

Abstract
Like type-2 diabetes mellitus (T2DM), neurodegenerative disorders and stroke are an ever increasing, health, social and economic burden for developed Westernized countries. Age is an important risk factor in all of these; due to the rapidly increasing rise in the elderly population T2DM and neurodegenerative disorders, both represent a looming threat to healthcare systems. Whereas several efficacious drugs are currently available to ameliorate T2DM, effective treatments to counteract pathogenic processes of neurodegenerative disorders are lacking and represent a major scientific and pharmaceutical challenge. Epidemiological data indicate an association between T2DM and most major neurodegenerative disorders, including Alzheimer's and Parkinson's diseases. Likewise, there is an association between T2DM and stroke incidence. Studies have revealed that common pathophysiological features, including oxidative stress, insulin resistance, abnormal protein processing and cognitive decline, occur across these. Based on the presence of shared mechanisms and signalling pathways in these seemingly distinct diseases, one could hypothesize that an effective treatment for one disorder could prove beneficial in the others. Glucagon-like peptide-1 (GLP-1)-based anti-diabetic drugs have drawn particular attention as an effective new strategy to not only regulate blood glucose but also to reduce apoptotic cell death of pancreatic beta cells in T2DM. Evidence supports a neurotrophic and neuroprotective role of GLP-1 receptor (R) stimulation in an increasing array of cellular and animal neurodegeneration models as well as in neurogenesis. Herein, we review the physiological role of GLP-1 in the nervous system, focused towards the potential benefit of GLP-1R stimulation as an immediately translatable treatment strategy for acute and chronic neurological disorders.
Keywords: glucagon-like peptide-1, exendin-4, liraglutide, type-2 diabetes mellitus, neurodegeneration, Alzheimer's disease, stroke, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, peripheral neuropathy, traumatic brain injury, neurogenesis, neuroprotection, neuroplasticity
Introduction
Neurodegenerative and cerebrovascular disorders are an ever increasing, health, social and economic burden for developed Westernized countries. While certain neurological conditions can present after episodes of clearly defined pathological causes, such as stroke or head trauma, some of the more common neurodegenerative conditions are insidious in nature. Of the more insidious forms of neurodegenerative diseases are Alzheimer's and Parkinson's disease (AD and PD, respectively) and amyotrophic lateral sclerosis (ALS) along with Huntington's disease (HD). These disorders can present with a mixture of motor function and cognitive function abnormalities and are primarily due to a selective type of neurological pathology, derived from quite distinct areas of the brain. Commonalities between many neurodegenerative disorders exist on multiple levels. For example, one tends to find abnormal protein processing, and deposition takes place in those areas of the brain that are classically associated with each disease. Some conditions can manifest as either sporadic or genetic in their origins. An example of a purely genetic disorder is HD. In HD, genetic abnormalities lie in the existence of multiple repeats of a tri-nucleotide sequence (CAG, glutamine) on chromosome 4 in the Huntington gene. The number of CAG repeats appears to be pivotal to the development of the disease condition, as under normal healthy conditions there are 1 to 35 repeats within the gene, while in HD there are 40 to 100 repeats (Huntington Disease Collaborative Research Group, 1993). In PD, there is also a genetic component, and while the majority of cases are sporadic in nature, a small part of PD is caused by various genetic mutations. Mutations encoding a dysfunctional protein called α-synuclein were the first discovered PD disease-causing mutations (Singleton et al., 2003). In AD, there are two well-defined abnormal proteins widely regarded to be pivotal to the pathology of the disease; amyloid-β (Aβ) and hyperphosphorylated microtubule-associated protein (commonly referred to as tau protein), both of which can form insoluble neurotoxic aggregates (Flament et al., 1990; Chartier-Harlin et al., 1991; Citron et al., 1991; Goate et al., 1991; Murrell et al., 1991). As it has been observed in HD and PD, a genetic association is also present in AD. Another commonality lies in the shared nature of the final biochemical effectors of neurodegeneration (Mattson, 2000; Greig et al., 2004). These can be mediated by the transmission of signals through cell surface receptors, or mitochondrial-derived mechanisms that ultimately lead to the activation of apoptotic pathways, leading to cell death and neurological disorder. Needless to say, the pathways leading to cell death are not exclusive to the CNS and are extensively found in tissues organism-wide.
Recently, a striking resemblance has been described between the altered metabolic status of the AD brain, with that of type 2 diabetes mellitus (T2DM) (Arvanitakis et al., 2004; Craft, 2007; Akter et al., 2011). In T2DM, peripheral tissues display a reduced sensitivity to insulin-dependent signalling events leading to a subsequent insufficiency of the secreted insulin, which with progressive disease eventually leads to decline in pancreatic beta-cell insulin synthesis and secretion. The brain is a highly insulin-sensitive organ, and some of these biochemical features are seen in the AD brain. The stark similarity between T2DM and AD has given rise to the hypothesis and new term for describing AD as, namely, ‘type 3 diabetes’ (Steen et al., 2005; de la Monte, 2009; Kroner, 2009; Akter et al., 2011). This being the case, it opens up a potential new strategy for treating AD as a metabolic disease where the utilization of successful therapeutic approaches similar to those used in T2DM may be of benefit to the AD patient.
Normal regulation of insulin secretion and function is driven by the detection of glucose in the blood. When blood glucose is elevated, pancreatic beta cells detect this increase and secret insulin in an attempt to regulate glucose levels by facilitating the transport of glucose into cells where it is utilized in cellular metabolism.
An alternative form of insulin regulation originates from cells found in the gastrointestinal tract. These cells produce a group of biologically active proteins called incretins that stimulate pancreatic beta cells to release insulin after eating. One incretin of particular interest is glucagon-like peptide-1 (GLP-1) (Figure 1). GLP-1 is a small peptide produced by enteroendocrine L cells that are distributed throughout the small intestine, with most of them located at ileum and colon (Drucker and Nauck, 2006; Lovshin and Drucker, 2009). The peptide was first identified in the early 1980s (Bell et al., 1983). GLP-1 can be released from L cells as a consequence of the ingestion of food. The immediate functions of GLP-1 are to induce the release of insulin from pancreatic beta cells, to slow gut emptying and also to inhibit the secretion of glucagon. The role of the incretin, GLP-1, on insulin signalling is quite well defined, and its effects on regulating glucose metabolism and thus blood glucose levels have been exploited in T2DM (Drucker and Nauck, 2006; Lovshin and Drucker, 2009). In addition to the benefits of the peptide on glucose regulation; GLP-1 receptor activation in pancreatic cells incites the proliferation and the anti-apoptotic protection of beta cells (Baggio and Drucker, 2006; Drucker and Nauck, 2006; Lovshin and Drucker, 2009). The peptide also stimulates the differentiation of ductal precursor cells into functional pancreatic beta cells. A small amount of GLP-1 is also produced in the brain, particularly from the nucleus of the solitary tract, area postrema and caudal brain stem (Campos et al., 1994; Calvo et al., 1995a,b; Alvarez et al., 1996; 2005; Larsen et al., 1997; Hamilton and Hölscher, 2009). GLP-1 acts through a GLP-1 receptor (R), which belongs to the class B family of seven-transmembrane-spanning, heterotrimic GPCRs. Upon GLP-1R activation, the major signalling pathway involves activation of adenylyl cyclase via stimulatory Gα, leading to increased intracellular cAMP levels and a series of subsequent signalling events that regulate various cell functions. GLP-1R is distributed in many peripheral tissues, and it is also widely expressed in many regions of the CNS. As stated, GLP-1-based drugs have proved beneficial in T2DM patients. It is noteworthy that recent epidemiological data indicate an association between T2DM and stroke, as well as between T2DM and neurodegenerative disorders such as AD and PD. Thus, it is conceivable that these beneficial effects of GLP-1 receptor activation may be relevant in other forms of disease, including those described in the CNS (Figure 2).
Figure 1.
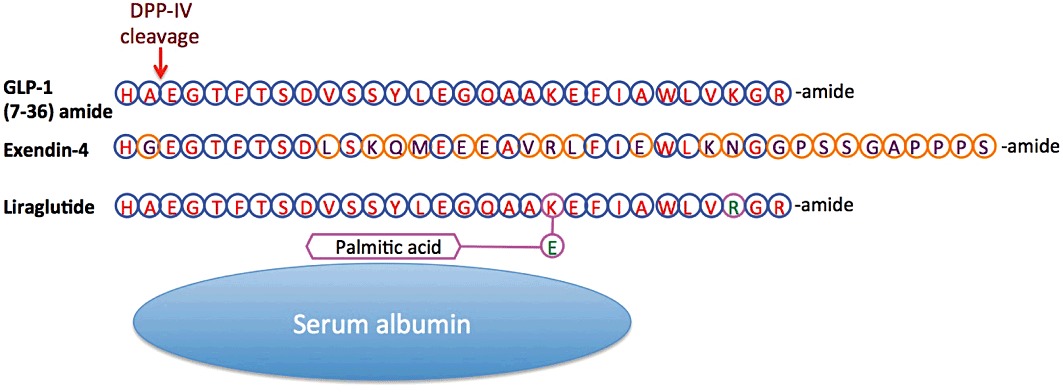
Amino acid sequence of GLP-1 and the long-acting GLP-1 analogs, Ex-4 and liraglutide. Ex-4 is known clinically as Byetta and Bydureon for twice a day and extended release (once weekly) dosing, respectively, whereas liraglutide is known clinically as Victoza (once daily). Amino acid homology (blue circles) and differences (orange circles) are highlighted. Peptidase cleavage by DPP-IV is shown. Of note, liraglutide possesses a C-16 fatty acid (palmitic acid) with a glutamic acid spacer attached to the lysine residue at position 26, permitting its binding to albumin.
