterial load in CXCR2-deficient compared with wild-type mice, since infection of mice lacking RAG1, IL-6, or CXCR3 also led to high bacterial colonization after 2 wk (Figs. 3A and 7A), but these mice showed no signs of diarrhea after infection (Fig. 7B). In stark contrast to CXCR2-deficient mice, the three groups of knockout mice without diarrhea had increased neutrophil numbers and/or MPO levels in the colon after C. rodentium infection (Figs. 3C and 7C) (16), suggesting that neutrophils can protect against infection-associated diarrhea beyond their antimicrobial function and regardless of the underlying immune defect in the different mice. Furthermore, mucosal neutrophil influx in RAG1-deficient mice was associated with increased colonic mRNA expression for CXCL1, CXCL2, and CXCL5 and increased protein production of at least one of these chemokines, CXCL5 (Fig. 7D). These data highlight that local chemokine induction and the accompanying CXCR2-dependent neutrophil influx represent innate responses to infection.
FIGURE 7.

Infection-induced diarrhea is unique to Cxcr2−/− mice and not related to bacterial load. C57BL/6J (WT) and the indicated gene-targeted mice were infected with C. rodentium. A, Bacterial numbers in the stool were determined by CFU assay at weeks 1 and 2. B, Fecal water analysis in infected and uninfected (week 0) mice. For A and B, data are means ± SEM (n ≥6/group); *, p < 0.05 by rank sum test compared with week 1 (A) or by t test relative to week 0 (B) of the same genotype. C, MPO activity was determined in colon extracts of wild-type and Rag1−/− mice at different times after infection (left panel), and neutrophils were detected by immunostaining of frozen colon sections for MPO in infected and uninfected (week 0) Rag1−/− mice (right panel). Images were taken with a ×20 lens and processed equally for the panel. D, Levels of mRNA in the colon of Rag1−/− mice were determined by real-time PCR, and protein levels in tissue extracts were measured by ELISA, at the indicated times after infection. Results are means ± SEM in C and D (n ≥ 3/group). *, p < 0.05 by t test relative to uninfected mice of the same genotype.
Discussion
Infections with EPEC are a major worldwide cause of diarrheal disease in children, yet the pathophysiologic mechanisms of disease and host defense are not well understood. Much is known about the interactions between the bacteria and host cells, particularly regarding their ability to adhere to epithelial cells (29), but these bacteria do not produce classical enterotoxins, suggesting that a complex cross-talk between bacteria and host cells, and the ensuing host responses, are important for determining the disease manifestations. The analysis of such responses often proceeds by testing individual host factors and processes in hypothesis-driven approaches. Important progress has been made with these approaches, particularly when combined with the commonly used murine infection model with the A/E lesion-forming pathogen C. rodentium. For example, the cytokines IFN-γ, TNF-α, and IL-6 were shown to have indispensable and distinct roles in host defense against C. rodentium (14–16). However, these studies were limited to a single host factor, precluding evaluation of their relative overall importance and interdependence with other host factors and processes. As an alternative, discovery-driven approaches are becoming increasingly prominent in obtaining a broader genomic view of the host response to infection. Our data and those in other infection models (20, 22) demonstrate that such genomic approaches can yield unbiased information about dominant features of the host response to infection. For example, analysis of susceptible and resistant strains of mice yielded key differences in the colonic expression of ion transporters after C. rodentium infection (20). We found here that a specific array of chemokines were among the most strongly induced genes in response to C. rodentium infection. Although chemokines are often early response genes in bacterial infections, the particular set of chemokines induced by C. rodentium could not have been easily predicted. Furthermore, gene-targeted mice for the common receptors of these chemokines had marked phenotypes in regard to bacterial clearance and mucosal responses, which validates the physiological importance of the genes identified by genome-wide expression studies and thereby the value of the genomics approach.
