

Abstract
BACKGROUND AND PURPOSE
Vascular smooth muscle cell (SMC) migration within the arterial wall is a crucial event in atherogenesis and restenosis. Monocyte chemotactic protein-1/CC-chemokine receptor 2 (MCP-1/CCR2) signalling is involved in SMC migration processes but the molecular mechanisms have not been well characterized. We investigated the role of PI3Kγ in SMC migration induced by MCP-1.
EXPERIMENTAL APPROACHES
A pharmacological PI3Kγ inhibitor, adenovirus encoding inactive forms of PI3Kγ and genetic deletion of PI3Kγ were used to investigate PI3Kγ functions in the MCP-1 and platelet-derived growth factor (PDGF) signalling pathway and migration process in primary aortic SMC.
KEY RESULTS
The γ isoform of PI3K was shown to be the major signalling molecule mediating PKB phosphorylation in MCP-1-stimulated SMC. Using a PI3Kγ inhibitor and an adenovirus encoding a dominant negative form of PI3Kγ, we demonstrated that PI3Kγ is essential for SMC migration triggered by MCP-1. PDGF receptor stimulation induced MCP-1 mRNA and protein accumulation in SMCs. Blockade of the MCP-1/CCR2 pathway or pharmacological inhibition of PI3Kγ reduced PDGF-stimulated aortic SMC migration by 50%. Thus PDGF promotes an autocrine loop involving MCP-1/CCR2 signalling which is required for PDGF-mediated SMC migration. Furthermore, SMCs isolated from PI3Kγ-deficient mice (PI3Kγ−/−), or mice expressing an inactive PI3Kγ (PI3KγKD/KD), migrated less than control cells in response to MCP-1 and PDGF.
CONCLUSIONS AND IMPLICATIONS
PI3Kγ is essential for MCP-1-stimulated aortic SMC migration and amplifies cell migration induced by PDGF by an autocrine/paracrine loop involving MCP-1 secretion and CCR2 activation. PI3Kγ is a promising target for the treatment of aortic fibroproliferative pathologies.
Keywords: PI3Kγ, aortic smooth muscle cells, migration, chemokines, platelet-derived growth factor
Introduction
Vascular smooth muscle cells (SMCs) play important functions in normal and diseased blood vessels. In normal adult arteries, SMC displays a contractile phenotype to maintain vascular tone. In response to vascular injury, however, they switch to a synthetic phenotype characterized by increased proliferation and migration, two major steps in atherosclerosis progression and neointimal thickening after angioplasty (Zargham, 2008; Orr et al., 2009). Among cytokines and growth factors released by injured vessels and inflammatory cells, the monocyte chemotactic protein-1 (MCP-1) plays a critical role in pathological vascular remodelling. MCP-1 amounts are particularly high in experimental mice during atherogenesis and after wire injury of the carotid artery. Monocyte recruitment is reduced when MCP-1 signalling is blocked as in MCP-1 knockout mice (Boring et al., 1998; Gu et al., 1998). The same applies to chemokine C-C motifs receptor 2 (CCR2) knockout mice, a GPCR, selectively activated by MCP-1. Importantly, the impaired MCP-1/CCR2 signal transduction axis reduces atherosclerotic lesions and prevents restenosis. These findings have prompted the design of anti-MCP-1/CCR2 therapeutic approaches, using for instance antibodies against MCP-1, CCR2 or an N-terminal truncated form of MCP-1. These treatments successfully prevented intimal hyperplasia in mice, rabbits and primates, suggesting that these therapies might have some potential to prevent arterial restenosis in humans (Egashira et al., 2002; Horvath et al., 2002; Mori et al., 2002; Zhong et al., 2008). The biological effects mediated by MCP-1 and its receptor, CCR2, are not restricted to the regulation of inflammatory processes but can also modify SMC functions. CCR2 mRNA has been detected in human vascular SMC (Hayes et al., 1998). In addition, CCR2 itself is involved in the functional switch of SMC to the synthetic phenotype (Denger et al., 1999) and induces migration of aortic SMC in rabbits and rats (Ma et al., 2007; Grassia et al., 2009). Moreover, in rats, aortic MCP-1 and CCR2 increase with age, as does the invasive ability of aortic SMC, suggesting that MCP-1/CCR2 signalling might play a role in age-associated vascular remodelling (Spinetti et al., 2004). While MCP-1/CCR2 signalling pathways leading to cell migration have been studied intensively in monocytes, molecular mechanisms involved in MCP-1-induced SMC migration are poorly understood.