Figure 2.
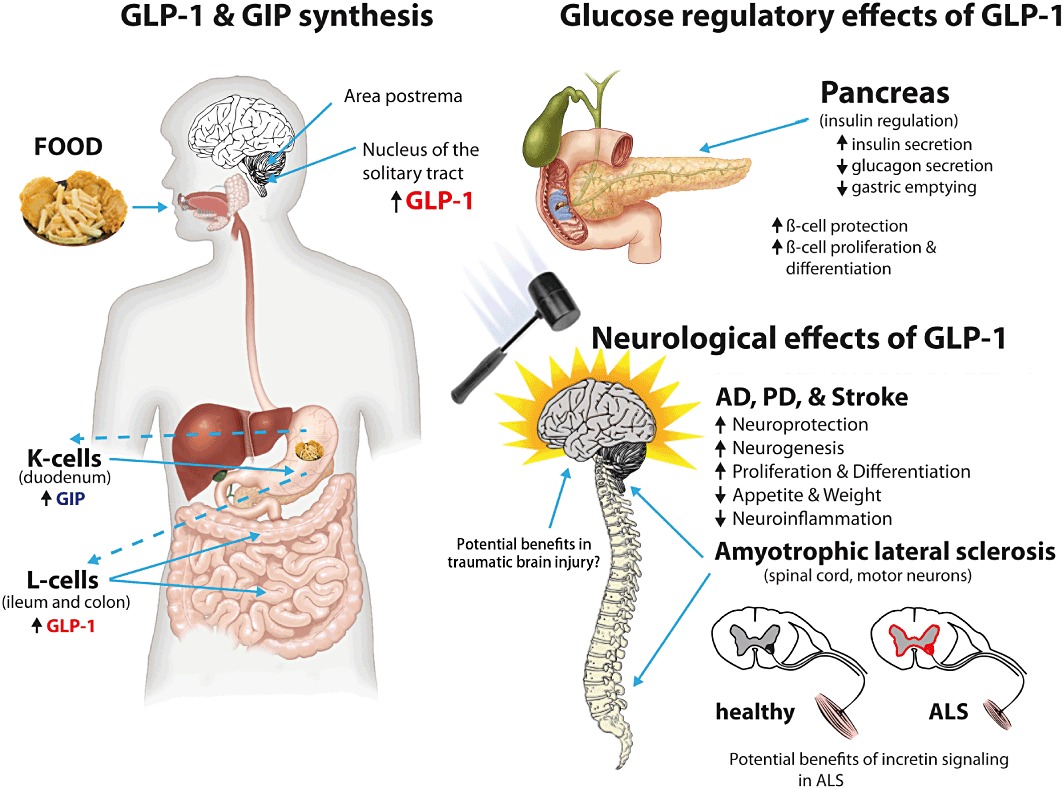
The release of incretins from the intestinal tract can be activated by the ingestion of food. Such release of GLP-1 and GIP into the blood stream results in actions on peripheral systems as well as in the central and peripheral nervous systems that both peptides appear to readily access. Primary systemic effects are on blood glucose regulation, via insulin. GLP-1 can additionally be synthesized and released from certain brain areas; however, the mechanisms responsible for such an action are not fully understood and require further study. Interestingly, putative actions of GLP-1 signalling within the CNS may be responsible for the suppression of appetite and, subsequently, weight loss. Known beneficial actions of GLP-1 signalling in brain may be responsible for the benefits described in the settings of AD and PD and stroke, as described in in vivo models. Likely, benefits of GLP-1 signalling in ALS, HD and brain and spinal trauma need to be fully addressed. Similarly, animal models of diabetic retinopathy and optic nerve crush models show improved outcomes of cellular function when treated locally with GLP-1 receptor analogs (not shown); hence, effects in human optic conditions warrant elucidation.
The utilization of recombinant endogenous proteins, such as GLP-1, has been invaluable in many fields of biomedical research and medicine. However, one caveat of the use of endogenous proteins is that biological systems usually possess effective mechanisms to limit the resulting biological effects in an organism. This is true for the GLP-1 peptide. A very effective protease exists that rapidly cleaves the circulating GLP-1 peptide, rendering it inert at the GLP-1 receptor. GLP-1 is rapidly digested by dipedylpeptidase-IV (DPP-IV) (Figure 1); a few minutes after its release into the blood stream, the majority of the incretin protein will have been degraded by this protease (Vilsbøll et al., 2003). As it happens, it is interesting to note that nature often tries to find ways to circumvent these self-limiting measures. This is well illustrated by the existence of a naturally occurring GLP-1R agonist, which has resistance to DPP-IV. The naturally occurring GLP-1R agonist, exendin-4 (Ex-4), was discovered in the saliva of a reptile native to the United States (Gila monster, Heloderma suspectum). The DPP-IV-resistant features of the protein have been identified, and synthetic forms of Ex-4 and GLP-1 have been generated (Figure 1). These analogs allow for substantially longer half-lives, and some forms are now in clinical use as agents for T2DM. However, in recent years, research involving GLP-1R stimulation has shifted from T2DM to focus upon neurodegenerative disorders. A relatively small but growing body of evidence now exists to support neuroprotective functions of GLP-1R stimulation. Activating the incretin pathway in neurons can produce cellular protection, proliferation and the differentiation of precursor cells into neurons, thus drawing striking similarities between cellular responses in pancreatic beta cells and neurons. Additionally, behavioural and functional benefits to GLP-1R stimulation in rodent models have been described.
In addition to GLP-1, there is a further but less well-studied member of the incretin family, gastric inhibitory peptide (GIP), that is also known as glucose-dependent insulinotropic polypeptide. GIP is similarly generated in gastrointestinal cells called K cells (Brown et al., 1975; Ross et al., 1977), and it is postulated to possess similar properties to that of GLP-1. Presently, there is sparse documentation of the effects of this peptide in connection with neurodegeneration. For this reason, we will primarily focus upon the actions of GLP-1 receptor (GLP-1R) stimulation in relation to the peripheral and central nervous system. Thus, the following article attempts to focus on existing scientific literature that documents inroads to addressing the potential beneficial role of GLP-1 receptor stimulation in neurodegenerative diseases.
GLP-1R activation in brain and neuronal cell model systems (physiological conditions)
Neuronal cell proliferation
GLP-1R activators stimulate neuronal proliferation. Investigations into this biological response have relied heavily on immunohistochemical methods that employ the use of stains that label various markers of cell division. Typically, increased levels of nuclear DNA are measurable by incorporation of the thymidine/uracil analog, bromodeoxyuridine (BrdU), or the abundance of a specific nuclear protein found only in actively dividing cells, the protein Ki-67. Alterations in these markers can be visualized and utilized to indicate cell proliferation occurring in both in vitro and in vivo models systems. With this in mind, investigators have used these markers to examine the effects of GLP-1R activation in various rodent-based models of neuronal proliferation. When primary adult mouse hypothalamic cultures were challenged with Ex-4, an elevated level of BrdU staining was observed (Belsham et al., 2009). Evidence exists that indirectly implicates GLP-1R-mediated signalling events in ciliary neurotrophic factor (CNTF)-dependent cell proliferation. The administration of CNTF in vivo has been shown to induce an increased expression of GLP-1 immunoreactivity in the hypothalamus of adult wild-type mice, thus indicating a role of GLP-1R signalling in CTNF-regulated cell proliferation. Furthermore, primary hypothalamic cultures derived from GLP-1R knockout mice failed to display CNTF-induced BrdU incorporation, implicating a functional requirement of GLP-1R signalling events in the induction of CNTF-dependent proliferation (Belsham et al., 2009). Similarly, when cells in vitro were administered a GLP-1R antagonist under basal conditions, Ex-(9–39), these cultures showed a decreased level of Ki-67 staining, indicating a role of GLP-1R stimulation in cell division. Chronic in vivo treatment of adult rodents with Ex-4 incited cell proliferation in the rat hippocampus dentate gyrus, as indicated by increased BrdU and doublecortin (DCX) staining, a marker of immature neuronal tissue. Also, the levels of gene transcripts for Ki-67 were elevated (Isacson et al., 2011), further demonstrating the proliferative properties of Ex-4 in adult brain. Long-term administration of Ex-4 in vivo (21 days) led to higher levels of proliferation (based upon Ki-67 staining) in the subgranular zone of adult mouse dentate gyrus (Li et al., 2010c). Additionally, in mouse models of diabetes, chronic treatment with GLP-1 analogues for 4–10 weeks significantly increased the number of progenitor cells or DCX-positive young neurons in the dentate gyrus, as measured by BrdU or DCX immunohistostaining. Conversely and not surprisingly, the GLP-1 receptor antagonist exendin(9–36) reduced progenitor cell proliferation in these mice (Hamilton et al., 2011), confirming that this action is GLP-1R mediated.
Interestingly, findings related to cellular proliferation in vitro appear to be somewhat dependent upon the model used to study the phenomenon. As the use of the PC12 cell line, which are derived from a form of tumour located in rat adrenal tissues, failed to show any significant levels of proliferation after GLP-1 treatment (Perry et al., 2002a). Yet SH-SY5Y cells, a human neuroblastoma cell line and another commonly used cell-based model system, showed evidence of proliferation at physiologically relevant doses of either GLP-1 or Ex-4 (Li et al., 2010b). This likely relates to the level of endogenous GLP-1R expression, which can be measured both in cell culture as well as in specific brain areas. Such levels can, additionally, be enhanced by GLP-1R overexpression in the former to aid characterize GLP-1 signalling mechanisms. In a primary rodent culture system, GLP-1R activation (Ex-4 and GLP-1) significantly increased mouse neural stem cell numbers in vitro, as indicated by BrdU incorporation and ATP level measurements (Bertilsson et al., 2008). The same findings were observed in a rat striatal embryonic cell line, ST14A cells (Bertilsson et al., 2008).