Of the four most strongly induced chemokines, three (CXCL1/5/9) were activated predominantly in CD11b+ dendritic cells, while one (CXCL2) was increased mostly in macrophages. These data suggest that specific cell types have distinct chemokine expression profiles (30, 31), despite the fact that many chemokine genes share important regulatory elements, particularly binding sites for NF-κB, in their promoter regions (32). Intestinal epithelial cells were not major producers of the prominent chemokines after C. rodentium infection, which contrasts with the prominent CXCL5 production by the cells in chemically induced colitis (33). These cells were previously shown to respond to EPEC infection with NF-κB activation and chemokine expression (34), although the relative induction was modest compared with infection with invasive enteric pathogens such as Salmonella (35). Moreover, C. rodentium infection activates NF-κB in the colonic epithelium in mice (36). However, our studies revealed only modest epithelial chemokine induction after C. rodentium infection, casting doubts on the relative importance of epithelial NF-κB in controlling the host chemokine response under these conditions. In addition to local intestinal responses, we observed significant chemokine induction in systemic sites of spleen and liver. These organs were colonized with C. rodentium at low levels, which may be sufficient to explain the chemokine induction at those sites, although increased intestinal uptake, systemic circulation, and host recognition of bacterial products (e.g., LPS) could also contribute.
The chemokines CXCL9, CXCL10, and, to a lesser extent, CXCL11 were markedly increased after C. rodentium infection and played a role in host defense against the bacteria, since loss of their common receptor, CXCR3, delayed bacterial eradication and compromised the normal development of antibacterial IgG. These results indicate that CXCR3 and its cognate ligands have a physiological function in mucosal defense against at least one bacterial pathogen of the intestinal tract. Such function was previously recognized mostly in immune defense against respiratory and skin pathogens. For example, CXCR3 deficiency delayed eradication of Bordetella bronchiseptica from the lungs (37) and compromised clearance of Leishmania major in the skin (38). CXCR3 is prominently expressed on activated T cells, particularly Th1 cells, and helps to orchestrate Th1-dependent immune responses (39). This function may explain the role of CXCR3 in controlling C. rodentium, since the Th1 signature cytokine IFN-γ, and thus presumably Th1 cells, is required for effective defense against C. rodentium (14). IFN-γ stimulates the bacterial killing capacity of phagocytic cells (40), which we also observed for neutrophil killing of C. rodentium. Additionally, Th1 cells can provide help to B cells in the generation of specific IgG responses, and CXCR3 on B cells may further promote such responses (41). Accordingly, we observed a diminished early production of antibacterial IgG, but not IgM, in the absence of CXCR3, which is likely to be significant since IgG contributes to clearance of C. rodentium (12, 13). Beyond its role in mucosal defense, CXCR3 was disproportionally important for preventing systemic bacterial spread to liver and spleen, and in protecting against necrotic liver damage. Such spread may occur secondarily to increased mucosal damage and epithelial barrier loss, although none was apparent histologically and deficiency in at least one of the key CXCR3 ligands, CXCL10, is associated with enhanced rather than diminished epithelial barrier function in the colon (42). Instead, CXCR3 may exert a unique immunological function in the liver or spleen. For example, the receptor is required for effective recruitment of effector T cells to the liver and is found on most T cells in different inflammatory conditions of the liver (43). If hepatic T cell-dependent defenses fail to develop normally in the absence of CXCR3, it may lead to enhanced bacterial proliferation and hepatic necrosis. Despite the importance of CXCR3 in controlling C. rodentium in intestine and liver, it was ultimately not required for eradication, emphasizing that redundancy of immune mechanisms in general, and chemokines in particular, is central to the development of robust host defenses against pathogenic microbes (32).
