

Abstract
BACKGROUND AND PURPOSE
Flupirtine is a non-opioid analgesic that has been in clinical use for more than 20 years. It is characterized as a selective neuronal potassium channel opener (SNEPCO). Nevertheless, its mechanisms of action remain controversial and are the purpose of this study.
EXPERIMENTAL APPROACH
Effects of flupirtine on native and recombinant voltage- and ligand-gated ion channels were explored in patch-clamp experiments using the following experimental systems: recombinant KIR3 and KV7 channels and α3β4 nicotinic acetylcholine receptors expressed in tsA 201 cells; native voltage-gated Na+, Ca2+, inward rectifier K+, KV7 K+, and TRPV1 channels, as well as GABAA, glycine, and ionotropic glutamate receptors expressed in rat dorsal root ganglion, dorsal horn and hippocampal neurons.
KEY RESULTS
Therapeutic flupirtine concentrations (≤10 µM) did not affect voltage-gated Na+ or Ca2+ channels, inward rectifier K+ channels, nicotinic acetylcholine receptors, glycine or ionotropic glutamate receptors. Flupirtine shifted the gating of KV7 K+ channels to more negative potentials and the gating of GABAA receptors to lower GABA concentrations. These latter effects were more pronounced in dorsal root ganglion and dorsal horn neurons than in hippocampal neurons. In dorsal root ganglion and dorsal horn neurons, the facilitatory effect of therapeutic flupirtine concentrations on KV7 channels and GABAA receptors was comparable, whereas in hippocampal neurons the effects on KV7 channels were more pronounced.
CONCLUSIONS AND IMPLICATIONS
These results indicate that flupirtine exerts its analgesic action by acting on both GABAA receptors and KV7 channels.
Keywords: flupirtine, analgesic, KIR channels, KV7 channels, GABAA receptors
Introduction
Chronic pain affects up to 50% of the general population and thus constitutes a major health problem (Elliott et al., 1999). Therefore, a large number of analgesic drugs with defined mechanisms of action have been developed. These are opioids acting at opioid receptors, non-steroidal anti-inflammatory drugs that inhibit COX, antiepileptic drugs and local anaesthetics that inhibit voltage-gated Na+ and Ca2+ channels, and cannabinoids acting at homonymous receptors. Nevertheless, due to either lack of efficacy or untoward effects of these families of drugs, there is still an unmet need for analgesics with alternative mechanisms of actions (Guindon et al., 2007).
One such analgesic with an atypical mechanism of action is flupirtine. This triaminopyridine derivative exerts analgesic effects in acute and chronic pain states in animal models as well as humans (Friedel and Fitton, 1993); its pain-relieving properties are comparable with or even superior to those of dihydrocodeine, pentazocine, tramadol or diclofenac (Moore et al., 1983; Galasko et al., 1985; Scheef, 1987; Mastronardi et al., 1988; Luben et al., 1994). Nevertheless, its underlying mechanisms of action remain controversial, if not elusive (Devulder, 2010). In fact, flupirtine has been reported to bind to adrenoceptors, 5-HT, opioid, NMDA and benzodiazepine receptors (Kornhuber et al., 1999a). However, these effects required flupirtine concentrations >10 µM [for a summary see (Kornhuber et al., 1999a)], although therapeutic plasma concentrations are in the range of up to 5 µM only (Hummel et al., 1991; Devulder, 2010). Later on, flupirtine was found to activate G protein-gated inward rectifier K+ (KIR3) channels in hippocampal neurons with half maximal effects at concentrations ≤1 µM (Jakob and Krieglstein, 1997). As a consequence, flupirtine was introduced as a SNEPCO, standing for selective neuronal potassium channel opener (Kornhuber et al., 1999b). More recently, flupirtine was demonstrated to activate recombinant KV7.2 K+ channels (Martire et al., 2004) and native KV7 channels in nodose ganglion neurons with half maximal effects at about 10 µM (Wladyka and Kunze, 2006).
This study was initiated to elucidate the mechanisms of action that underlie the analgesic effects of flupirtine. The investigations first focus on KIR3 and KV7 channels, as flupirtine has been reported to activate these channels (Jakob and Krieglstein, 1997; Wladyka and Kunze, 2006), and activation of either of these two types of channels is known to mediate analgesic effects (Mitrovic et al., 2003; Passmore et al., 2003). Other obvious targets to be investigated were TRPV1 channels as well as voltage-gated Na+ and Ca2+ channels, as binding at these channels contributes to the analgesic effects of capsaicin, local anaesthetics and ziconotid, respectively (Basbaum et al., 2009). Finally, our experiments also included the ionotropic receptors for glutamate (Miller et al., 2011), GABA (Zeilhofer et al., 2009) and glycine (Xiong et al., 2011), as these ion channels are known to mediate the analgesic effects of a variety of drugs. As GABAA receptors and KV7 channels turned out to be the targets with the highest sensitivity towards flupirtine, their modulation was assessed in more detail using rat dorsal root ganglion and dorsal horn neurons. Effects observed in these neurons of the pain pathways were compared with those observed in hippocampal neurons. The results indicate that the modulation of both, KV7 channels and GABAA receptors, contributes to the analgesic effects of flupirtine.
Methods
Primary cultures of native neurons
Primary cell cultures were prepared as previously described for hippocampal (Boehm and Betz, 1997) and superior cervical (Scholze et al., 2002) as well as for dorsal root ganglion (Yousuf et al., 2011) neurons. Tissue was obtained from Sprague-Dawley rats that had been killed by decapitation in full accordance with all rules of the Austrian animal protection law (see http://www.ris.bka.gv.at/GeltendeFassung.wxe?Abfrage=Bundesnormen&Gesetzesnummer=20003541) and the Austrian animal experiment by-laws (see http://www.ris2.bka.gv.at/Dokumente/BgblPdf/2000_198_2/2000_198_2.pdf). Briefly, hippocampi and dorsal horns were dissected from neonatal rats, cut into small pieces and incubated in papain (1 mg mL−1 in L-15 Leibovitz Medium, supplemented with 1 mM kynurenic acid) for 30 min at 37°C. Subsequently, the tissue was dissociated by trituration in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal calf serum and 5 µg mL−1 insulin, 5 µg mL−1 transferrin, 5 ng mL−1 Na-selenite, 18 µg mL−1 putrescine, 10 nM progesterone, 2 mM MgCl2, 25 000 IU L−1 penicillin and 25 mg L−1 streptomycin. About 50 000 cells were seeded into microchambers created by glass rings (inner diameter 10 mm) placed in the centre of 35 mm culture dishes (Nunc, Roskilde, Denmark) coated with poly d-lysine (0.1 mg m−1) and containing 2 mL culture medium. After 2–3 h, the rings were removed and the next day, medium was changed to 3 mL penicillin- and streptomycin-free culture medium. After 3–5 days, 1 µM cytosine arabinoside was added to the culture medium to inhibit further proliferation of non-neuronal cells.
Dorsal root ganglia (DRG) were dissected from 10 to 12- day old rats and superior cervical ganglia (SCG) from 4 to 6 day old rats. In both cases, collected ganglia were incubated first in collagenase (1.5 mg m−1) and dispase (3.0 mg mL−1) for 30 min and thereafter in 0.25% trypsin for 15 min at 37°C. After trituration, cells were resuspended in DMEM containing 4.5 g L−1 glucose, 10 mg L−1 insulin, 25 000 IU L−1 penicillin and 25 mg L−1 streptomycin, 50 µg L−1 nerve growth factor and 5% heat-inactivated FCS. Finally, dissociated cells were seeded onto 35-mm culture dishes coated with rat tail collagen (Biomedical Technologies Inc., Stoughton, MA, USA). All cultures were kept in a humidified 5% CO2 atmosphere at 37°C. DRG neurons were used for electrophysiological experiments after 2–4 days in culture, SCG neurons after 4–7 days, whereas hippocampal and dorsal horn neurons were used after 10–15 days.