Neuronal cell differentiation and neurite outgrowth
Similar to measuring levels of cell division, assessments of cell differentiation are chiefly determined by immunohistochemistry methods, which allow the identification of the different maturation stages of neuronal cells, and can also be used to determine the phenotype of a given cell. As indicated above, GLP-1R activators can induce the differentiation of neural stem cells into neurons. Treatment of adult mice with Ex-4 showed a 1.7-fold increase in DCX-positive cells in the medial striatum and doubled the number of BrdU-positive cells in the subventricular zone, a known pool of neuronal stem cells (Bertilsson et al., 2008). Isacson et al. (2011) demonstrated that the administration of Ex-4 (2 weeks) to adult rodent induced an elevation in DCX and Mash-1 gene transcripts, both markers of neurogenesis in the hippocampus. Li et al.'s (2010c) study also showed an enhanced level of neurogenesis and neuroblast differentiation in mouse dentate gyrus, as indicated by double staining with BrdU and DCX.
Another beneficial feature of incretin pathway signalling lies in the production of neurite outgrowths. Neurites are essential components in the formation of functional synapses between neurons and their surrounding microenvironment. The treatment of human SH-SY5Y cells with Ex-4 has been shown to increase the numbers of neurite-bearing cells, in addition to the actual number of neurites per cell (Luciani et al., 2010). Interestingly, when the morphology of Ex-4-induced neurites were compared with that of neurites produced by treatment with retinoic acid (RA), a well-defined promoter of neurite outgrowth, the overall morphology was similar to that of RA neurites; yet the Ex-4 neurites were shorter in length (Luciani et al., 2010). Likewise, in another widely used model of neuronal differentiation using PC12 cells, GLP-1R stimulation incited neurite outgrowth in a manner similar to nerve growth factor (NGF). Paralleling the finding in SH-SY5Y cells, Ex-4-induced PC12 cell neurites displayed comparable morphology and yet were shorter in length and smaller in number, with less branching when compared with NGF control cells (Perry et al., 2002a).
Enhanced synaptic plasticity and memory formation
Long-term potentiation (LTP) is widely held as the cellular correlate of memory formation, and it is defined as a long-lasting enhancement in signal transmission between two neurons, resulting from their synchronous activity. This phenomenon has a highly significant meaning when considering that several neurodegenerative conditions manifest clinical symptoms of abnormal memory (epitomized by AD and mild cognitive impairment). Under normal physiological conditions, GLP-1 receptor stimulation has been shown to promote LTP. Liraglutide (Figure 1), (Asp7) GLP-1, (Pro9) GLP-1 and N-glyc-GLP-1 are all GLP-1R agonists that also enhance LTP (McClean et al., 2011). Further evidence supporting a positive role of GLP-1R stimulation on LTP is described in studies where neurons from GLP-1R knockout mice (Glp-1r–/–) displayed impaired LTP (Abbas et al., 2009). When extrapolating the LTP phenomenon from neuronal cells to a whole organism, it becomes clear that GLP-1R-dependent benefits to LTP are in fact translated to rodent behavioural paradigms. Wild-type rodents treated with Ex-4 had improved reference memory over controls, in a radial maze paradigm (Isacson et al., 2011). GLP-1 and the incretin mimetic [Ser(2)]ex(1–9) administered to rats improved the treated animal performance in passive avoidance and Morris Water Maze paradigms, suggesting heightened hippocampal-dependent associative and spatial learning. Improvements were determined to be dependent on GLP-1R signalling as co-treatment with an antagonist, Ex-(9–39), reversed the effects. Examples further solidifying the beneficial role of GLP-1R signalling in in vivo behavioural studies are illustrated by the use of GLP-1R knockout mice. The animals demonstrated significant deficits in contextual hippocampal fear conditioning assessments when compared with heterozygous (Glp1r+/–) and wild-type animals. Additionally, phenotypically restored GLP-1R knockout animals performed better than (Glp1r–/–) animals. Likewise, GLP-1R overexpression in the hippocampus improved Morris Water Maze and contextual fear learning performance (During et al., 2003). Glp-1r–/– mice have been shown to fail to distinguish between new and familiar objects in novel object recognition assessments along with poor Morris Water Maze results; however, these deficiencies could not be explained by anxiety or exploratory differences between groups, so impaired memory formation was the likely cause (Abbas et al., 2009). EEG hippocampal θ waves are a measure of brain arousal states in rodents. These waves are also indicative of hippocampal memory function and have been linked to learning across mammalian species (Hasselmo, 2005; Kesner and Hopkins, 2006; Hyman et al., 2011). The wave forms can be subdivided into two types (types I and II), where type I waves are associated with voluntary behaviour and are suppressed under conditions of anaesthesia. In an anaesthetized rat study, when the animals were administered GLP-1, a transient elevation in hippocampal θ waves was observed, indicating an increase of predominately type II waves (Oka et al., 1999a), which are indicative of sedentary states of mental arousal associated with learning and memory retrieval and are believed to be vital to the induction of LTP (Hyman et al., 2003; Hasselmo, 2005; Kesner and Hopkins, 2006). In synopsis, it appears that GLP-1R signalling events possess desirable effects on neuronal tissues under healthy physiological conditions. The observed benefits of GLP-1R stimulation at the cellular level may be responsible for the associated enhancements in learning and memory, which may in turn be efficacious in the treatment of neurodegenerative disease as well as diabetes-associated cognitive impairments.
Neuroprotective action of GLP-1R stimulation in neurodegenerative disease
As indicated previously, specific neurodegenerative conditions display shared hallmark features in the forms of protein aggregates (Aβ peptide and hyperphosphorylated tau in AD and α-synuclein in PD), genetic predispositions (sporadic and familial disease origins) and shared mechanisms of neuronal cell death. Additional shared facets of neurodegenerative disease often include immune cell-derived pathophysiological events, such as damage induced by oxidative stress and the consequences of unregulated pro-inflammatory cytokine responses. As the term neurodegeneration implies, the pathological processes appear to follow a continuum of events where the destruction of existing neurons and synaptic connections ultimately cause the clinical manifestation of a specific human disease. Potential therapeutic strategies that are capable of limiting the destructive effects of pathological cellular processes on neuronal tissues while directly or indirectly promoting the creation of new functional neurons are highly desirable. Agents such as these will prove to be crucial in preserving healthy neuronal function, if not also possibly reversing existing neuronal dysfunction. The observed ability of GLP-1R stimulation to induce beneficial physiological effects on neural cell proliferation, neural stem cell differentiation with neurite outgrowth and improved memory in adult rodents indicates a highly exploitable feature of incretin signalling (Perry and Greig, 2002; 2003; 2005). If these physiological properties carry over under pathological conditions and then translate to the human brain, they could be of great benefit in neurodegenerative disease. The beneficial effects of GLP-1R signalling events under pathophysiological conditions in cell-based and animal model systems are described.
GLP-1R stimulation rescues memory dysfunction
Aβ and its insoluble aggregates found in post mortem human brain have been implicated as a key contributor to the pathology of AD. Amyloid-β precursor protein (APP) is proteolytically cleaved to generate a variety of products by the actions of three proteases, α-, β- and γ-secretase. Cleavage of APP by α- and γ-secretase tends to generate non-toxic forms of the protein, while cleavage of APP by β- and γ-secretase generates the formation of peptides that can form soluble or insoluble neurotoxic proteins (Sambamurti et al., 2002; 2006; O'Brien and Wong, 2011). The insoluble version of the peptide can form aggregated protein deposits that are associated with cellular toxicity; while the soluble form of the peptide, particularly in the form of oligomeric assemblies or Aβ–derived diffusible ligands (ADDLs), has been described to target synapses, induce neuronal dysfunction and impair cognition (Lacor et al., 2007; Shankar et al., 2008; Ashe and Zahs, 2010; Marchetti and Marie, 2011). Thus, the formation of Aβ peptides from β- and γ-secretase cleavage in brain are considered to be highly undesirable. Based on in vitro tissue culture and from transgenic mouse models of AD, the toxic form of Aβ has been shown to promote cellular dysfunction within the CNS, which is associated with cognitive impairments. Under such conditions, GLP-1R stimulation has been shown to alter cellular production and accumulation of Aβ deposits in association with reduced toxicity (Qin et al., 2008). Ex-4 and GLP-1 pretreatment of cultured hippocampal neuronal cells in vitro reduced both Aβ- and Fe2+-induced cell death. While GLP-1R stimulation failed to reduce elevated levels of Aβ in NGF-stimulated PC12 cells, GLP-1 treatment reduced the mouse brain-derived Aβ (Perry et al., 2003). Exposure of PC12 cells to Ex-4 and GLP-1 resulted in reduced levels of both secreted APP and cellular APP (Perry et al., 2003). Ex-4 protected rodent primary cortical neurons and SH-SY5Y cells from Aβ- and oxidative stress-induced toxic insult respectively. Also, GLP-1R stimulation by Ex-4 decreased the levels of secreted Aβ in human neuroblastoma cultures under euglycaemic and hyperglycaemic conditions (Li et al., 2010a). Administration of streptozocin to rodents generates a diabetic pathology; in addition, this agent has been demonstrated to increase APP and soluble Aβ levels, and to a lesser extent τ, measured within the brain of triple transgenic AD mice that express both Aβ and τ pathology and, thereby, mimic human AD (Li et al., 2010a). Chronic administration of systemic Ex-4 at a dose that compared favourably with that administered to humans fully ameliorated streptozocin-mediated changes in brain AD protein levels and may have reduced the number of amyloid plaques, localized in the hippocampus (Li et al., 2010a). In addition, it appeared to lower levels of Aβ in AD transgenic mice without streptozocin. The use of a different long-acting GLP-1R agonist, Val(8)GLP-1, likewise reduced the number of Congo red-positive amyloid plaques as well as activated microglia in an elderly cohort of a transgenic (APP/PS1) AD mouse line (Gengler et al., 2012); and recent studies of Val(8)GLP-1 in streptozocin-challenged rats effectively lowered brain levels of τ and ameliorated associated learning impairments (Li et al., 2012a), both validating and strengthening reported beneficial actions of GLP-1R activation on AD-associated proteins and their action on cognition.