The chemokines CXCL1, CXCL2, and CXCL5 were among the most highly induced genes in the C. rodentium infection model. All three are cognate ligands for CXCR2, and deficiency of the receptor compromised bacterial clearance, indicating that these chemokines constitute a critical component of the intestinal defense against the bacteria. Similarly, CXCR2 was previously shown to be indispensable in immune defense against pathogenic bacteria in other tissues, including Pseudomonas aeruginosa and Nocardia asteroides in the lungs (44, 45), and uropathogenic E. coli in the urinary tract (46). The protective functions of CXCR2 are by no means universal, since the receptor is not required for clearance of influenza virus in the lungs (47), and is in fact detrimental in models of septic peritonitis (48). CXCR2 is strongly expressed on neutrophils, and its cognate ligands are potent neutrophil chemoattractants. Consistent with this, we found that most Gr-1+ cells that invaded the mucosa in response to C. rodentium were positive for CXCR2. Conversely, practically all CXCR2+ cells were Gr-1+ neutrophils. Importantly, loss of CXCR2 abrogated neutrophil influx into the colon mucosa, indicating that the receptor and its cognate ligands were critical for orchestrating this process. Neutrophil attraction to sites of infection and damage mediated by CXCR2 and its ligands is dependent on the tissue context and stimulation conditions (49, 50). Irrespective of the means of chemoattraction, neutrophils typically exert their defense functions by direct microbial killing of obligate extracellular microbes or facultative intracellular microbes during transient passage between host cells (51). C. rodentium and EPEC reside predominantly outside host cells in the intestinal lumen and at the epithelial surface, although they can invade host cells and low numbers are found deeper in the mucosa and in systemic sites. Infiltrating neutrophils were localized around the crypts throughout the colonic mucosa and were able to kill C. rodentium when activated, suggesting that they contribute to antibacterial defense by forming an active defense barrier against bacterial spread across the crypts (11), similar to their function in protecting against systemic spread of orally ingested Salmonella (52, 53). Moreover, lack of CXCR2 and the accompanying mucosal neutrophil influx led to heavy and deep bacterial colonization of the crypts, suggesting that neutrophils are critical for preventing the build-up of bacterial repertoires in the crypts. Such microbial colonies in the crypts are presumably protected against removal in the luminal bulk flow, which could allow them to serve as seed sites for continued luminal infection. We note that neutrophils are necessary for mucosal defense against C. rodentium (11), but they are not sufficient since mice lacking T and B cells or IL-6 have high mucosal neutrophil numbers but exhibit bacterial clearance defects (10, 12, 16).
Watery diarrhea and the accompanying dehydration are hallmarks of EPEC-induced disease in humans. Diarrhea can occur within hours of infection, suggesting that bacterial factors drive these early events (54). Disruption of epithelial tight junctions, increased paracellular permeability, dysregulation of critical ion transporters, and loss of microvilli have been reported to occur rapidly in response to translocation of EPEC effector proteins into host cells (2–4, 55). However, diarrhea can be variable and persist over extended periods up to weeks, suggesting that host factors are likely to be decisive in later stages of the human disease. Infection is accompanied by influx of inflammatory cells that can release secretagogues, and it leads to mucosal up-regulation of receptors whose activation causes fluid secretion (56). The importance of host factors in determining infection-induced disease is clearly demonstrated by the finding that the genetic background of certain strains of mice can render them susceptible to massive diarrhea and dehydration-related mortality (57). Differential disease susceptibility in these mice is accompanied by altered expression of key solute transporters, particularly DRA and aquaporin 8 (20), although the underlying mechanisms are not understood. Our data show that CXCR2-dependent processes are critical determinants of infection-induced diarrhea, providing strong support for the role of host factors in controlling disease outcome upon infection with an A/E lesion-forming pathogen. These murine findings suggest that similar principles may also underlie the variable disease manifestations in human EPEC infections. Mechanistically, CXCR2 was required in the mice for induction of key elements of ion transport pathways, including DRA, PAT1, and ATP1β2, which may be interpreted as a compensatory induction of fluid uptake processes in the colon to counteract any inflammation-associated increase in secretagogue activities. Loss of DRA causes a chloride-losing chronic diarrhea in patients and gene-targeted mice (27), suggesting that the failure to induce DRA normally in CXCR2-deficient mice is probably important in the diarrhea observed in these mice. Although our studies were not designed to examine the mechanisms by which CXCR2 mediates solute transporter induction, one could speculate that neutrophils have a key function, as these were the main CXCR2-expressing cells after infection. Their absence in CXCR2-deficient mice was accompanied by diarrhea, while their presence in several other models with high bacterial colonization levels was consistently associated with protection against diarrhea. These observations suggest that neutrophils have a role in preventing infection-associated diarrhea that goes beyond their function in antibacterial defense. Such protective neutrophil functions have also been demonstrated in different models of acute colitis (58). Thus, neutrophils must be viewed as multifunctional cells that contribute to intestinal inflammation and diarrhea under some conditions (59), but also protect against microbial growth and entry, and the accompanying diarrheal manifestations in other circumstances. Exploiting these protective functions may suggest an avenue for managing chronic diarrheal diseases in humans caused by EPEC and other A/E lesion-causing pathogens.