PI3Ks, a family of lipid kinases, are important regulators of cell survival and growth, as well as of migration through the direct binding of proteins to their lipid product. Based on their structural characteristic and substrate specificity, PI3Ks are divided into the three classes, I, II and III (Vanhaesebroeck et al., 2001; Wymann et al., 2003). Class IA and class IB of PI3K comprise a highly homologous 110 kDa catalytic subunit (α, β and δ for class IA; γ for class IB) and an associated regulatory subunit. Both these kinase classes act in vivo on phosphoinositol (4,5) bisphosphate (PI4, 5P2) substrate to produce phosphoinositol (3,4,5) trisphosphate (PIP3). Whereas class IA PI3Ks are typically recruited downstream of tyrosine kinase receptors, class IB is specifically activated by βγ subunits of heterotrimeric G proteins conferring on PI3Kγ specific functions downstream of GPCR activation (Wymann et al., 2003; Fougerat et al., 2008). A growing body of evidence suggests that PI3Ks are involved in SMC migration (Goncharova et al., 2002; Irani et al., 2002; Radhakrishnan et al., 2008; Zhou et al., 2009). In this study, we identified PI3Kγ as the major PI3K involved in PKB (Akt) phosphorylation and migration downstream of CCR2 activation by MCP-1 in aortic SMC. Furthermore, we found that PI3Kγ could also amplify platelet-derived growth factor receptor (PDGF)-induced migration by an autocrine/paracrine loop involving MCP-1/CCR2 signalling. This provides a new insight into the molecular mechanisms of, and signalling involved in, the aortic SMC migration process.
Methods
Materials
Cell culture reagents were purchased from Invitrogen (Grand Island, NY, USA). Human recombinant EGF was from Peprotech (Rocky Hill, NJ, USA), human recombinant PDGF-BB and mouse MCP-1 were from R&D Systems (Lille, France), and swine recombinant MCP-1 was from Kingfisher Biotechnologies (Euromedex, Mundolsheim, France). The inhibitors AS-252424, wortmannin, RS-102895, AG1478, AG1296 and DAPI were from Sigma-Aldrich (Saint-Quentin Fallavier, France). The PKB (Akt) inhibitor (PKB inhibitor VIII, Isozyme-Selective, PKBi-1/2) was from Tebu Bio (Le Perray en Yvelines, France). The pig MCP-1-neutralizing antibody was from Bethyl Laboratories, Inc. (Euromedex, Souffelweyersheim, France). Rabbit monoclonal antibody against phosphorylated PKB (serine 473), rabbit monoclonal antibody against phosphorylated PKB (threonine 308), rabbit polyclonal antibody against PKB and HRP-conjugated anti-rabbit antibody were from Cell Signaling Technologies (Danvers, MA, USA); anti-cmyc clone 9E10 was from BD Biosciences (Le Pont de Claix, France), anti-GFP was from Roche (Meylan, France), cyanin-2 conjugated secondary antibody was from Jackson Immunoresearch Laboratories (Suffolk, UK) and HRP-conjugated anti-mouse antibody was from Sigma-Aldrich. The enhanced chemiluminescence (ECL) system was from Amersham Biosciences (Munich, Germany).
Animals
PI3Kγ-deficient mice (PI3Kγ−/−) and PI3KγKD/KD mice were generated as previously described (Hirsch et al., 2000; Patrucco et al., 2004). Both mutant and control mice were derived from 15 generation backcrosses to the C57Bl/6 genetic background.