As accumulations of abnormal Aβ peptide levels, particularly soluble oligomeric forms, are considered to be pivotal to the progression of AD in humans (Sambamurti et al., 2002; 2006; O'Brien and Wong, 2011), reductions in peptide levels and protection from Aβ peptide by GLP-1R-mediated processes should be beneficial. Current studies show this to be the case and suggest a strong candidate for the treatment of AD and possibly other dementia-related human disorders.
Functional evidence illustrating the benefits of GLP-1R stimulation in conditions of memory dysfunction is presented (D'Amico et al., 2010). The significance of this feature of GLP-1R signalling becomes clear when examining the effects of GLP-1R stimulation on abnormalities in LTP as observed in models of dementia. This is specifically relevant to the setting of AD, as animal studies involving Aβ toxic insult can induce a severe inhibition of LTP, while the impairment has been shown to be reversed by treatment with the GLP-1R agonist Val(8)GLP-1 (Gault and Hölscher, 2008; Wang et al., 2010). Moreover, control (wild-type) mice displayed an enhanced LTP as the result of administration of GLP-1 and Val(8)GLP-1 (Gault and Hölscher, 2008). AD transgenic mice, which overexpressed human APP, were also found to have a diminished or no LTP; yet treatment with Val(8)GLP-1 rescued the observed LTP deficit. In addition to this, wild-type mice displayed an enhanced LTP, more specifically subtypes of LTP associated with short-term memory formation (Gengler et al., 2012). Rodents challenged with central (i.c.v. injected) Aβ displayed deficits in spatial learning and memory capabilities, as assessed by the Morris Water Maze. However, pretreatment with Val(8)-GLP-1 improved performance in both parameters (Wang et al., 2010).
Conversely, Aβ peptide treated rats that displayed impaired learning and memory displayed increased expression of endogenous GLP-1 peptide (Oka et al., 1999b). The relevance of this compensatory action remains to be determined. In the same animal model, electrophysiological studies suggested that endogenous GLP-1 lowered hippocampal activity in the presence of Aβ, whereas a GLP-1R antagonist (Ex-(9–39)) elevated spontaneous neuronal firing and ameliorated Aβ-induced memory impairments (Oka et al., 2000). These findings are at odds with the direction of the majority of the more recent literature, and their significance currently remains unknown. As accumulations of abnormal Aβ peptide levels, particularly soluble oligomeric forms, are considered to be pivotal to the progression of AD in humans (Sambamurti et al., 2002; 2006; O'Brien and Wong, 2011), reductions in peptide levels and protection from Aβ peptide by GLP-1R-mediated processes should be beneficial. Current studies show this to be the case and suggest that GLP-1R activation in brain represents a strategy worth assessing for the treatment of AD and possibly other dementia-related human disorders.
Interestingly, when Ex-4 was administered over an extended period in rodents, studies indicated a decreased immobility observed in the forced swim test, a classic measure of depression. This effect was not reproduced when Ex-4 was administered 1 h before behavioural assessment, suggesting a possible neurogenic mechanism (Isacson et al., 2011). Hence, Ex-4 may well have antidepressant functions mediated by possible neurogenic properties, but further research is required to confirm such function, which certainly would be of value in light of the accompaniment of depression with neurodegenerative disorders (Aarsland et al., 2011).
GLP-1R stimulation decreases neuromotor impairment
PD is characterized by a loss of dopaminergic neurons and cellular degeneration of the striatum. It is associated with deficits in motor function, which are often the initial indicators of the disease in humans. To study PD and possible therapeutics that may ameliorate the disease symptoms and progression, investigators invariably utilize toxins that selectively kill dopaminergic cells or transgenic animals models that possess mutations in genes associated with the human disease (for review, see Jackson-Lewis et al., 2012; McDowell and Chesselet, 2012). Presently, data obtained regarding GLP-1R stimulation in models of PD are derived from studies that utilize neurotoxins in animals, or in neuronal tissue culture studies. A toxin widely used as a basic research tool for PD in humans and other mammals is 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). This agent, following its metabolism to 1-methyl-4-phenylpyridinium (MPP+) that is then selectively transported into the axons of dopaminergic neurons causes cell death by inhibiting mitochondrial complex 1 (Denton and Howard, 1987; Nicklas et al., 1987; Chan et al., 1991; Kang et al., 1997; Marini and Nowak, 2000) and, possibly as a consequence of this, by the formation of reactive oxygen species (Kovacic et al., 1991; Przedborski et al., 1992). When MPTP was administered to rats or mice, substantial losses of dopaminergic neurons occurred, which was associated with a heightened inflammatory response. The use of GLP-1R agonists has been shown to protect animals against MPTP toxic insult. The MPTP toxicity was fully reversed by Ex-4, which increased numbers of viable dopaminergic neurons and tempered the level of inflammation (Kim et al., 2009; Li et al., 2009). TH is a key enzyme important for the production of dopamine; it converts tyrosine into L-DOPA, a direct precursor of dopamine. Cell culture studies have demonstrated that Ex-4 elevates endogenous TH levels in primary dopaminergic neurons (Li et al., 2009). Studies in TH expressing catecholamine neurons in the area postrema have, likewise, demonstrated that Ex-4 significantly elevates TH levels and suggest this is mediated by Ex-4 induction of TH gene expression through the TH promoter (Yamamoto et al., 2003). In contrast, MPTP treatment reduces TH-positive neurons, decreases concentrations of dopamine as well as its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), and increases the ratio of dopamine metabolites to dopamine. These effects were fully prevented by treatment with Ex-4 (Li et al., 2009). Similarly, MPTP administration in rats resulted in severe impairments in motor function, yet treatment with Ex-4 restored the observed deficits (Li et al., 2009).
6-Hydroxydopamine (6-OHDA) is a neurotoxin that, similar to MPTP, kills dopaminergic neurons; likewise, GLP-1R stimulation has been shown to protect neurons from exposure to 6-OHDA. GLP-1 and Ex-4 dose-dependently protected SH-SY5Y cells against 6-OHDA-induced cell death (Li et al., 2009; 2010b). In mesencephalic neuronal cultures that are rich in dopaminergic neurons, 6-OHDA lowered the number of TH-positive cells, indicating dopaminergic neuronal death. Treatment with Ex-4 was not only shown to rescue dopaminergic neurons but induced a 60% increase in TH-positive cells over control values (Li et al., 2009). In rats injected with 6-OHDA or LPS, nigrostriatal dopamine levels and the L-DOPA synthesizing capabilities of these cells were markedly diminished, in line with reduced levels of TH-positive neurons. These deficits were reversed by Ex-4 treatment (Harkavyi et al., 2008). Similarly, in 6-OHDA brain-lesioned rats, lower levels of TH-positive and vesicular monoamine transporter 2 (VMAT2)-positive cells were observed; while these changes were halted by Ex-4 treatment (Bertilsson et al., 2008). VMAT2 has regulatory functions involved in the storage and processing of dopamine into synaptic vesicles.
As stated previously, PD is associated with selective neuronal dopaminergic cell degeneration and consequently disturbances in motor function. Apomorphine, a dopaminergic drug, has been used to induce circling behaviour in PD animals; the degree of circling behaviour indicates the severity of PD-like damage in the striatum. In 6-OHDA/LPS-challenged rodents, the apomorphine circling behaviour was alleviated by Ex-4 in a dose-dependent manner (Harkavyi et al., 2008). Additional experiments using 6-OHDA to induce lesions and abnormal behaviour have reinforced these findings. The measurement of an animal's circling behaviour before and after Ex-4 treatment indicated a near complete normalization in behaviour (Bertilsson et al., 2008). These data illustrate GLP-1R stimulation benefits in the setting of neurotoxin-derived models of PD in terms of dopamine cell survival, cell functionality and the resolution of abnormal behaviour. These findings strengthen the hypothesis that GLP-1R stimulation may have therapeutic value in the setting of human PD.