Supplementary Material
Acknowledgments
We thank Yolanda Andersen, Dustin Hammond, John J. Quinlan III, and Lucia Hall for expert technical support.
Footnotes
This work was supported by National Institutes of Health Grants DK70867, DK35108, AI56075, and RR17030, and the University of California, San Diego Digestive Diseases Research Development Center (DK80506). M.E.S. was supported by a fellowship from the MFG Educative Science, S.M.D. by a fellowship from the Crohn’s and Colitis Foundation of America, and P.H. by a fellowship from the Swiss National Science Foundation (SSMBS; PASMA 114623).
Abbreviations used in this paper: EPEC, enteropathogenic Escherichia coli; A/E, attaching-and-effacing; Isc, short-circuit current; MPO, myeloperoxidase.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Clarke SC, Haigh RD, Freestone PP, Williams PH. Virulence of enteropathogenic Escherichia coli, a global pathogen. Clin Microbiol Rev. 2003;16:365–378. doi: 10.1128/CMR.16.3.365-378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen HD, Frankel G. Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol Rev. 2005;29:83–98. doi: 10.1016/j.femsre.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cell Microbiol. 2008;10:1735–1745. doi: 10.1111/j.1462-5822.2008.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, Zaheer A, Ramaswamy K, Hecht G, Dudeja PK. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenicE coli. J Clin Invest. 2007;117:428– 437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson E, Barthold SW. The ultrastructure of transmissible murine colonic hyperplasia. Am J Pathol. 1979;97:291–313. [PMC free article] [PubMed] [Google Scholar]
- 6.Schauer DB, Falkow S. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect Immun. 1993;61:2486–2492. doi: 10.1128/iai.61.6.2486-2492.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 8.Borenshtein D, McBee ME, Schauer DB. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol. 2008;24:32–37. doi: 10.1097/MOG.0b013e3282f2b0fb. [DOI] [PubMed] [Google Scholar]
- 9.Bry L, Brenner MB. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol. 2004;172:433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 10.Simmons CP, Clare S, Ghaem-Maghami M, Uren TK, Rankin J, Huett A, Goldin R, Lewis DJ, MacDonald TT, Strugnell RA, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogenCitrobacter rodentium. Infect Immun. 2003;71:5077–5086. doi: 10.1128/IAI.71.9.5077-5086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils toCitrobacter rodentium. J Immunol. 2007;179:566–577. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 12.Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, Schreiber JR, Varki NM, Kagnoff MF, Eckmann L. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. 2004;72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, Kaser A, Nagaishi T, Higgins DE, Mizoguchi E, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. 2006;116:2142–2151. doi: 10.1172/JCI27821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins LM, Frankel G, Connerton I, Goncalves NS, Dougan G, MacDonald TT. Role of bacterial intimin in colonic hyperplasia and inflammation. Science. 1999;285:588–591. doi: 10.1126/science.285.5427.588. [DOI] [PubMed] [Google Scholar]
- 15.Goncalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor α in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun. 2001;69:6651–6659. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E, Eckmann L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J Immunol. 2008;180:6816– 6826. doi: 10.4049/jimmunol.180.10.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen. Citrobacter rodentium, in mice lacking IL-12 or IFN-γ. J Immunol. 2002;168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 19.