Cell lines and transfections
tsA 201 cells (a subclone of HEK 293 cells stably expressing the SV40 large T-antigen) were cultured in DMEM containing 1 g L−1 glucose and l-glutamine, supplemented with 10% FCS and 25 000 IU L−1 penicillin plus 25 mg L−1 streptomycin. For patch-clamp experiments, cells were plated in 35 mm culture dishes coated with poly-d-lysine. Cells were transfected with human concatenated KIR3.1/KIR3.2 (kindly provided by A. Karschin, Wuerzburg, Germany), human KV7.2 and KV7.3 (kindly provided by M.S. Shapiro, San Antonio, TX, USA), rat α3 and β4 subunits of nicotinic acetylcholine receptors (kindly provided by S. Huck, Vienna, Austria), and rat P2Y12 receptors (Schicker et al., 2009) using either ExGen 500 (Fermentas, St.Leon-Rot, Germany) or Lipofectamine 2000 (Invitrogen, Lofer, Austria) according to the manufacturers' recommendations. To identify successfully transfected cells, the pEYFP-N1 vector (Becton Dickinson, Heidelberg, Germany) was cotransfected, and patch-clamp recordings were carried out with fluorescent cells 24 to 48 h after transfection.
Electrophysiology
All currents were recorded at room temperature (20 to 24°C) using an Axopatch 200B amplifier and the pCLAMP 8.1 hard- and software (Molecular Devices, Sunnyvale, CA, USA). All DRG neurons included were characterized as capsaicin-sensitive (see Figure 3C). Currents through KV7 channels and ligand-gated ion channels were determined in the perforated patch-clamp mode, whereas voltage-activated Ca2+ and Na+ currents, as well as currents through KIR channels, were measured in the whole-cell configuration. Signals were low-pass filtered at 5 kHz, digitized at 10 to 50 kHz and stored on an IBM-compatible computer. Traces were analysed off-line by the Clampfit 8.2 program (Molecular Devices). Patch electrodes were pulled (Flaming-Brown puller, Sutter Instruments, Novato, CA, USA) from borosilicate glass capillaries (Science Products, Frankfurt/Main, Germany).
Figure 3.
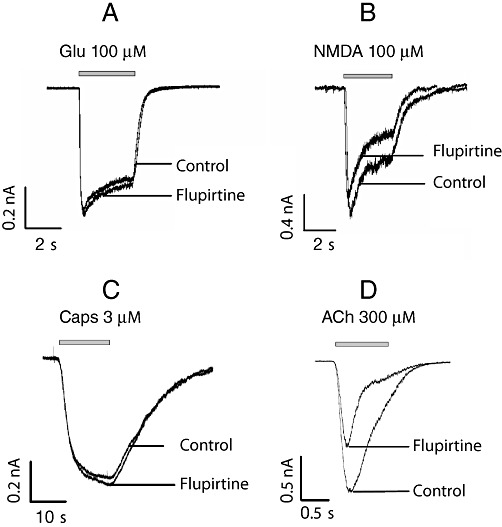
Effects of flupirtine on various ligand-gated ion channels. The indicated concentrations of (A) glutamate (Glu) or (B) NMDA were applied to hippocampal neurons. (C) Capsaicin was applied to DRG neurons. (D) Rat α3 and β4 subunits of nicotinic acetylcholine receptors were expressed in tsA cells, and the indicated concentration of ACh was applied. All cells were clamped at a voltage of −70 mV using the perforated patch-clamp technique. The representative current traces shown were obtained in the presence of either solvent (control) or 30 µM flupirtine. A summary of the effects of flupirtine is given in Table 2.
The internal solution for perforated patch recordings contained (mM): KCl (140), CaCl22H2O (1.6), EGTA (10) and HEPES (10), adjusted to pH 7.3 with KOH, for currents through ligand-gated ion channels and K2SO4 (75), KCl (55), MgCl2 (8) and HEPES (10), adjusted to pH 7.3 with KOH, for recordings of currents through KV7 channels. The recording electrodes were first front-filled with the internal solution and then back-filled with the same solution containing 200 µg mL−1 amphotericin B (in 0.8% dimethylsulfoxide). For whole-cell recordings of currents through KIR channels, the pipette solution contained (mM): KCl (140), CaCl22H2O (1.6), EGTA (10), Mg-ATP (2), Na-GTP (2) and HEPES (10), adjusted to pH 7.3 with KOH, whereas for Ca2+ and Na+ currents, the pipette solution consisted of (mM): CsCl (130), tetraethylammonium chloride (20), CaCl2 (0.24), glucose (10), HEPES (10), EGTA (5), Mg-ATP (2) and Na-GTP (2), adjusted to pH 7.3 with CsOH.
The external (bathing) solution for perforated patch routinely contained (mM): NaCl (140), KCl (3.0), CaCl2 (3.0), MgCl2 (2.0), glucose (20), HEPES (10), adjusted to pH 7.4 with NaOH. For the recordings of currents through KV7 channels and ligand-gated ion channels, TTX (0.5 µM) was included to suppress voltage-activated Na+ currents. In addition, CNQX (10 µM) was used for GABA- and glycine-evoked currents to block ionotropic glutamate receptors, whereas bicuculline methiodide (30 µM) was added when glutamate-evoked currents were measured in order to block GABAA receptors (Boehm and Betz, 1997). For NMDA-evoked currents, the solution lacked MgCl2, but contained glycine (10 µM), strychnine (0.1 µM) and NBQX (10 µM) to facilitate the gating of NMDA receptors, but block inhibitory glycine receptors (Boehm and Betz, 1997). For whole cell recordings of Ca2+ currents, the external solution contained (mM): NaCl (120), tetraethylammonium chloride (20), KCl (3.0), CaCl2 (5.0), MgCl2 (2.0), TTX (0.0005), glucose (20) and HEPES (10), adjusted to pH 7.4 with KOH. For Na+ currents, the same solution excluding TTX, but including CdCl2 (0.1) was used. For currents through KIR channels, the external solution contained (mM): NaCl (23), KCl (120), CaCl2 (2.0), MgCl2 (2.0), TTX (0.0005), glucose (20) and HEPES (10) (adjusted to pH 7.4 with KOH); for currents through KIR channels in hippocampal neurons, CNQX (10 µM) and bicuculline methiodide (30 µM) were added to block ionotrpic glutamate and GABAA receptors, respectively. Tip resistances were between 1.5 and 3.5 MΩ. The resulting liquid junction potentials were below ±10 mV and were not taken into further consideration during experimentation or data evaluation. Drugs were applied via a DAD-12 drug application device (Adams & List, Westbury, NY, USA), which permits a complete exchange of solutions surrounding the cells being investigated within less than 100 ms (Boehm and Betz, 1997).
Currents through ligand-gated ion channels were elicited by application of the agonists for one to five s to neurons clamped at −70 mV and were quantified by determining peak current amplitudes. Unless indicated otherwise, non inactivating currents through KV7 channels were elicited by depolarizing cells to −30 mV; once every 10 s, cells were hyperpolarized to −55 mV for 1 s periods to let the channels close; the de-activation current observed during these 1 s hyperpolarizations is specific for the KV7 channels (Boehm, 1998); currents were quantified by measuring amplitudes observed at −30 mV. Ca2+ and Na+ currents were elicited by depolarizations from a holding potential of −80 to +10 mV at a frequency of 4 min-1; leakage currents were corrected for by an online leak subtraction protocol that applies 4 hyperpolarizing pulses before the depolarization to +10 mV. Currents were quantified through peak current amplitudes. Currents through KIR channels were activated by voltage ramps from −140 to +40 mV; for quantification, current amplitudes at −140 mV were determined. In hippocampal neurons, currents through KIR channels were evoked by 200 ms hyperpolarizations from a holding potential of −70 mV to potentials ranging from −80 to −140 mV with 10 mV increments as published previously (Jakob and Krieglstein, 1997).