In line with findings obtained from CNS-derived Parkinsonian motor dysfunction, a form of peripheral neuropathy induced by over-consumption of vitamin B6 (pyridoxine) has displayed benefits of GLP-1R activation. In a model of pyridoxine-induced peripheral neuropathy where the systemic treatment of rodents with high concentrations of pyridoxine induces a severe sensory neuropathy that bears some similarities to diabetic neuropathy and is accompanied by abnormal nerve fibre geometry, axonal wasting and electrophysiological abnormalities (Perry et al., 2004), treatment of animals with a GLP-1R agonist showed cellular and behavioural benefits compared to control pyridoxine treated animals (Perry et al., 2007). The sciatic nerve and dorsal root ganglia have been indicated as central to the pathology of peripheral neuropathy. Targeting these tissues with vitamin B6 produced fewer large-diameter fibres, a greater number of small-diameter fibres, a smaller number of myelinated fibres and a larger area of connective tissue between fibres. Morphological changes of this nature indicate neuropathy, leading to functional impairments. Pyridoxine insult presented functionally as hind limb deficits, which quickly spread to all four limbs, culminating in a severe walking impediment. Measurements of motor coordination, rotarod and inclined screen tests also underscored these deficiencies in pyridoxine-treated rats. GLP-1 and Ex-4 improved vitamin B6-induced functional and behavioural deficits in addition to morphological normalization of the sciatic nerve and dorsal root ganglia (Perry et al., 2007).
This peripheral neuropathy research has recently been extended to diabetes, where Ex-4 treatment has been shown to attenuate streptozotocin-induced reductions of motor nerve conduction velocity and paw intraepidermal fibre density evident in diabetic mice (Jolivalt et al., 2011), as well as to significantly ameliorate diabetic polyneuropathy likewise in streptozotocin-induced diabetic mice (Himeno et al., 2011). The pleiotropic effects of GLP-1-based therapies with regard to their potential clinical utility in vascular complications in diabetes have recently been reviewed (Yamagishi and Matsui, 2011).
HD is a genetic disorder attributed to an expansion of CAG trinucleotide repeats within the huntingtin protein gene. This mutation results in abnormal, intracellular aggregations of this protein in pancreatic and CNS tissues, with the latter leading to a loss in motor coordination. Mouse models of HD employing the mutant huntingtin protein, in addition to reproduction of the motor dysfunction pathology, also exhibit dysregulated blood sugar control. In an age-dependent manner, animals display poor performance on a rotarod with increasingly diminished motor function over time, which was associated with increased levels of mutant huntingtin protein aggregates. The use of GLP-1R stimulation as an intervention in HD, in the HD mouse strain N171-82Q, induced amelioration of abnormal blood sugar levels along with reduced quantities of mutant huntingtin protein accumulations in pancreas and brain. Ex-4-treated animals exhibited reduced motor function deficits and enhanced the animal survival time by 18% compared with control N171-82Q mouse life span (Martin et al., 2009).
A recent study has focused on the potential benefits of GLP-1R activation in cellular and mouse models of ALS, as motor neurons express the GLP-1R (Li et al., 2012b). ALS, also known as Lou Gehrig's disease, is characterized by selective and progressive death of motor neurons within the brain and spinal cord, which leads to paralysis of voluntary muscles and, eventually, death within 5 years of clinical onset (Habib and Mitsumoto, 2011). Whereas most cases of ALS occur sporadically with unknown aetiology, some 10% are inherited in an autosomal-dominant manner (Pasinelli and Brown, 2006). Of these, 20% are caused by mutations within the gene encoding the superoxide dismutase 1 (SOD-1) protein, an enzyme involved in the scavenging and detoxification of superoxide radicals. Transgenic mice expressing the same SOD-1 mutations as humans exhibit similar histopathological and clinical phenotypes as ALS patients (Kato, 2008), and together with neuronal cultures with and without SOD-1 mutations have been widely used to elucidate mechanisms inducing ALS pathology as well as to screen for potential therapeutics. In line with recent studies indicating that GLP-1 protects cultured motor neurons against glucosamine-induced cytotoxicity (Lim et al., 2010), both GLP-1 and Ex-4 have been shown in NSC19 cells to be neurotrophic, elevating cholinergic markers and neuroprotective against oxidative stress and apoptosis induced by serum deprivation (Li et al., 2012b). These actions largely translated to SOD-1 (G93A) mice, in which s.c. Ex-4 not only mitigated the dysregulation of glucose evident in animals but also proved neuroprotective at the level of the spinal cord, preserving spinal cord structure and neuron density, and mitigating apoptosis and loss of cholinergic markers (Li et al., 2012). Albeit that survival time was not a focus of the study, this appeared to be unaltered by the beneficial Ex-4 actions, suggesting that GLP-1R activation in ALS mice may provide quality of life improvements rather than impacting survival at the chosen dose studies. Nevertheless, results from this recent study encourage further research of GLP-1R activation in ALS to define potential for clinical translation.
Although functional improvements in motor coordination within these paradigms may be the result of changes in neuronal morphology and decreases in protein aggregation, other factors may also account for these enhancements. GLP-1R activation increases cellular cAMP levels, which may promote motor neuron survival in response to nutrient deprivations (Hanson et al., 1998). Similar to that observed with cognitive impairments, an increasing body of preclinical evidence exists that supports a beneficial role of GLP-1R signalling events in the setting of motor function degenerative diseases.
GLP-1R stimulation ameliorates stroke
In the event of a cerebral stroke, a blood vessel can be blocked or a blood vessel may rupture, which causes a disruption in blood flow to the surrounding tissues. The tissues in the area of the stroke can become irreversibly damaged and ultimately become necrotic and die. In this situation, there is nothing that can be done to prevent the cell death and resulting brain damage. However, in the area surrounding the damaged stroke infarct zone, there is a region of tissue that may be amenable to therapeutic manipulation. Studies have indicated alterations in GLP-1R expression associated with areas of stroke and cellular damage, in which receptor expression was elevated surrounding the area of damage and was associated with glial cells (astrocytes and microglia) (Chowen et al., 1999; Lee et al., 2011), in contrast to its predominant association with neuronal cells in healthy brain. The use of rodent transient middle cerebral artery occlusion (MCAo) models of stroke has shown that both pretreatment and post-treatment with the GLP-1R agonist Ex-4 possess marked beneficial effects on infarct size, which is absent in GLP-1R knock out mice (Li et al., 2009; Teramoto et al., 2011). Benefits in rodent motor activity were evident in activity chambers (Li et al., 2009) and improvement in neurological deficits observed (Teramoto et al., 2011). Interestingly, a component of the damage caused by stroke to the surrounding tissues of the infarct area may be due the phenomenon of excitotoxicity.
Cellular damage due to excitotoxicity results from rapid, heavy influxes of Na+ and Ca2+ upon activation of metabotropic and NMDA receptors. These ion flux alterations produce cellular enlargement and eventual lysis, as well as changes to the endoplasmic reticulum and mitochondria, leading to apoptosis (Mattson, 2008). GLP-1 treatment of cultured neurons can protect against glutamate-induced alterations in calcium currents (Gilman et al., 2003). Ibotenic acid, a glutamatergic agonist (Bunch and Krogsgaard-Larsen, 2009), is typically used to lesion the basal forebrain's basal nucleus in mice; this results in extensive loss of cholinergic neurons. Treatment with GLP-1 and Ex-4 effectively protected the animals against this form of cell loss, as measured by increased choline acetyltransferase immunoreactivity in the region. Moreover, neurons challenged with glutamate, an NMDA receptor agonist, exhibited cell death marked by apoptosis, which was decreased with Ex-4 treatment (Perry et al., 2002b). Kainic acid, a glutamate receptor agonist, can cause seizures and neuronal death and has been used to generate rodent models of epilepsy. When GLP-1R knockout mice (Glp1r–/–) were administered kainic acid, they displayed decreased latency to seizure onset, increased seizure severity and increased hippocampal apoptosis as compared with wild-type animals (Glp1r+/+). Furthermore, if wild-type mice were treated with [Ser(2)]exendin(1–9), another incretin mimetic, a reduction in kainic acid-induced hippocampal apoptosis was detected (During et al., 2003).
In addition to promoting excitotoxicity, a stroke can induce environmental deprivations in oxygen and glucose, and the lack of blood flow allows for the build up of harmful metabolites of cell function. GLP-1R activation has been shown to counteract some of these environmental deficits. Primary cerebral cortical neurons were protected from hypoxia-triggered cell death by GLP-1 and Ex-4. Supporting the involvement of GLP-1R stimulation, neurons from GLP-1R knockout mice (Glp1r–/–) and normal mice co-treated with the GLP-1R antagonist Ex-(9–39) were not protected from hypoxia-induced damage by Ex-4 treatment (Li et al., 2009). Currently, the only human epidemiological data available on a neurological condition and the use of GLP-1 agonists in humans is a retrospective study on the effects of Exenatide (Ex-4) and cardiovascular disease, including stroke. The study indicated that daily treatment of Exenatide in T2DM patients lowered the occurrence of cardiovascular disease (Best et al., 2012). Another example of a lethal environmental deprivation is the removal of the neurotrophic factor, NGF, from cells in vitro. Administration of GLP-1 was shown to stop cell death and morphological degeneration in PC12 cells and in cultured sympathetic neurons as a response to NGF removal (Biswas et al., 2008).