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Borenshtein D, Fry RC, Groff EB, Nambiar PR, Carey VJ, Fox JG, Schauer DB. Diarrhea as a cause of mortality in a mouse model of infectious colitis. Genome Biol. 2008;9:R122. doi: 10.1186/gb-2008-9-8-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costello CM, Mah N, Hasler R, Rosenstiel P, Waetzig GH, Hahn A, Lu T, Gurbuz Y, Nikolaus S, Albrecht M, et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005;2:e199. doi: 10.1371/journal.pmed.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 23.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukocyte Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 24.Cash HL, Whitham CV, Hooper LV. Refolding, purification, and characterization of human and murine RegIII proteins expressed inEscherichia coli. Protein Expression Purif. 2006;48:151–159. doi: 10.1016/j.pep.2006.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 27.Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M. Slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem. 2006;281:37962–37971. doi: 10.1074/jbc.M607527200. [DOI] [PubMed] [Google Scholar]
- 28.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 29.Frankel G, Phillips AD. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell Microbiol. 2008;10:549–556. doi: 10.1111/j.1462-5822.2007.01103.x. [DOI] [PubMed] [Google Scholar]
- 30.Volpe E, Cappelli G, Grassi M, Martino A, Serafino A, Colizzi V, Sanarico N, Mariani F. Gene expression profiling of human macrophages at late time of infection withMycobacterium tuberculosis. Immunology. 2006;118:449– 460. doi: 10.1111/j.1365-2567.2006.02378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaharik ML, Nayar T, White R, Ma C, Vallance BA, Straka N, Jiang X, Rey-Ladino J, Shen C, Brunham RC. Genetic profiling of dendritic cells exposed to live- or ultraviolet-irradiated Chlamydia muridarum reveals marked differences in CXC chemokine profiles. Immunology. 2007;120:160–172. doi: 10.1111/j.1365-2567.2006.02488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 33.Kwon JH, Keates AC, Anton PM, Botero M, Goldsmith JD, Kelly CP. Topical antisense oligonucleotide therapy against LIX, an enterocyte-expressed CXC chemokine, reduces murine colitis. Am J Physiol. 2005;289:G1075–G1083. doi: 10.1152/ajpgi.00073.2005. [DOI] [PubMed] [Google Scholar]
- 34.Savkovic SD, Koutsouris A, Hecht G. Activation of NF-κB in intestinal epithelial cells by enteropathogenicEscherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- 35.Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, Kagnoff MF. NF-κB is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol. 1999;163:1457–1466. [PubMed] [Google Scholar]
- 36.Wang Y, Xiang GS, Kourouma F, Umar S. Citrobacter rodentium-induced NF-κB activation in hyperproliferating colonic epithelia: role of p65 (Ser536) phosphorylation. Br J Pharmacol. 2006;148:814– 824. doi: 10.1038/sj.bjp.0706784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Widney DP, Hu Y, Foreman-Wykert AK, Bui KC, Nguyen TT, Lu B, Gerard C, Miller JF, Smith JB. CXCR3 and its ligands participate in the host response to Bordetella bronchiseptica infection of the mouse respiratory tract but are not required for clearance of bacteria from the lung. Infect Immun. 2005;73:485–493. doi: 10.1128/IAI.73.1.485-493.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosas LE, Barbi J, Lu B, Fujiwara Y, Gerard C, Sanders VM, Satoskar AR. CXCR3−/− mice mount an efficient Th1 response but fail to control Leishmania major infection. Eur J Immunol. 2005;35:515–523. doi: 10.1002/eji.200425422. [DOI] [PubMed] [Google Scholar]