All animal care and experimental procedures were in accordance with institutional guidelines on Animal Experimentation and with the French Ministry of Agriculture license. Moreover, they conformed to US NIH Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and our institution's ethical policy is listed as A5326-01 on office of laboratory animal welfare (NIH website).
Adenoviral production and infection
The PI3Kγ KR construct was PCR modified from ADNc PI3Kγ KR (Bondeva et al., 1998) to insert a myc-tag and inserted into the multiple cloning sites of the pShuttle-CMV. Then, PI3Kγ KR-myc adenoviruses were generated by homologous recombination using the pAdEasy system (He et al., 1998).
Infection with adenovirus was achieved by incubating a cell suspension with 150 IP per cell in serum-free medium. After 1 h, the cells were incubated in 10% fetal calf serum (FCS) culture medium for 72 h. Cells were then placed in serum-free medium for 24 h before stimulation by different agonists or seeded in a 96-well plate for migration experiments.
Cell culture
Aortic SMCs were prepared from the thoracic aorta of a 6-week-old pig. First, the pig was sedated using ketamine hydrochloride (25 mg·kg−1 i.m.) and anaesthesia was induced by i.v. injection of sodium pentobarbital (20–30 mg·kg−1). After a thoracotomy, the aorta was dissected and removed. Then, cells were isolated by using an explant technique and were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FCS and antibiotics. SMCs at 80% confluence from passages 3 to 6 were used in the experiments. Before stimulation, cells were incubated for 48 h in serum-free medium. Human aortic SMCs were from ATCC (Manassas, VA, USA; CRL-1999).
Mouse aortic SMCs were isolated from 8-week-old PI3Kγ+/+ and PI3Kγ−/− or PI3KγKD/KD mice. First, mice were killed using sodium pentobarbital in a 1 mg·mL−1 solution at 0.10 mL·10 g−1 body weight dosage injected into the left peritoneal cavity. Then, aortas were dissected out from their origin at the left ventricle to the iliac bifurcation. After being flushed with sterile PBS, the aortas were placed in DMEM supplemented with 10% FCS and 1 µg·mL−1 Fungizone. The adventitia was then totally removed from the aorta under a dissecting microscope. The aorta was placed in DMEM supplemented with 10% FCS and cut into small pieces of 1–2 mm. These pieces were then digested in DMEM supplemented with 10% FCS containing 1.36 mg·mL−1 collagenase type II (LS004174, Worthington Biochemical Corporation, Serlabo technologies, Entraigues, France) for 4 h in a humidified 5% CO2 atmosphere at 37°C. Cells were centrifuged and resuspended in DMEM, 10% FCS, in a single well of a 24-well plate. After 5 days, the medium was replaced systematically until confluence was reached. Mouse SMCs from the 5th to 10th passages were used in experiments.
Western blot analysis
Proteins from whole cells were dissolved in Laemmli buffer, boiled, separated by SDS-PAGE and transferred onto PVDF membrane. Immunodetection was achieved using the relevant primary antibody, anti-cmyc (1:10 000), anti-GFP (1:1000), anti-phosphorylated Ser473PKB (1:2000), anti-phosphorylated Thr308PKB (1:1000) or anti-PKB (1:1000) overnight at 4°C. Horseradish peroxidase–conjugated secondary antibody (anti-rabbit 1:5000 and anti-mouse 1:10 000) was incubated for 1 h at room temperature and immunoreactive proteins were detected with ECL reagents according to the manufacturer's instructions.