Interestingly, in some conditions, dietary energy restriction has been shown to protect against neuronal degeneration (Mattson, 2010). Clinical studies utilizing the administration of GLP-1 or Ex-4 have found that patients experience a decrease in caloric/food intake, hunger and body weight (Drucker and Nauck, 2006; Lovshin and Drucker, 2009; Vilsbøll et al., 2012). In addition, subjects had increased feelings of satiety/fullness and an increased resting metabolic rate (Bradley et al., 2010). Aspects of these studies are recapitulated in rodent studies (Hayes et al., 2011). However, it is important not to confuse abnormal energy availability and metabolism with dietary energy caloric restriction, as the former tends to be associated with age-associated disease and neurodegenerative conditions (Blass et al., 1988); and unintentional weight loss in the elderly should in general be avoided (Huffman, 2002; Amella, 2004). Caloric restriction has been shown to reduce Aβ and τ accumulation in a mouse AD model (Halagappa et al., 2007). Interestingly, the neurotrophic factor brain-derived neuroptophic factor (BDNF) promotes neural progenitor cell differentiation and subsequent survival; it enhances neurite outgrowth as well as synaptogenesis, thereby promoting long-term potentiation and protecting neurons from excitoxicity (Mattson, 2008; Zuccato and Cattaneo, 2009; Nagahara and Tuszynski, 2011). Contrasting these effects, reductions in BDNF add to cognitive impairments seen in diabetes and enhance PD pathology. Impacting energy intake and expenditure may contribute to neuroprotective functions seen in incretin pathway stimulation, and these functions are possibly mediated by BDNF. However, the described neurotrophic/neuroprotective actions of GLP-1R activation can be considered to be chiefly ‘direct’ actions, rather than secondary ones consequent to reduced food intake. Not surprisingly, given their wide range of trophic activities, GLP-1 and its analogs join a list of neurotrophic factors that protect against neuronal degeneration. The protective effects of BDNF, insulin-like growth factor, NGFs and in some conditions TNF-α are such examples (Pezet and Malcangio, 2004). Neurotrophic protection is essential, as many neurodegenerative states are accompanied by decreased cellular energy availability, as exemplified by AD and conditions of increased oxidative stress.
GLP-1R stimulation reduces oxidative stress
Oxidative stress is a common feature of several neurodegenerative conditions, and oxidative stress plays a key contributory role in the progressive nature of diseases like PD, AD and ALS, as well as in acute disorders such as stroke and traumatic brain injury. Principal sources of reactive factors responsible for oxidative cell damage are mitochondria. These organelles generate reactive oxygen species by the normal cellular processes of mitochondrial function; however, when there is an imbalance between the processes that generate and then eliminate the highly active chemicals, problems can arise. Additional to oxidative stress induced by abnormal cellular activities, as is seen in ALS, a further source of oxidative damage can originate from activated peripheral macrophages or the brain resident glial cells in response to microenvironmental activators, such as Aβ and α-synuclein protein and circulating cytokines; all of which are observed in the setting of neurodegenerative disease. GLP-1R stimulation has been shown to attenuate the synthesis of a pro-inflammatory cytokine (IL-1β) in activated astrocytes (Iwai et al., 2006). A classic in vitro model of oxidative stress involves the use of hydrogen peroxide (H2O2) added to culture media. It has been shown that GLP-1R stimulation is able to ameliorate the detrimental cellular changes induced by this form of stress. GLP-1 and Ex-4 dose-dependently protected SH-SY5Y cells from H2O2 induced cell death (Li et al., 2009; 2010b). It is likewise known that 6-OHDA exerts it toxicity through oxidative stress. As mentioned before, GLP-1R stimulation is able to reduce 6-OHDA-induced cell death in SH-SY5Y cells (Li et al., 2010b) as well as primary ventral mesencephalic (dopaminergic) neurons (Li et al., 2009). Similarly, the incretin mimetic geniposide has been described to mitigate H2O2-mediated death in PC12 cells (Liu et al., 2007; 2009). Furthermore, GLP-1R activation can protect PC12 cells against the pro-apoptotic effects of methylglyoxal, an agent associated with oxidation-induced neural damage in diabetes mellitus and more recently with AD dementias (Kimura et al., 2009). Ischaemia–reperfusion injury can also cause oxidative stress, which results in cell damage and death. In this regard, in a MCAo stroke mouse model, treatment with Ex-4 significantly reduced markers of oxidative stress, namely 8-hydroxy deoxyguanosine and 4-hydroxy 2-hexenal (Teramoto et al., 2011).
GLP-1R stimulation ameliorates retinal degeneration
The retina is considered part of the CNS. It has been demonstrated that GLP-1R is expressed in rat retina (Zhang et al., 2009b) and in a specialized eye cell type, the Müller cell/glia cell line rMc-1 cells (Zhang et al., 2011a). Diabetic retinopathy, as a neurodegenerative disease of the eye, is the leading cause of blindness in patients aged 20 to 70 years in the United States. Using a streptozotocin-induced diabetic rat model, whose retinal pathology resembles that observed in the early stage of human diabetic retinopathy, Zhang Y et al. demonstrated that an Ex-4 analog called E4a can protect retinal cells when administered s.c. (Zhang et al., 2009) or intravitreally (Zhang et al., 2011). Blood glucose levels were normalized in rats when E4a was administered subcutaneously but not intravitreally. Electroretinogram (ERG) is used as an objective method to evaluate the retinal function. While both B-wave amplitudes and oscillatory potentials are decreased in diabetic rats, E4a treatments significantly rescued these deficits. Moreover, E4a treatment also protected retinal neurons from death, as demonstrated by a significant rescue of the thickness of different retinal layers (Zhang et al., 2009a; 2011). In an alternative optic nerve crush model of eye neurodegeneration in a rat model in which GLP-1 was delivered via an intraocular cell based implantation, neuroprotection was observed that resulted in a higher survival rate of retinal ganglion cells following the crush procedure (Zhang et al., 2009b; 2011).
Conclusion
In summary, activation of the GLP-1R-driven incretin pathway has been shown to be neurotrophic, neuroprotective, and functionally and behaviourally beneficial under numerous physiological and pathophysiological paradigms. As GLP-1R stimulation has proven to be beneficial and well tolerated in the setting of human T2DM, one can hypothesize that these types of agents would likely perform similarly in human neurological diseases. This hypothesis requires validation in humans by clinical studies. In this regard, a phase II clinical trial of Ex-4 for the treatment of AD is currently recruiting participants (clinicaltrial.gov identifier: NCT01255163). A pilot clinical study using Ex-4 to treat PD is also ongoing (clinicaltrial.gov identifier: NCT01174810), as is a further one to evaluate efficacy of the agent in diabetics with peripheral neuropathy (clinical trial.gov identifier: NCT00855439). However, based on research data currently available, obtained from models of AD, PD and HD, this new potential treatment strategy shows great promise as a candidate treatment of human neurodegenerative disorders. Benefits in cellular pathology have been described in rodents administered with GLP-1 peptide secreting cells after brain injury (Heile et al., 2009). Although such cells secrete a number of factors in addition to GLP-1, these elegant studies nonetheless point towards a further area of potential GLP-1 clinical utility. Presently, fully published data are lacking exploring the utility of this approach in animal models of head trauma; however, studies into these areas are ongoing, and Ex-4 has been recently described as promising in models of both blunt injury (weight drop) and blast injury (controlled explosive detonation) (Rubovitch et al., 2011) (Chaim Pick, Tel Aviv University, pers. comm.). This is of relevance to neurodegenerative disorders as traumatic brain injury is considered a conduit to their later development (Johnson et al., 2010; Costanza et al., 2011). In the meantime, research will continue to identify benefits and possible drawbacks of using GLP-1 peptide analogs for neurological disorders. Hopefully, the promising preliminary characterizations of GLP-1R pathway functions in neurons (Li et al., 2010b) will combine with preclinical and clinical results to culminate in efficacious treatments for some of the most devastating human diseases in aging patients. The ever-expanding elderly population is of increasing social and medical relevance; thus, we must continue to explore options to support healthy aging and alleviate deteriorations that occur consequent to age-associated diseases, particularly those impacting the nervous system.
Acknowledgments
This work was supported by the Intramural Research Program, National Institute on Aging, National Institutes of Health. The authors declare no conflicts of interest regarding the contents of this manuscript.
Glossary
- Aβ
amyloid-β
- AD
Alzheimer's disease
- APP
amyloid-β precursor protein
- ALS
amyotrophic lateral sclerosis
- BDNF
brain-derived neurotrophic factor
- BrdU
bromodeoxyuridine
- CNTF
ciliary neurotrophic factor
- DCX
doublecortin
- DOPAC
3,4-dihydroxyphenylacetic acid
- DPP-IV
dipedylpeptidase-IV
- ERG
electroretinogram
- Ex-4
exendin-4
- GIP
gastric inhibitor peptide
- GLP-1
glucagon-like peptide-1
- GLP-1R
glucagon-like peptide-1 receptor
- HVA
homovanillic acid
- HD
Huntington's disease
- LTP
long term potentiation
- MCAo
middle cerebral artery occlusion
- MPTP
1-methyl-4-phenyl-1,2,3,4,6-tetrahydroxypyridine
- MPP+
1-methyl-4-phenylpyridinum ion
- NGF
nerve growth factor
- 6-OHDA
6-hydroxydopamine
- PD
Parkinson's disease
- RA
retinoic acid
- VMAT2
vesicle monoamine transporter 2
Conflicts of interest
None.