- 39.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu YK, Arkins S, Li YM, Dantzer R, Kelley KW. Reduction in superoxide anion secretion and bactericidal activity of neutrophils from aged rats: reversal by the combination of γ interferon and growth hormone. Infect Immun. 1994;62:1–8. doi: 10.1128/iai.62.1.1-8.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, Zhang HH, Gebeyehu S, Wright TM, et al. The CXC chemokine murine monokine induced by IFN-γ (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J Immunol. 2002;169:1433–1443. doi: 10.4049/jimmunol.169.3.1433. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki S, Yoneyama H, Suzuki K, Suriki H, Aiba T, Watanabe S, Kawauchi Y, Kawachi H, Shimizu F, Matsushima K, et al. Blockade of CXCL10 protects mice from acute colitis and enhances crypt cell survival. Eur J Immunol. 2002;32:3197–3205. doi: 10.1002/1521-4141(200211)32:11<3197::AID-IMMU3197>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 43.Zeremski M, Petrovic LM, Chiriboga L, Brown QB, Yee HT, Kinkhabwala M, Jacobson IM, Dimova R, Markatou M, Talal AH. Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology. 2008;48:1440–1450. doi: 10.1002/hep.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore TA, Newstead MW, Strieter RM, Mehrad B, Beaman BL, Standiford TJ. Bacterial clearance and survival are dependent on CXC chemokine receptor-2 ligands in a murine model of pulmonary Nocardia asteroides infection. J Immunol. 2000;164:908–915. doi: 10.4049/jimmunol.164.2.908. [DOI] [PubMed] [Google Scholar]
- 45.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. 2000;68:4289– 4296. doi: 10.1128/iai.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svensson M, Irjala H, Svanborg C, Godaly G. Effects of epithelial and neutrophil CXCR2 on innate immunity and resistance to kidney infection. Kidney Int. 2008;74:81–90. doi: 10.1038/ki.2008.105. [DOI] [PubMed] [Google Scholar]
- 47.Wareing MD, Shea AL, Inglis CA, Dias PB, Sarawar SR. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 2007;20:369–378. doi: 10.1089/vim.2006.0101. [DOI] [PubMed] [Google Scholar]
- 48.Ness TL, Hogaboam CM, Strieter RM, Kunkel SL. Immunomodulatory role of CXCR2 during experimental septic peritonitis. J Immunol. 2003;171:3775–3784. doi: 10.4049/jimmunol.171.7.3775. [DOI] [PubMed] [Google Scholar]
- 49.Johnston RA, Mizgerd JP, Shore SA. CXCR2 is essential for maximal neutrophil recruitment and methacholine responsiveness after ozone exposure. Am J Physiol. 2005;288:L61–L67. doi: 10.1152/ajplung.00101.2004. [DOI] [PubMed] [Google Scholar]
- 50.Coates NJ, McColl SR. Production of chemokines in vivo in response to microbial stimulation. J Immunol. 2001;166:5176–5182. doi: 10.4049/jimmunol.166.8.5176. [DOI] [PubMed] [Google Scholar]
- 51.Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol. 2007;15:87–92. doi: 10.1016/j.tim.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 52.Vassiloyanakopoulos AP, Okamoto S, Fierer J. The crucial role of polymorphonuclear leukocytes in resistance to Salmonella dublin infections in genetically susceptible and resistant mice. Proc Natl Acad Sci USA. 1998;95:7676–7681. doi: 10.1073/pnas.95.13.7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheminay C, Chakravortty D, Hensel M. Role of neutrophils in murine salmonellosis. Infect Immun. 2004;72:468– 477. doi: 10.1128/IAI.72.1.468-477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet. 1978;1:1119–1122. doi: 10.1016/s0140-6736(78)90299-4. [DOI] [PubMed] [Google Scholar]
- 55.McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enter-opathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hecht G, Marrero JA, Danilkovich A, Matkowskyj KA, Savkovic SD, Koutsouris A, Benya RV. Pathogenic Escherichia coli increase Cl-secretion from intestinal epithelia by upregulating galanin-1 receptor expression. J Clin Invest. 1999;104:253–262. doi: 10.1172/JCI6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borenshtein D, Nambiar PR, Groff EB, Fox JG, Schauer DB. Development of fatal colitis in FVB mice infected withCitrobacter rodentium. Infect Immun. 2007;75:3271–3281. doi: 10.1128/IAI.01810-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuhl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, Loddenkemper C, Heimesaat MM, Grollich K, Zeitz M, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 59.Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, Mrsny RJ. 5′-Adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.