Calculations and statistics
To evaluate the effects of flupirtine on various types of currents, chosen drug concentrations and the appropriate solvent were applied alternately; current amplitudes obtained in the presence of either flupirtine or solvent were then normalized to the arithmetic mean of control current amplitudes recorded before and after the application of both, drug and solvent (normalized current amplitudes). For statistical purposes, amplitudes in the presence of flupirtine were compared with those in the presence of solvent. Unless indicated otherwise, significances of differences were evaluated using a Kruskal–Wallis ANOVA followed by a Dunn's multiple comparison test; for pair wise comparisons, a Mann-Whitney test or a paired Wilcoxon signed rank test were used instead.
For concentration-response curves of GABA-induced currents in the presence of solvent or flupirtine, current amplitudes evoked by different GABA concentrations in solvent or flupirtine were normalized to those evoked by 30 µM GABA in solvent in the very same neuron.
Materials
Flupirtine, GABA, bicuculline methiodide (BMI), kynurenic acid, cyano-2,3-dihydroxy-7-nitroquinoxaline (CNQX), ADP, glutamate, glycine, NMDA, acetylcholine, capsaicin, strychnine, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), putrescine, progesterone, poly-D-lysine, cytosine arabinoside, amphotericin B, Mg-ATP, Na-GTP, as well as the bulk chemicals were obtained from Sigma-Aldrich (Vienna, Austria). Insulin, transferrin, Na-selenite and dispase were obtained from Roche (Mannheim, Germany); Dulbecco's modified Eagle's medium (DMEM), Leibovitz L-15 medium, penicillin, streptomycin and l-glutamine were purchased from PAA Laboratories (Pasching, Austria); papain and trypsin were bought from Worthington (Lakewood, NJ, USA); nerve growth factor from R&D Systems Inc. (Minneapolis, MN, USA); heat-inactivated fetal calf serum was obtained from Invitrogen (Lofer, Austria) and tetrodotoxin (TTX) from Latoxan (Rosans, France).
Nomenclature
The drug/molecular target nomenclature conforms with BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Results
Flupirtine does not affect currents through KIR3 channels
Currents through G protein-gated inward rectifier K+ (KIR3) channels in rat hippocampal neurons have been reported to be augmented in the presence of flupirtine at submicromolar to low micromolar concentrations (Jakob and Krieglstein, 1997). Such currents are mainly carried by KIR3.1 and 3.2 subunits (Koyrakh et al., 2005). Therefore, we tested the effects of flupirtine on currents in tsA cells expressing concatenated pairs of KIR3.1/3.2 subunits (O'Connor et al., 1999). However, currents evoked by voltage ramps from −140 to +40 mV were not different whether obtained in the presence of either 30 µM flupirtine or solvent (Figure 1A and B). To be able to further activate these channels, Gi coupled rat P2Y12 receptors (Schicker et al., 2009) were co-expressed together with these concatemers; activation of these receptors by 100 µM ADP significantly enhanced the currents evoked by the ramp depolarizations, but flupirtine still failed to cause significant alterations when compared with solvent (Figure 1A and B). Nevertheless, these currents were largely reduced by either 150 nM tertiapin-Q or 1 mM Ba2+ (not shown), both being blockers of KIR3 channels (Kubo et al., 2005).
Figure 1.
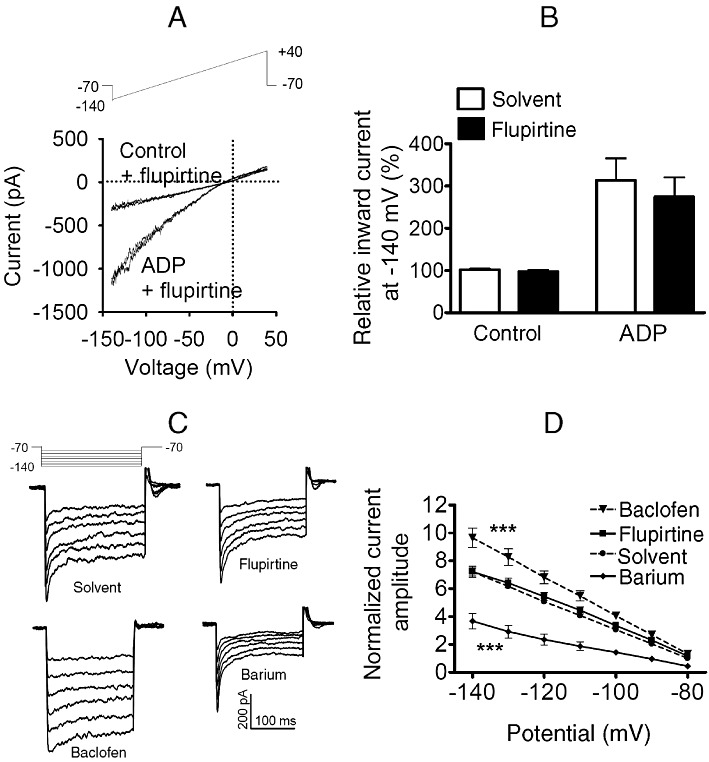
Effects of flupirtine on recombinant and native G-protein coupled inwardly rectifying K channels. (A, B) KIR3.1/3.2 concatemers and P2Y12 receptors were co-expressed in tsA cells. Currents were evoked by ramp depolarizations from −140 to 40 mV during periods of 200 ms. (A) Representative traces of recordings in the absence (control) or presence of ADP 100 µM, either with solvent or with 30 µM flupirtine. (B) Amplitudes of inward currents were determined at −140 mV; normalized amplitudes of currents obtained either in solvent or in 30 µM flupirtine, either alone (control) or together with 100 µM ADP, are shown (n = 7). (C, D) Currents were evoked in hippocampal neurons by 200 ms hyperpolarizations from a holding potential of −70 mV to potentials ranging from −80 to −140 mV with 10 mV increments as published previously (Jakob and Krieglstein, 1997). This pulse protocol was applied in the presence of either solvent or 100 µM baclofen, 10 µM flupirtine and 1 mM BaCl2, respectively. (C) Representative original traces. (D) Summary of results obtained in nine different neurons; for each neuron, all amplitude values were normalized to the amplitude determined in the presence of solvent at a potential of −80 mV. *** indicates statistically significant differences versus currents in the presence of solvent at P < 0.001 as determined by a two way ANOVA.
To obtain data directly comparable with those of Jakob and Krieglstein, currents through KIR3 channels in hippocampal neurons were determined by the previously published protocol (Jakob and Krieglstein, 1997). These currents were attenuated by 1 mM Ba2+ and enhanced in the presence of 100 µM of the GABAB receptor agonist baclofen (Figure 1C and D). This current enhancement by baclofen was reduced by the GABAB antagonist CGP35348 (100 µM) (Olpe et al., 1990) by 47.1 ± 0.8% (n = 8; P < 0.01; Wilcoxon signed rank test). In the presence of 10 µM flupirtine, however, these currents remained unchanged (Figure 1C and D).
Flupirtine enhances currents through KV7 channels
Sensory neurons predominantly express KV7.2 and KV7.3 subunits and only small amounts of KV7.5 (Rose et al., 2011). Therefore, KV7.2 and KV7.3 were co-expressed in tsA cells, and currents were elicited by slow voltage ramps from −100 to −20 mV. The resulting outward rectifying currents were shifted to more negative potentials in a concentration-dependent manner in the presence of either 3 or 30 µM flupirtine (Figure 2A). To obtain full concentration–response curves, non-inactivating outward currents were measured at –30 mV, and their enhancement in the presence of 0.1 to 100 µM flupirtine was determined. Flupirtine augmented outward current amplitudes by up to 70.9 ± 8.8% with half maximal effects at 4.6 ± 1.2 µM (Figure 2B). Virtually identical results were obtained with currents through KV7 channels endogenously expressed in rat superior cervical ganglion (SCG) neurons: 30 µM flupirtine shifted the current-voltage curve by about 20 mV in a hyperpolarizing direction, and currents at −30 mV were enhanced by up to 70.9 ± 6.8% with half maximal effects occurring at 4.6 ± 3.9 µM flupirtine (n = 6).