References
- Aarsland D, Påhlhagen S, Ballard CG, Ehrt U, Svenningsson P. Depression in Parkinson disease-epidemiology, mechanisms and management. Nat Rev Neurol. 2011;8:35–47. doi: 10.1038/nrneurol.2011.189. [DOI] [PubMed] [Google Scholar]
- Abbas T, Faivre E, Hölscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer's disease. Behav Brain Res. 2009;205:265–271. doi: 10.1016/j.bbr.2009.06.035. [DOI] [PubMed] [Google Scholar]
- Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer's disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71:365–376. doi: 10.1111/j.1365-2125.2010.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez E, Roncero I, Chowen JA, Thorens B, Blázquez E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J Neurochem. 1996;66:920–927. doi: 10.1046/j.1471-4159.1996.66030920.x. [DOI] [PubMed] [Google Scholar]
- Alvarez E, Martínez MD, Roncero I, Chowen JA, García-Cuartero B, Gispert JD, et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J Neurochem. 2005;92:798–806. doi: 10.1111/j.1471-4159.2004.02914.x. [DOI] [PubMed] [Google Scholar]
- Amella EJ. Feeding and hydration issues for older adults with dementia. Nurs Clin North Am. 2004;39:607–623. doi: 10.1016/j.cnur.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med. 2006;57:265–281. doi: 10.1146/annurev.med.57.110104.115624. [DOI] [PubMed] [Google Scholar]
- Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature. 1983;302:716–718. doi: 10.1038/302716a0. [DOI] [PubMed] [Google Scholar]
- Belsham DD, Fick LJ, Dalvi PS, Centeno ML, Chalmers JA, Lee PK, et al. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009;23:4256–4265. doi: 10.1096/fj.09-133454. [DOI] [PubMed] [Google Scholar]
- Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson's disease. J Neurosci Res. 2008;86:326–338. doi: 10.1002/jnr.21483. [DOI] [PubMed] [Google Scholar]
- Best JH, Lavillotti K, Deyoung MB, Garrison LP. The effects of exenatide BID on metabolic control, medication use, and hospitalization in patients with type 2 diabetes mellitus in clinical practice: a systematic review. Diabetes Obes Metab. 2012;14:387–398. doi: 10.1111/j.1463-1326.2011.01533.x. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Buteau J, Greene LA. Glucagon-like peptide-1 (GLP-1) diminishes neuronal degeneration and death caused by NGF deprivation by suppressing Bim induction. Neurochem Res. 2008;33:1845–1851. doi: 10.1007/s11064-008-9646-4. [DOI] [PubMed] [Google Scholar]
- Blass JP, Sheu K, Cederbaum JM. Energy metabolism in disorders of the nervous system. Rev Neurol (Paris) 1988;144:543–563. [PubMed] [Google Scholar]
- Bradley DP, Kulstad R, Schoeller DA. Exenatide and weight loss. Nutrition. 2010;26:243–249. doi: 10.1016/j.nut.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Brown JC, Dryburgh JR, Ross SA, Dupre' J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res. 1975;31:487–532. doi: 10.1016/b978-0-12-571131-9.50017-7. [DOI] [PubMed] [Google Scholar]
- Bunch L, Krogsgaard-Larsen P. Subtype selective kainic acid receptor agonists: discovery and approaches to rational design. Med Res Rev. 2009;29:3–28. doi: 10.1002/med.20133. [DOI] [PubMed] [Google Scholar]
- Calvo JC, Gisolfi CV, Blazquez E, Mora F. Glucagon-like peptide-1(7-36)amide induces the release of aspartic acid and glutamine by the ventromedial hypothalamus of the conscious rat. Brain Res Bull. 1995a;38:435–439. doi: 10.1016/0361-9230(95)02010-o. [DOI] [PubMed] [Google Scholar]
- Calvo JC, Yusta B, Mora F, Blázquez E. Structural characterization by affinity cross-linking of glucagon-like peptide-1(7-36)amide receptor in rat brain. J Neurochem. 1995b;64:299–306. doi: 10.1046/j.1471-4159.1995.64010299.x. [DOI] [PubMed] [Google Scholar]
- Campos RV, Lee YC, Drucker DJ. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology. 1994;134:2156–2164. doi: 10.1210/endo.134.5.8156917. [DOI] [PubMed] [Google Scholar]
- Chan P, DeLanney LE, Irwin I, Langston JW, Di Monte D. Rapid ATP loss caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mouse brain. J Neurochem. 1991;57:348–351. doi: 10.1111/j.1471-4159.1991.tb02134.x. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- Chowen JA, de Fonseca FR, Alvarez E, Navarro M, García-Segura LM, Blázquez E. Increased glucagon-like peptide-1 receptor expression in glia after mechanical lesion of the rat brain. Neuropeptides. 1999;33:212–215. doi: 10.1054/npep.1999.0757. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1991;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Costanza A, Weber K, Gandy S, Bouras C, Hof PR, Giannakopoulos P, et al. Review: contact sport-related chronic traumatic encephalopathy in the elderly: clinical expression and structural substrates. Neuropathol Appl Neurobiol. 2011;37:570–584. doi: 10.1111/j.1365-2990.2011.01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S. Insulin resistance and Alzheimer's disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- D'Amico M, Di Filippo C, Marfella R, Abbatecola AM, Ferraraccio F, Rossi F, et al. Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer's prone mice. Exp Gerontol. 2010;45:202–207. doi: 10.1016/j.exger.2009.12.004. [DOI] [PubMed] [Google Scholar]
- de la Monte SM. Insulin resistance and Alzheimers's disease. BMB Rep. 2009;42:475–481. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton T, Howard BD. A dopaminergic cell line variant resistant to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem. 1987;49:622–630. doi: 10.1111/j.1471-4159.1987.tb02909.x. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9:1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- Flament S, Delacourte A, Mann DM. Phosphorylation of Tau proteins: a major event during the process of neurofibrillary degeneration. A comparative study between Alzheimer's disease and Down's syndrome. Brain Res. 1990;516:15–19. doi: 10.1016/0006-8993(90)90891-e. [DOI] [PubMed] [Google Scholar]
- Gault VA, Hölscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol. 2008;587:112–117. doi: 10.1016/j.ejphar.2008.03.025. [DOI] [PubMed] [Google Scholar]
- Gengler S, McClean PL, McCurtin R, Gault VA, Hölscher C. Val(8)GLP-1 rescues synaptic plasticity and reduces dense core plaques in APP/PS1 mice. Neurobiol Aging. 2012;33:265–276. doi: 10.1016/j.neurobiolaging.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87:1137–1144. doi: 10.1046/j.1471-4159.2003.02073.x. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, et al. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann N Y Acad Sci. 2004;1035:290–315. doi: 10.1196/annals.1332.018. [DOI] [PubMed] [Google Scholar]
- Habib AA, Mitsumoto H. Emerging drugs for amyotrophic lateral sclerosis. Expert Opin Emerg Drugs. 2011;16:537–558. doi: 10.1517/14728214.2011.604312. [DOI] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Hölscher C. Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system. Neuroreport. 2009;20:1161–1166. doi: 10.1097/WNR.0b013e32832fbf14. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Patterson S, Porter D, Gault VA, Holscher C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res. 2011;89:481–489. doi: 10.1002/jnr.22565. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson's disease. J Neuroinflammation. 2008;5:19. doi: 10.1186/1742-2094-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME. What is the function of hippocampal theta rhythm?–Linking behavioral data to phasic properties of field potential and unit recording data. Hippocampus. 2005;15:936–949. doi: 10.1002/hipo.20116. [DOI] [PubMed] [Google Scholar]
- Hayes MR, Leichner TM, Zhao S, Lee GS, Chowansky A, Zimmer D, et al. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab. 2011;13:320–330. doi: 10.1016/j.cmet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heile AMB, Wallrapp C, Klinge PM, Samii A, Kassem M, Silverberg G, et al. Cerebral transplantation of encapsulated mesenchymal stem cells improves cellular pathology after experimental traumatic brain injury. Neurosci Lett. 2009;463:176–181. doi: 10.1016/j.neulet.2009.07.071. [DOI] [PubMed] [Google Scholar]
- Himeno T, Kamiya H, Naruse K, Harada N, Ozaki N, Seino Y, et al. Beneficial effects of exendin-4 on experimental polyneuropathy in diabetic mice. Diabetes. 2011;60:2397–2406. doi: 10.2337/db10-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman GB. Evaluating and treating unintentional weight loss in the elderly. Am Fam Physician. 2002;65:640–650. [PubMed] [Google Scholar]
- Huntington Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Hyman JM, Wyble BP, Goyal V, Rossi CA, Hasselmo ME. Stimulation in hippocampal region CA1 in behaving rats yields LTP when delivered to the peak of theta and LTD when delivered to the trough. J Neurosci. 2003;23:11725–11731. doi: 10.1523/JNEUROSCI.23-37-11725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman JM, Hasselmo ME, Seamans JK. What is the functional relevance of prefrontal cortex entrainment to hippocampal theta rhythms? Front Neurosci. 2011;5:24. doi: 10.3389/fnins.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacson R, Nielsen E, Dannaeus K, Bertilsson G, Patrone C, Zachrisson O, et al. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur J Pharmacol. 2011;650:249–255. doi: 10.1016/j.ejphar.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci Res. 2006;55:352–360. doi: 10.1016/j.neures.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Blesa J, Przedborski S. Animal models of Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl. 1):S183–S185. doi: 10.1016/S1353-8020(11)70057-8. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Fineman M, Deacon CF, Carr RD, Calcutt NA. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab. 2011;13:990–1000. doi: 10.1111/j.1463-1326.2011.01431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Miyako K, Kuribayashi F, Hasegawa E, Mitsumoto A, Nagano T, et al. Changes of energy metabolism induced by 1-methyl-4-phenylpyridinium (MPP+)-related compounds in rat pheochromocytoma PC12 cells. Arch Biochem Biophys. 1997;337:75–80. doi: 10.1006/abbi.1996.9727. [DOI] [PubMed] [Google Scholar]