Figure 2.
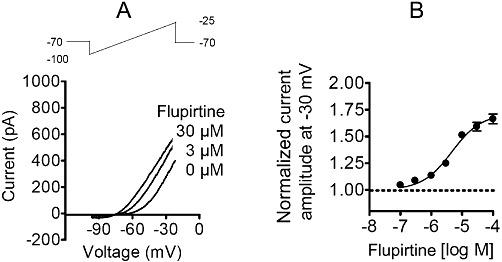
Effects of flupirtine on heterologously expressed KV7 channels. Human KV7.2 and 7.3 were co-expressed in tsA cells. Currents were evoked by ramp hyperpolarizations from −25 to −100 mV during periods of 1 s. (A) Representative current traces in the presence of solvent and 3 or 30 µM flupirtine, respectively. (B) Current amplitudes were determined at −30 mV in the presence of solvent or increasing concentrations of flupirtine; the average of normalized current amplitudes is shown for seven cells.
Voltage-activated Na+ and Ca2+ channels are blocked by flupirtine at concentrations >10 µM
Blockade of voltage-activated Na+ and Ca2+ channels is also a pharmacotherapeutic principle in analgesic therapy (Basbaum et al., 2009). In order to assess the effects of flupirtine on such channels expressed in the pain pathways, Na+ and Ca2+ currents were elicited in rat DRG neurons by depolarizations from −80 to +10 mV in the presence of 10 or 30 µM flupirtine. For comparison with results obtained in non pain-related neurons, these experiments were repeated in hippocampal neurons. As can be seen from Table 1, 10 µM flupirtine failed to cause any significant changes, and 30 µM flupirtine led to significant reductions only on Na+ currents in DRG and the Ca2+ currents in hippocampal neurons.
Table 1.
Effects of flupirtine on voltage gated Na+ and Ca2+ channels
| 10 µM flupirtine | 30 µM flupirtine | ||||||
|---|---|---|---|---|---|---|---|
| Current | Neuron | Normalized amplitude | n | P | Normalized amplitude | n | P |
| INa | DRG | 0.95 ± 0.05 | 5 | >0.05 | 0.77 ± 0.04 | 5 | <0.05 |
| HC | 1.00 ± 0.03 | 4 | >0.05 | 0.99 ± 0.03 | 4 | >0.05 | |
| ICa | DRG | 0.84 ± 0.05 | 7 | >0.05 | 0.78 ± 0.08 | 7 | >0.05 |
| HC | 0.98 ± 0.02 | 6 | >0.05 | 0.72 ± 0.11 | 6 | <0.05 | |
Na+ and Ca2+ currents were elicited by depolarizations from −80 to +10 mV in either hippocampal or DRG neurons. Normalized peak current amplitudes in the presence of 10 or 30 µM flupirtine were compared with those in the presence of solvent (not shown). P values for significances of differences versus solvent were calculated by a Kruskal–Wallis ANOVA followed by Dunn's post hoc test.
GABAA receptors, but no other ligand-gated ion channels are modulated by low micromolar concentrations of flupirtine
The capsaicin-sensitive TRPV1 channel is activated by various noxious stimuli and leads to the excitation of nociceptors (Basbaum et al., 2009). When currents through TRPV1 channels in DRG neurons were activated by 0.3 or 3 µM capsaicin in the presence of either solvent or 10 or 30 µM flupirtine, there were no significant differences in the resulting peak current amplitudes (Figure 3C and Table 2).
Table 2.
Effects of flupirtine on TRPV1 channels, as well as ionotropic glutamate, GABAA, glycine and nicotinic acetylcholine receptors
| 10 µM flupirtine | 30 µM flupirtine | ||||||
|---|---|---|---|---|---|---|---|
| Agonist | Concentration | Normalized amplitude | n | P | Normalized amplitude | n | P |
| Capsaicin | 0.3 µM | 1.09 ± 0.05 | 5 | >0.05 | 0.98 ± 0.01 | 4 | >0.05 |
| 3 µM | 0.95 ± 0.06 | 5 | >0.05 | 1.03 ± 0.05 | 4 | >0.05 | |
| Glutamate | 3 µM | 0.98 ± 0.05 | 9 | >0.05 | 0.96 ± 0.07 | 9 | >0.05 |
| 100 µM | 1.05 ± 0.03 | 12 | >0.05 | 0.98 ± 0.04 | 12 | >0.05 | |
| NMDA | 10 µM | 0.89 ± 0.03 | 10 | >0.05 | 0.82 ± 0.03 | 10 | <0.05 |
| 100 µM | 0.87 ± 0.03 | 10 | >0.05 | 0.82 ± 0.04 | 10 | <0.01 | |
| GABA | 3 µM | 1.29 ± 0.09 | 6 | <0.05 | 1.45 ± 0.13 | 7 | <0.05 |
| 100 µM | 1.00 ± 0.04 | 8 | >0.05 | 0.98 ± 0.03 | 8 | >0.05 | |
| Glycine | 30 µM | 1.02 ± 0.05 | 5 | >0.05 | 1.04 ± 0.06 | 5 | >0.05 |
| 300 µM | 1.03 ± 0.02 | 5 | >0.05 | 0.97 ± 0.03 | 5 | >0.05 | |
| ACh | 10 µM | 0.88 ± 0.05 | 5 | >0.05 | 0.54 ± 0.12 | 5 | <0.01 |
| 300 µM | 0.98 ± 0.01 | 7 | >0.05 | 0.42 ± 0.04 | 7 | <0.001 | |
Currents were evoked by application of the indicated agonist concentrations in hippocampal neurons (glutamate, NMDA, GABA, glycine), DRG neurons (capsaicin), or in tsA cells expressing α3β4 subunits of nicotinic acetylcholine receptors (ACh). Normalized peak current amplitudes in the presence of 10 or 30 µM flupirtine were compared with those in the presence of solvent (not shown). P values for significances of differences versus solvent were calculated by a Kruskal–Wallis ANOVA followed by Dunn's post hoc test.
Among the transmitter-gated ion channels, ionotropic glutamate, GABAA and glycine receptors are of utmost importance in pain perception (Basbaum et al., 2009). Therefore, the effects of flupirtine on these receptors were investigated using primary rat hippocampal neurons that express all these receptors. To be able to identify potential effects on either partially or fully activated receptors, just suprathreshold and near saturating concentrations of agonists were applied as follows: 3 and 100 µM glutamate (in the presence of 2 mM Mg2+ to block NMDA receptors) to activate non-NMDA glutamate receptors, 10 and 100 µM NMDA in the continuous presence of 10 µM glycine to activate NMDA receptors, 3 and 100 µM GABA to activate GABAA receptors, and 30 and 300 µM glycine to activate inhibitory glycine receptors. These agonist concentrations were applied either in solvent or together with 10 or 30 µM flupirtine. For glutamate (Figure 3A) and glycine (Figure 4C), current amplitudes determined in the presence of either flupirtine concentration were not different from those in solvent (Table 2). However, NMDA-induced current amplitudes (Figure 3B), irrespective of the agonist concentration used, were significantly reduced by 30 µM, but not by 10 µM, flupirtine as compared with solvent (Table 2). In contrast, amplitudes of currents evoked by 3 µM GABA (Figure 4A) were significantly enhanced by 10 as well as 30 µM flupirtine, whereas the currents evoked by 100 µM GABA were not affected (Table 2).
Figure 4.
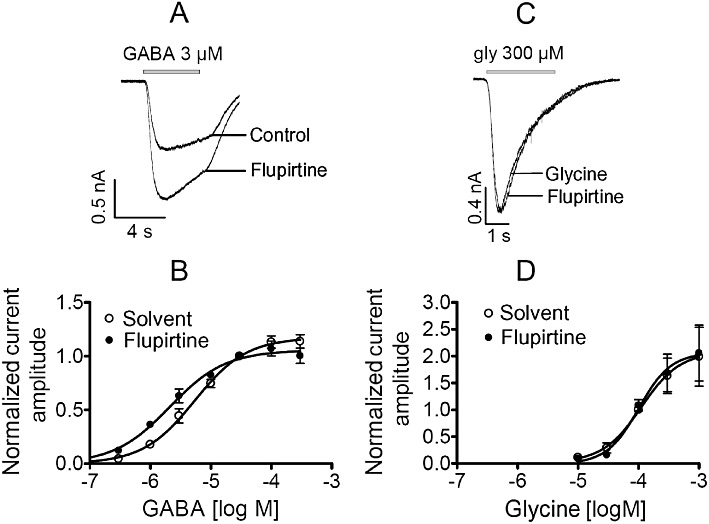
Effects of flupirtine on GABAA and glycine receptors in hippocampal neurons. Neurons were clamped at −70 mV and increasing concentrations of GABA or glycine were applied for 3 to 5 s in the presence of either solvent or 30 µM flupirtine. (A and C) Original traces evoked by 3 µM GABA and 300 µM glycine, respectively. (B and D) Concentration–response curves for currents induced by GABA (n = 5 to 11) and glycine (n = 5), respectively. Amplitudes evoked by different agonist concentrations in solvent or flupirtine were normalized to those evoked in solvent in the very same neuron by 30 µM GABA and 100 µM glycine, respectively. Values for statistical differences (F-test) between EC50 values in the presence of either solvent or flupirtine are P < 0.001 for GABA and P > 0.8 for glycine.
Peptides that block peripheral neuronal nicotinic acetylcholine receptors are known to alleviate neuropathic pain (Clark et al., 2006). Therefore, currents through recombinant nicotinic acetylcholine receptors composed of α3 and β4 subunits were triggered by 10 or 300 µM acetylcholine and were tested for their sensitivity towards flupirtine. At a concentration of 10 µM, flupirtine did not cause significant changes when compared with solvent, but 30 µM led to a significant reduction in peak current amplitudes (Table 2).
As flupirtine effects on GABAA receptors depended on the concentration of the receptor agonist used to induce currents, entire concentration–response curves for GABA-evoked currents were determined in hippocampal neurons. The GABA concentration required to achieve half-maximal activation was 5.4 ± 0.8 µM. In the presence of 30 µM flupirtine, this concentration–response curve was shifted to the left by a factor of 2.5 without a significant alteration in maximal currents (Figure 4A and B).
GABAA and glycine receptors of hippocampal neurons have been shown to interact with each other (Grassi, 1992), but flupirtine appeared to act on GABAA receptors only (Table 2). To confirm this selectivity, two types of experiments were performed: (i) full concentration–response curves were also obtained for glycine-induced currents in hippocampal neurons, again in the presence of either 30 µM flupirtine or solvent: in this case, flupirtine failed to cause any alteration (Figure 4C and D); (ii) approximate EC50 concentrations of GABA (5 µM) and glycine (100 µM) were applied in the absence and presence of the selective antagonists bicuculline methiodide and strychnine, respectively. Bicuculline methiodide (10 µM) reduced GABA-induced currents (normalized current amplitude 0.09 ± 0.01; n = 6; P < 0.05; Wilcoxon signed rank test), but left glycine-evoked currents unaltered (normalized current amplitude 1.07 ± 0.04; n = 6). Vice versa, strychnine (0.3 µM) reduced glycine-induced currents (normalized current amplitude 0.13 ± 0.04; n = 6; P < 0.05; Wilcoxon signed rank test), but left GABA-evoked currents unaltered (normalized current amplitude 1.02 ± 0.04; n = 6). All together, these data indicate that flupirtine acts on GABAA, but not on glycine, receptors in hippocampal neurons.
One additional indirect mechanism might contribute to the apparently direct effect of flupirtine on GABAA receptors: an action via GABAB receptors that might get activated as soon as GABA is applied to the neurons. To exclude this possibility, currents were evoked by low GABA concentrations (0.4 µM) in the presence of either the GABAB agonist baclofen (10 µM) or the GABAB antagonist CGP35348 (100 µM) (Olpe et al., 1990), which clearly attenuated the activation of KIR3 channels by 100 µM baclofen (see earlier discussion). However, amplitudes of currents evoked by 0.4 µM GABA in the presence of baclofen (normalized current amplitude 0.98 ± 0.04; n = 5) were not significantly different from those in the presence of CGP35348 (normalized current amplitude 0.97 ± 0.08; n = 5). Hence, GABA-evoked currents were not affected by concomitant GABAB receptor activation, and flupirtine thus must have acted independently of GABAB receptors.
Effects of flupirtine on KV7 channels in pain-related and non-related neurons
So far, flupirtine at therapeutic concentrations [i.e. 10 µM or below; see (Hummel et al., 1991)] had been found to affect KV7 channels and GABAA receptors. Therefore, our further experiments focused on these and used DRG, dorsal horn and hippocampal neurons to compare effects in neurons involved and not involved in pain sensation, respectively. To obtain an estimate as to how many KV7 channels contribute to outward currents in the near threshold voltage range, neurons were depolarized to −30 mV and shortly hyperpolarized to −55 mV to determine the de-activation of these channels; using this voltage protocol, 30 µM linopirdine, which blocks currents through KV7 channels in sensory neurons by more than 90% (Passmore et al., 2003), was used to quantify the contribution of these channels to the overall currents. The inhibition of standing outward currents at −30 mV by 30 µM linopirdine was similar for the three types of neurons and ranged between 30 and 60% (Figure 5A and B). In the presence of flupirtine, the outward currents seen at −30 mV were enhanced in a concentration-dependent manner, but this enhancement was more pronounced in hippocampal than in DRG or dorsal horn neurons (Figure 5A and C).
Figure 5.
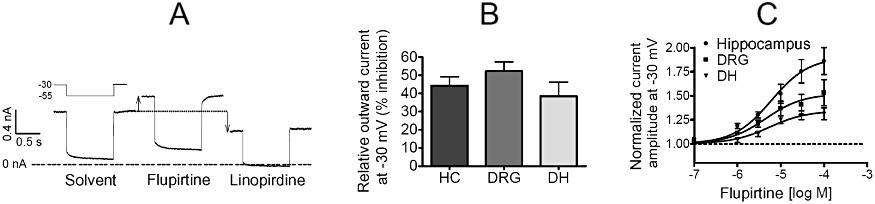
Comparison of the effects of flupirtine on KV7 channels in hippocampal, DRG and dorsal horn neurons. Cultured hippocampal (HC), DRG or dorsal horn (DH) neurons were clamped at −30 mV and hyperpolarized to −55 mV for 1 s periods every 10 s in order to de-activate KV7 channels. Drugs were present for at least 8 s before their effects on the currents were determined. (A) Representative traces measured in a DRG neuron in the presence of solvent, 30 µM flupirtine or 30 µM linopirdine. The shift in the outward current at −30 mV is indicated by the arrows. (B) Normalized amplitudes of outward currents at −30 mV were measured in the presence of solvent or 30 µM linopirdine in hippocampal (n = 5), DRG (n = 6) and dorsal horn (n = 4) neurons. (C) Normalized amplitudes of currents measured at −30 mV in the absence and presence of increasing concentrations of flupirtine were determined in hippocampal (n = 9), DRG (n = 10) and dorsal horn (n = 8) neurons, respectively. EC50 values were 6.1, 4.4 and 5.4 µM in hippocampal, DRG and dorsal horn neurons, respectively. The maximal effects observed in hippocampal neurons were significantly different from those in DRG or dorsal horn neurons at P < 0.01.