- Kato S. Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:97–114. doi: 10.1007/s00401-007-0308-4. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Hopkins RO. Mnemonic functions of the hippocampus: a comparison between animals and humans. Biol Psychol. 2006;73:3–18. doi: 10.1016/j.biopsycho.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson's disease. J Endocrinol. 2009;202:431–439. doi: 10.1677/JOE-09-0132. [DOI] [PubMed] [Google Scholar]
- Kimura R, Okouchi M, Fujioka H, Ichiyanagi A, Ryuge F, Mizuno T, et al. Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience. 2009;162:1212–1219. doi: 10.1016/j.neuroscience.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Kovacic P, Edwards WD, Ming G. Theoretical studies on mechanism of MPTP action: ET interference by MPP+ (1-methyl-4-phenylpyridinium) with mitochondrial respiration vs. oxidative stress. Free Radic Res Commun. 1991;14:25–32. doi: 10.3109/10715769109088938. [DOI] [PubMed] [Google Scholar]
- Kroner Z. The relationship between Alzheimer's disease and diabetes: type 3 diabetes? Altern Med Rev. 2009;14:373–379. [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77:257–270. doi: 10.1016/s0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- Lee CH, Yan B, Yoo KY, Choi JH, Kwon SH, Her S, et al. Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J Neurosci Res. 2011;89:1103–1113. doi: 10.1002/jnr.22596. [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci USA. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J Alzheimers Dis. 2010a;19:1205–1219. doi: 10.3233/JAD-2010-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem. 2010b;113:1621–1631. doi: 10.1111/j.1471-4159.2010.06731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lee CH, Yoo KY, Choi JH, Park OK, Yan BC, et al. Chronic treatment of exendin-4 affects cell proliferation and neuroblast differentiation in the adult mouse hippocampal dentate gyrus. Neurosci Lett. 2010c;486:38–42. doi: 10.1016/j.neulet.2010.09.040. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol. 2012a;674:280–286. doi: 10.1016/j.ejphar.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, et al. Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. PLoS ONE. 2012b;7:e32008. doi: 10.1371/journal.pone.0032008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JG, Lee JJ, Park SH, Park JH, Kim SJ, Cho HC, et al. Glucagon-like peptide-1 protects NSC-34 motor neurons against glucosamine through Epac-mediated glucose uptake enhancement. Neurosci Lett. 2010;479:13–17. doi: 10.1016/j.neulet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Liu JH, Yin F, Zheng X, Jing J, Hu Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem Int. 2007;51:361–369. doi: 10.1016/j.neuint.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Liu JH, Yin F, Guo LX, Deng XH, Hu YH. Neuroprotection of geniposide against hydrogen peroxide induced PC12 cells injury: involvement of PI3 kinase signal pathway. Acta Pharmacol Sin. 2009;30:159–165. doi: 10.1038/aps.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- Luciani P, Deledda C, Benvenuti S, Cellai I, Squecco R, Monici M, et al. Differentiating effects of the glucagon-like peptide-1 analogue exendin-4 in a human neuronal cell model. Cell Mol Life Sci. 2010;67:3711–3723. doi: 10.1007/s00018-010-0398-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models? Rev Neurosci. 2011;22:373–402. doi: 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- Marini AM, Nowak TS., Jr Metabolic effects of 1-methyl-4-phenylpyridinium (MPP(+)) in primary neuron cultures. J Neurosci Res. 2000;62:814–820. doi: 10.1002/1097-4547(20001215)62:6<814::AID-JNR8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington's disease. Diabetes. 2009;58:318–328. doi: 10.2337/db08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. The impact of dietary energy intake on cognitive aging. Front Aging Neurosci. 2010;2:5. doi: 10.3389/neuro.24.005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell K, Chesselet MF. Animal models of the non-motor features of Parkinson's disease. Neurobiol Dis. 2012 doi: 10.1016/j.nbd.2011.12.040. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Youngster SK, Kindt MV, Heikkila RE. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721–729. doi: 10.1016/0024-3205(87)90299-2. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka JI, Goto N, Kameyama T. Glucagon-like peptide-1 modulates neuronal activity in the rat's hippocampus. Neuroreport. 1999a;10:1643–1646. doi: 10.1097/00001756-199906030-00004. [DOI] [PubMed] [Google Scholar]
- Oka JI, Suzuki E, Goto N, Kameyama T. Endogenous GLP-1 modulates hippocampal activity in beta-amyloid protein-treated rats. Neuroreport. 1999b;10:2961–2964. doi: 10.1097/00001756-199909290-00016. [DOI] [PubMed] [Google Scholar]
- Oka JI, Suzuki E, Kondo Y. Endogenous GLP-1 is involved in beta-amyloid protein-induced memory impairment and hippocampal neuronal death in rats. Brain Res. 2000;878:194–198. doi: 10.1016/s0006-8993(00)02741-4. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Perry T, Greig NH. The glucagon-like peptides: a new genre in therapeutic targets for intervention in Alzheimer's disease. J Alzheimers Dis. 2002;4:487–496. doi: 10.3233/jad-2002-4605. [DOI] [PubMed] [Google Scholar]
- Perry T, Greig NH. The glucagon-like peptides: a double-edged therapeutic sword? Trends Pharmacol Sci. 2003;24:377–383. doi: 10.1016/S0165-6147(03)00160-3. [DOI] [PubMed] [Google Scholar]
- Perry T, Greig NH. Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer's disease. Curr Alzheimer Res. 2005;2:377–385. doi: 10.2174/1567205054367892. [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Chen D, Zhou J, Shaw KT, Egan JM, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002a;300:958–966. doi: 10.1124/jpet.300.3.958. [DOI] [PubMed] [Google Scholar]
- Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002b;302:881–888. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603–612. doi: 10.1002/jnr.10611. [DOI] [PubMed] [Google Scholar]
- Perry T, Holloway HW, Weerasuriya A, Mouton PR, Duffy K, Mattison JA, et al. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp Neurol. 2007;203:293–301. doi: 10.1016/j.expneurol.2006.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry TA, Weerasuriya A, Mouton PR, Holloway HW, Greig NH. Pyridoxine-induced toxicity in rats: a stereological quantification of the sensory neuropathy. Exp Neurol. 2004;190:133–144. doi: 10.1016/j.expneurol.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M. Brain-derived neurotrophic factor as a drug target for CNS disorders. Expert Opin Ther Targets. 2004;8:391–399. doi: 10.1517/14728222.8.5.391. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci. 1992;12:1658–1667. doi: 10.1523/JNEUROSCI.12-05-01658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Sun Z, Huang J, Hu Y, Wu Z, Mei B. Mutated recombinant human glucagon-like peptide-1 protects SH-SY5Y cells from apoptosis induced by amyloid-beta peptide (1-42) Neurosci Lett. 2008;444:217–221. doi: 10.1016/j.neulet.2008.08.047. [DOI] [PubMed] [Google Scholar]
- Ross SA, Brown JC, Dupre' J. Hypersecretion of gastric inhibitory polypeptide following oral glucose in diabetes mellitus. Diabetes. 1977;26:525–529. doi: 10.2337/diab.26.6.525. [DOI] [PubMed] [Google Scholar]
- Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol. 2011;232:280–289. doi: 10.1016/j.expneurol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambamurti K, Greig NH, Lahiri DK. Advances in the cellular and molecular biology of the beta-amyloid protein in Alzheimer's disease. Neuromolecular Med. 2002;1:1–31. doi: 10.1385/NMM:1:1:1. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Suram A, Venugopal C, Prakasam A, Zhou Y, Lahiri DK, et al. A partial failure of membrane protein turnover may cause Alzheimer's disease: a new hypothesis. Curr Alzheimer Res. 2006;3:81–90. doi: 10.2174/156720506775697142. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease-is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Teramoto S, Miyamoto N, Yatomi K, Tanaka Y, Oishi H, Arai H, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2011;31:1696–1705. doi: 10.1038/jcbfm.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilsbøll T, Agersø H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab. 2003;88:220–224. doi: 10.1210/jc.2002-021053. [DOI] [PubMed] [Google Scholar]
- Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ. 2012;344:d7771. doi: 10.1136/bmj.d7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Li L, Hölscher C, Pan YF, Chen XR, Qi JS. Val8-glucagon-like peptide-1 protects against Aβ1-40-induced impairment of hippocampal late-phase long-term potentiation and spatial learning in rats. Neuroscience. 2010;170:1239–1248. doi: 10.1016/j.neuroscience.2010.08.028. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Matsui T. Pleiotropic Effects of Glucagon-like Peptide-1 (GLP-1)-Based Therapies on Vascular Complications in Diabetes. Curr Pharm Des. 2011;17:4379–4385. doi: 10.2174/138161211798999456. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kishi T, Lee CE, Choi BJ, Fang H, Hollenberg AN, et al. Glucagon-like peptide-1–responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J Neurosci. 2003;23:2939–2946. doi: 10.1523/JNEUROSCI.23-07-02939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Packard BA, Tauchi M, D'Alessio DA, Herman JP. Glucocorticoid regulation of preproglucagon transcription and RNA stability during stress. Proc Natl Acad Sci USA. 2009a;106:5913–5918. doi: 10.1073/pnas.0808716106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang Q, Zhang J, Lei X, Xu GT, Ye W. Protection of exendin-4 analogue in early experimental diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2009b;247:699–706. doi: 10.1007/s00417-008-1004-3. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang H, Xu L, Ma K, Wallrapp C, Jonas JB. Neuroprotective effect of intravitreal cell-based glucagon-like peptide-1 production in the optic nerve crush model. Acta Ophthalmol. 2011a;89:e320–e326. doi: 10.1111/j.1755-3768.2010.02044.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Wang Q, Lei X, Chu Q, Xu GT, et al. Intravitreal injection of exendin-4 analogue protects retinal cells in early diabetic rats. Invest Ophthalmol Vis Sci. 2011b;52:278–285. doi: 10.1167/iovs.09-4727. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]