Effects of flupirtine on GABAA receptors in pain-related and non-related neurons
The data presented earlier indicate that the effects of flupirtine on KV7 channels are more pronounced in hippocampal and SCG neurons than in DRG or dorsal horn neurons. The effects of flupirtine on GABAA receptors in hippocampal neurons are shown in Figure 4. For DRG, dorsal horn and SCG neurons, such experiments were repeated, and the results are shown in Figure 6. The GABA concentration required to achieve half-maximal activation was much higher in dorsal horn (30.0 ± 11.4 µM) and DRG (32.5 ± 3.5 µM) neurons (Figure 6A and B) than in SCG (7.8 ± 0.6 µM; Figure 6c) or hippocampal (5.4 ± 0.8 µM; Figure 4B) neurons. In the presence of 30 µM flupirtine, the concentration–response curves obtained in each of these types of neurons were shifted to the left, but the extent of leftward shift was more pronounced in DRG (4.1-fold shift) and dorsal horn (3.7-fold shift) neurons (Figure 6A and B) than in SCG (2.5-fold shift; Figure 6C) or hippocampal (2.5-fold shift; Figure 4B) neurons. Furthermore, 30 µM flupirtine had marginal effects on maximal amplitudes in dorsal horn, SCG and hippocampal neurons, but clearly suppressed amplitudes of currents induced by high (≥100 µM) GABA concentrations in DRG neurons (Figure 6A). Hence, the effects of flupirtine on GABAA receptors were clearly different in these three types of neurons investigated. The results obtained in SCG neurons can further be used to exclude a possible contribution of GABAB receptors to the flupirtine effects observed, as these neurons are known not to express such receptors (Filippov et al., 2000).
Figure 6.
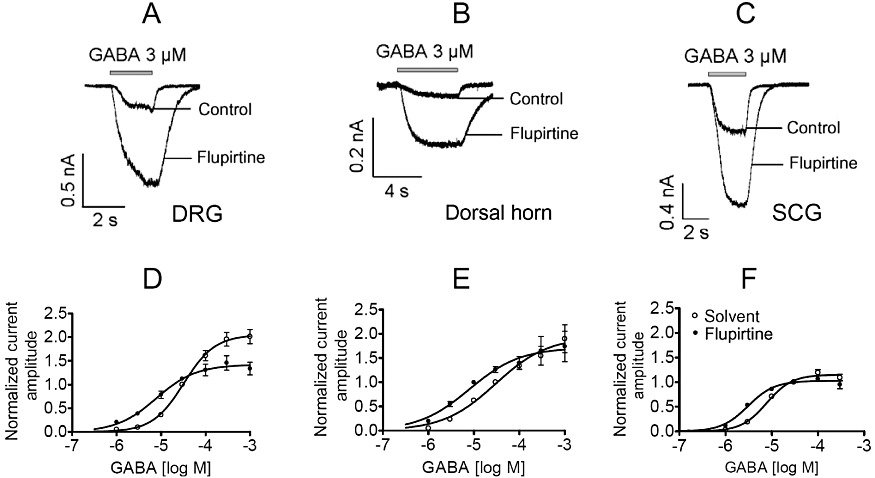
Comparison of the effects of flupirtine on GABAA receptors in DRG, dorsal horn and SCG neurons. Neurons were clamped at −70 mV and increasing GABA concentrations were applied for 3 to 5 s in the presence of either solvent or 30 µM flupirtine. (A–C) Original traces evoked by 3 µM GABA in DRG, dorsal horn and SCG neurons, respectively. (D–F) Concentration–response curves obtained in DRG (n = 5 to 15), dorsal horn (n = 6 to 16) and SCG (n = 5 to 13) neurons. Amplitudes evoked by different GABA concentrations in solvent or flupirtine were normalized to those evoked by 30 µM GABA in solvent in the very same neuron. Values for statistical differences (F test) between EC50 values in the presence of either solvent or flupirtine are P < 0.001 for DRG, P < 0.05 for dorsal horn and P < 0.001 for SCG neurons, respectively.
Concentration-dependence of the effects of flupirtine on GABAA receptors in pain-related and non-related neurons; comparison with the effects on KV7
To obtain information about the flupirtine concentrations required to enhance currents evoked by low GABA concentrations, concentration–response curves for flupirtine were constructed for each neuron type using GABA concentrations equivalent to the EC5 value: this was 0.4 µM in hippocampal, 1 µM in DRG and 3 µM in dorsal horn neurons (Figure 7). The results show that flupirtine acted more potently on GABA-induced currents in DRG neurons (half maximal effects at 22 ± 3 µM) than in dorsal horn (half maximal effects at 53 ± 10 µM; P < 0.01) or hippocampal (half maximal effects at 65 ± 20 µM; P < 0.01) neurons. Moreover, maximal enhancement of GABA-induced currents by flupirtine was more pronounced in dorsal horn (15-fold increase) than in DRG neurons (6-fold increase; P < 0.001), but was not significantly different from that in hippocampal neurons (7-fold increase; P > 0.05). With 30 µM flupirtine, the enhancement of GABA-induced currents in hippocampal neurons was significantly different from that observed in either DRG or dorsal horn neurons (P < 0.05).
Figure 7.
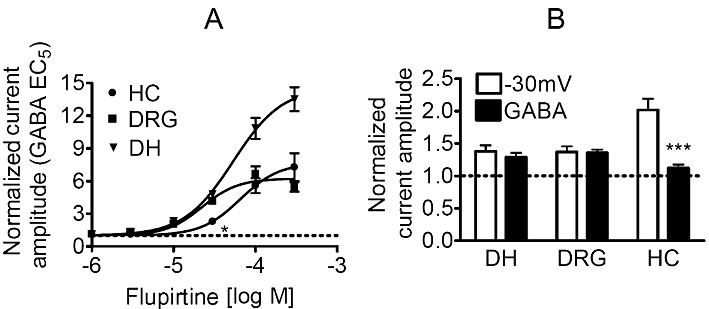
Comparison of the effects of therapeutic flupirtine concentrations in hippocampal, DRG and dorsal horn neurons. (A) The concentration-dependence of the effects of flupirtine on GABAA receptors was investigated in hippocampal (HC; n = 6), DRG (n = 6 to 8) and dorsal horn (n = 5 to 8) neurons clamped at −70 mV using GABA concentrations corresponding to the calculated EC5 values to induce currents; these EC5 values were 0.4, 3 and 1 µM for hippocampal (HC), DRG and dorsal horn (DH) neurons, respectively. * indicates a significant difference versus the corresponding result in DRG and dorsal horn neurons at P < 0.05. (B) Outward currents at −30 mV or currents induced by GABA at the above EC5 concentrations were determined in hippocampal (n = 6), DRG (n = 6 to 8) and dorsal horn (n = 4 to 5) neurons in the presence of either solvent or 3 µM flupirtine. The graph shows normalized current amplitudes in the presence of flupirtine; *** indicates a significant difference between the effects on the two types of currents as determined by a Kruskal–Wallis ANOVA followed by Dunn's multiple comparison test.
When comparing the effects of flupirtine on KV7 channels (Figure 4) with those on GABAA receptors (Figure 7A), it is obvious that flupirtine is more potent at enhancing currents through KV7 channels (half maximal effects: 5 µM vs. >20 µM), but more efficacious at augmenting GABA-induced currents (maximal increases: <twofold vs. >sixfold). To directly compare the effects of flupirtine at therapeutic concentrations, outward currents at −30 mV and currents induced by GABA EC5 concentrations were determined in the presence of either 3 µM flupirtine or solvent. In dorsal horn and DRG neurons, currents through KV7 channels and GABAA receptors were enhanced to about the same extent. In hippocampal neurons, in contrast, the enhancement of KV7 currents was much more pronounced than the enhancement of GABA-induced currents (Figure 7B). This indicates that therapeutic concentrations of flupirtine facilitate the gating of KV7 channels and GABAA receptors involved in pain sensation to a similar extent.
Discussion
Flupirtine has been used as an analgesic for more than 20 years and is marketed as SNEPCO [Selective NEuronal Potassium Channel Opener (Kornhuber et al., 1999b)]. This characterization reflects the fact that this drug has been suggested to activate KIR3 channels (Jakob and Krieglstein, 1997). However, later on, flupirtine was shown to facilitate the gating of KV7 channels (Wladyka and Kunze, 2006), which further supports its characterization as SNEPCO. The present results confirm its action on KV7 channels, but demonstrate that currents through KIR3 channels are not affected, and reveal, for the first time, that GABAA receptors are modulated by flupirtine in a neuron-specific manner.
A facilitating effect of flupirtine on inward rectifier K+ currents had been observed in primary cultures of rat hippocampal neurons (Jakob and Krieglstein, 1997). Using the same preparation, inward currents in response to hyperpolarizing voltage steps (from −80 to −140 mV) were enhanced by the activation of GABAB receptors and reduced by the KIR3 channel blocker Ba2+, but not altered by 30 µM flupirtine. The reason for this discrepancy is unfortunately not known, but is most likely to be explained by differences in the culture conditions or by differences between the two rat strains that have been used. In the previous experiments, the effects of flupirtine were abolished by pertussis toxin, thus suggesting an involvement of a receptor coupled to inhibitory G proteins (Jakob and Krieglstein, 1997). Thus, differences in the pattern and levels of receptor expression could offer an explanation for the discrepancies observed, but were not further investigated here.
In hippocampal neurons, KIR3.1 and 3.2 channel subunits provide the majority of G protein gated inwardly rectifying K+ currents (Koyrakh et al., 2005). Therefore, we performed experiments on concatemers of these subunits expressed in tsA cells, and currents through the resulting channels were determined. These currents were enhanced by the activation of co-expressed Gi coupled P2Y12 receptors and inhibited by KIR3 channel blockers, such as Ba2+, but they were not altered by 30 µM flupirtine. Thus, G protein gated inward rectifier K+ channels are unlikely to be direct targets of flupirtine.
Another K+ channel family that has been proposed to be opened by flupirtine is the KV7 family: the voltage-dependence of recombinant KV7.2 channels (Martire et al., 2004) as well as of native KV7 channels in nodose ganglion neurons (Wladyka and Kunze, 2006) was found to be shifted in a hyperpolarizing direction. This was confirmed here using KV7.2/KV7.3 heteromers expressed in tsA cells and KV7 channels endogenously expressed in SCG, DRG, dorsal horn and hippocampal neurons of the rat; in all these systems, the effects of flupirtine were half maximal at about 5 µM, and outward currents determined at −30 mV were enhanced by up to 90%. The maximal potentiation of currents through KV7 channels by high flupirtine concentrations was more pronounced in hippocampal and SCG than in DRG or dorsal horn neurons.
For comparison, the effects of flupirtine on other voltage gated ion channels, in particular on Na+ and Ca2+ channels, were also tested in DRG and hippocampal neurons. The accompanying currents were not affected by flupirtine at concentrations up to 10 µM and were reduced in amplitude by less than 30% at 30 µM. Thus, therapeutic flupirtine concentrations [≤5 µM; (Hummel et al., 1991)] do not affect voltage gated Na+ or Ca2+ channels.
The previously reported inhibition of NMDA receptors by flupirtine (Kornhuber et al., 1999a) prompted us to investigate its effects on ligand gated ion channels in hippocampal neurons. In fact, NMDA receptors were blocked by 30 µM, but not by 10 µM flupirtine. Similar results were obtained for recombinant α3β4 nicotinic acetylcholine receptors. In contrast, non-NMDA ionotropic glutamate receptors, inhibitory glycine receptors, and TRPV1 channels were insensitive to flupirtine at both concentrations.
Unexpectedly, flupirtine did modulate currents through GABAA receptors by increasing the potency of GABA to induce currents without enhancing maximal current amplitudes. Although such an effect has never been reported for flupirtine itself, another KV7 channel opener with a highly similar structure, retigabine, has been found to potentiate GABA- induced currents (Rundfeldt and Netzer, 2000) as well as inhibitory synaptic currents (Otto et al., 2002) in cortical and hippocampal neurons, respectively. However, retigabine is not used as analgesic, but has been marketed recently as adjunctive treatment for partial-onset seizures (Stafstrom et al., 2011). When the effect of flupirtine on GABAA receptors was compared for DRG, dorsal horn, SCG and hippocampal neurons, it became obvious that the action was tissue-specific with respect to the shift of the concentration-response curves for GABA and to the changes in maximum current amplitudes. Moreover, at half maximal flupirtine concentrations the potentiation of GABA- induced currents was significantly greater in DRG and dorsal horn than in hippocampal neurons.
Reasoning that flupirtine acts preferentially on GABAA receptors in DRG and dorsal horn neurons, leads to the question as to which GABAA receptors could mediate analgesia. Benzodiazepines have been shown to cause analgesia via GABAA receptors located within the spinal cord and containing α2 or α3 in addition to γ2 subunits; the analgesic effects of gaboxadol require the presence of α4 and β3 subunits (Zeilhofer et al., 2009). Recent experiments suggest that the GABAA receptor α2 subunits in the spinal cord that mediate analgesia are located on the presynaptic nerve endings of DRG neurons rather than on postsynaptic dorsal horn neurons (Witschi et al., 2011). Nevertheless, whether flupirtine acts preferentially or exclusively via GABAA receptors containing certain combinations of subunits and being expressed on DRG or dorsal horn neurons remains open for future investigations.
Considering that flupirtine concentrations of 10 µM affected only KV7 channels and GABAA receptors, but no other ligand- or voltage-gated ion channel, the question remains as to whether both of these effects may contribute to the analgesic action. In fact, an enhancement in the activities of both, KV7 channels (Passmore et al., 2003) and GABAA receptors (Zeilhofer et al., 2009), is known to provide analgesic effects. However, therapeutic flupirtine plasma concentrations are in the range of about 5 µM only (Hummel et al., 1991; Devulder, 2010). In DRG and dorsal horn neurons, 3 µM flupirtine enhanced currents through KV7 channels and GABAA receptors to a similar extent; in hippocampal neurons, for comparison, the effects on KV7 channels were much more pronounced than those on GABAA receptors. Thus, in neurons involved in pain perception, KV7 channels and GABAA receptors appear to be facilitated by flupirtine to a similar extent, whereas in other regions of the CNS it is rather the action on KV7 channels that predominates. Notably, another analgesic employed in various pain states, meclofenamic acid, has also been reported to facilitate the gating of KV7 channels (Peretz et al., 2005) as well as GABAA receptors (Smith et al., 2004). Although this drug is known as a COX inhibitor, therapeutic plasma concentrations in the range of 10 µM (Conroy et al., 1991) are sufficient to mediate these ion channel effects. Hence, the concomitant potentiation of KV7 channels and GABAA receptors might be a more general mechanism of action for analgesic drugs.
In conclusion, the present results reveal GABAA receptors as novel sites of action for the non-opioid analgesic flupirtine. Thus, flupirtine is not a SNEPCO as inferred up to now, but may instead exert its analgesic actions by combining two therapeutic principles, the facilitation of both GABAA receptors and KV7 channels. As this combined action on two independent ion channels is also shared by at least one other analgesic, meclofenamic acid, it may well turn out to be a promising principle in the pharmacotherapy of pain.
Acknowledgments
This study was supported by grants P19710, P23658 and P23670 from the Austrian Science Funds (FWF); FK, GKC and IS are members of the doctoral programme CCHD supported by the FWF (W1205), the Medical University of Vienna and the Austrian Academy of Sciences. The perfect technical assistance of Gabi Gaupmann is gratefully acknowledged.
Glossary
- BMI
bicuculline methiodide
- CNQX
cyano-2,3-dihydroxy-7-nitroquinoxaline
- DRG
dorsal root ganglion
- NBQX
2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
- SCG
superior cervical ganglion
- SNEPCO
selective neuronal potassium channel opener
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th Edition) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. Br J Pharmacol. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. J Neurosci. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RJ, Fischer H, Nevin ST, Adams DJ, Craik DJ. The synthesis, structural characterization, and receptor specificity of the alpha-conotoxin Vc1.1. J Biol Chem. 2006;281:23254–23263. doi: 10.1074/jbc.M604550200. [DOI] [PubMed] [Google Scholar]
- Conroy MC, Randinitis EJ, Turner JL. Pharmacology, pharmacokinetics, and therapeutic use of meclofenamate sodium. Clin J Pain. 1991;7(Suppl. 1):S44–S48. [PubMed] [Google Scholar]
- Devulder J. Flupirtine in pain management: pharmacological properties and clinical use. CNS Drugs. 2010;24:867–881. doi: 10.2165/11536230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Elliott AM, Smith BH, Penny KI, Smith WC, Chambers WA. The epidemiology of chronic pain in the community. Lancet. 1999;354:1248–1252. doi: 10.1016/s0140-6736(99)03057-3. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Couve A, Pangalos MN, Walsh FS, Brown DA, Moss SJ. Heteromeric assembly of GABA(B)R1 and GABA(B)R2 receptor subunits inhibits Ca(2+) current in sympathetic neurons. J Neurosci. 2000;20:2867–2874. doi: 10.1523/JNEUROSCI.20-08-02867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel HA, Fitton A. Flupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs. 1993;45:548–569. doi: 10.2165/00003495-199345040-00007. [DOI] [PubMed] [Google Scholar]
- Galasko CS, Courtenay PM, Jane M, Stamp TC. Trial of oral flupirtine maleate in the treatment of pain after orthopaedic surgery. Curr Med Res Opin. 1985;9:594–601. doi: 10.1185/03007998509109640. [DOI] [PubMed] [Google Scholar]
- Grassi F. Cl(-)-mediated interaction between GABA and glycine currents in cultured rat hippocampal neurons. Brain Res. 1992;594:115–123. doi: 10.1016/0006-8993(92)91035-d. [DOI] [PubMed] [Google Scholar]
- Guindon J, Walczak JS, Beaulieu P. Recent advances in the pharmacological management of pain. Drugs. 2007;67:2121–2133. doi: 10.2165/00003495-200767150-00002. [DOI] [PubMed] [Google Scholar]
- Hummel T, Friedmann T, Pauli E, Niebch G, Borbe HO, Kobal G. Dose-related analgesic effects of flupirtine. Br J Clin Pharmacol. 1991;32:69–76. doi: 10.1111/j.1365-2125.1991.tb05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob R, Krieglstein J. Influence of flupirtine on a G-protein coupled inwardly rectifying potassium current in hippocampal neurones. Br J Pharmacol. 1997;122:1333–1338. doi: 10.1038/sj.bjp.0701519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Bleich S, Wiltfang J, Maler M, Parsons CG. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J Neural Transm. 1999a;106:857–867. doi: 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Maler M, Wiltfang J, Bleich S, Degner D, Ruther E. [Neuronal potassium channel opening with flupirtine] Fortschr Neurol Psychiatr. 1999b;67:466–475. doi: 10.1055/s-2007-994997. [DOI] [PubMed] [Google Scholar]
- Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, et al. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, et al. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57:509–526. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- Luben V, Muller H, Lobisch M, Worz R. [Treatment of tumor pain with flupirtine. Results of a double-blind study versus tramadol] Fortschr Med. 1994;112:282–286. [PubMed] [Google Scholar]
- Martire M, Castaldo P, D'amico M, Preziosi P, Annunziato L, Taglialatela M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronardi P, D'onofrio M, Scanni E, Pinto M, Frontespezi S, Ceccarelli MG, et al. Analgesic activity of flupirtine maleate: a controlled double-blind study with diclofenac sodium in orthopaedics. J Int Med Res. 1988;16:338–348. doi: 10.1177/030006058801600503. [DOI] [PubMed] [Google Scholar]
- Miller KE, Hoffman EM, Sutharshan M, Schechter R. Glutamate pharmacology and metabolism in peripheral primary afferents: physiological and pathophysiological mechanisms. Pharmacol Ther. 2011;130:283–309. doi: 10.1016/j.pharmthera.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, Basbaum AI. Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci U S A. 2003;100:271–276. doi: 10.1073/pnas.0136822100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RA, Bullingham RE, Simpson S, O'sullivan G, Evans PJ, Mcquay HJ, et al. Comparison of flupirtine maleate and dihydrocodeine in patients following surgery. Br J Anaesth. 1983;55:429–432. doi: 10.1093/bja/55.5.429. [DOI] [PubMed] [Google Scholar]
- O'connor V, El Far O, Bofill-Cardona E, Nanoff C, Freissmuth M, Karschin A, et al. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- Olpe HR, Karlsson G, Pozza MF, Brugger F, Steinmann M, Van Riezen H, et al. CGP 35348: a centrally active blocker of GABAB receptors. Eur J Pharmacol. 1990;187:27–38. doi: 10.1016/0014-2999(90)90337-6. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Rose K, Ooi L, Dalle C, Robertson B, Wood IC, Gamper N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain. 2011;152:742–754. doi: 10.1016/j.pain.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundfeldt C, Netzer R. Investigations into the mechanism of action of the new anticonvulsant retigabine. Interaction with GABAergic and glutamatergic neurotransmission and with voltage gated ion channels. Arzneimittelforschung. 2000;50:1063–1070. doi: 10.1055/s-0031-1300346. [DOI] [PubMed] [Google Scholar]
- Scheef W. Analgesic efficacy and safety of oral flupirtine in the treatment of cancer pain. Postgrad Med J. 1987;63(Suppl. 3):67–70. [PubMed] [Google Scholar]
- Schicker K, Hussl S, Chandaka GK, Kosenburger K, Yang JW, Waldhoer M, et al. A membrane network of receptors and enzymes for adenine nucleotides and nucleosides. Biochim Biophys Acta. 2009;1793:325–334. doi: 10.1016/j.bbamcr.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Scholze T, Moskvina E, Mayer M, Just H, Kubista H, Boehm S. Sympathoexcitation by bradykinin involves Ca2+-independent protein kinase C. J Neurosci. 2002;22:5823–5832. doi: 10.1523/JNEUROSCI.22-14-05823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Oxley B, Malpas S, Pillai GV, Simpson PB. Compounds exhibiting selective efficacy for different beta subunits of human recombinant gamma-aminobutyric acid A receptors. J Pharmacol Exp Ther. 2004;311:601–609. doi: 10.1124/jpet.104.070342. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Grippon S, Kirkpatrick P. Ezogabine (retigabine) Nat Rev Drug Discov. 2011;10:729–730. doi: 10.1038/nrd3561. [DOI] [PubMed] [Google Scholar]
- Witschi R, Punnakkal P, Paul J, Walczak JS, Cervero F, Fritschy JM, et al. Presynaptic alpha2-GABAA receptors in primary afferent depolarization and spinal pain control. J Neurosci. 2011;31:8134–8142. doi: 10.1523/JNEUROSCI.6328-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Cheng K, Cui T, Godlewski G, Rice KC, Xu Y, et al. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat Chem Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf A, Klinger F, Schicker K, Boehm S. Nucleotides control the excitability of sensory neurons via two P2Y receptors and a bifurcated signaling cascade. Pain. 2011;152:1899–1908. doi: 10.1016/j.pain.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Mohler H, Di Lio A. GABAergic analgesia: new insights from mutant mice and subtype-selective agonists. Trends Pharmacol Sci. 2009;30:397–402. doi: 10.1016/j.tips.2009.05.007. [DOI] [PubMed] [Google Scholar]