Cell migration assay
The wound-healing assay was performed using the Oris cell migration kit (Oris™ Cell Migration Assay-Fibronectin Coated, Platypus Technologies, Tebu Bio) according to the manufacturer's instructions. Formatted for a 96-well plate, this assay uses silicone stoppers, which restrict cell seeding to the outer annular regions of the well. When the stoppers are removed, cells can migrate in a 2 mm-diameter region in the centre of the well. We seeded 3.5 × 104 cells of pig SMC or 5 × 104 cells of mouse SMC per well. After attachment, cells were treated with PDGF-BB (10 ng·mL−1) or MCP-1 (10 ng·mL−1), alone or together with AS-252424 (100 nM), RS-102895 (5 µg·mL−1), PKB inhibitor (20 µM) or pig MCP-1-neutralizing antibody (5 µg·mL−1). Cells were allowed to migrate for 48 h at 37°C then stained with DAPI and counted under a fluorescence microscope. The number of fluorescent nuclei in each well was counted using Image J Software (NIH).
Real-time quantitative PCR
SMCs were treated with PDGF-BB (10 ng·mL−1) for 0.5–4 h. Total RNA was extracted with Trizol reagent (Invitrogen), phase separation was performed with chloroform and RNA precipitation with isopropanol. RNA concentrations were determined using a spectrophotometer. One microgram of total RNA was used for cDNA synthesis with hexamers. The cDNA was amplified by quantitative real-time PCR with SYBR Green (Roche). Primers to amplify MCP-1 RNA were designed using the Primer Express software (Life Technologies SAS, Villebon sur Yvette, France) with the following sequences: 5′-GCTGTGATCTTCAAGACCATTGTG-3′ (F) and 5′-GAA TCCTGAACCCACTTCTGCTT-3′ (R). All real-time PCR reactions were performed on a LC480 (Roche), with the following thermal cycling parameters: annealing at 60°C for 40 s, amplification at 95°C for 10 s and dissociation at 95°C for 10 s. The PCR programme was followed by a melting curve and values were normalized to the relative amounts of hypoxanthine-guanine phosphoribosyltransferase (HPRT). CT values of HPRT were constant in all samples.
Quantification of MCP-1 by ELISA
SMCs were stimulated by PDGF-BB (10 ng·mL−1) for 2–48 h. Cell culture supernatants were used to determine the concentrations of MCP-1 by the specific Pig CCL2 ELISA Kit (Euromedex, Souffelweyersheim, France) according to the manufacturer's instructions.
Immunofluorescence
Mouse aortic SMCs were fixed with ice-cold methanol, washed in PBS and incubated with anti-smooth muscle-α-actin primary antibody (1:1000, clone 1A4, Sigma, Saint-Quentin Fallavier, France) for 1 h. After being washed three times with PBS, cells were incubated with a secondary cyanin-2 conjugated anti-mouse antibody and DAPI (1 µg·mL−1). Pictures were obtained using a fluorescence microscope (TE2000-E, Eclipse, NIKON, Melville, NY, USA).
Statistical analysis
For cell migration experiments, each individual experiment was performed in 8 wells for each condition and repeated at least three times. Experimental data were analysed by Mann–Whitney test. Results are expressed as means ± SEM. Differences were considered significant at P < 0.05.
Results
MCP-1-induced PKB phosphorylation does not require tyrosine kinase receptor transactivation
To investigate the involvement of the PI3K/PKB pathway in MCP-1-induced activation of aortic SMC, we first analysed the phosphorylation of PKB after MCP-1 stimulation in primary pig aortic SMC. Because most signalling pathways activated by GPCR agonists in SMC require tyrosine kinase receptor activation, such as EGF receptor or PDGF receptor (Kalmes et al., 2001; Voisin et al., 2002; Tanimoto et al., 2004), we first analysed the effect of MCP-1 on PKB phosphorylation in the presence of the EGF receptor inhibitor AG1478 and PDGF receptor inhibitor AG1296. We observed that AG1478 and AG1296 did not modify MCP-1-induced PKB phosphorylation on both serine 473 and threonine 308 (Figure 1A). However, AG1478 effectively inhibited the EGF pathway and AG1296 similarly inhibited PDGF (Figure 1B). This indicates that PI3K activation by MCP-1 does not require tyrosine kinase receptor transactivation. The same results were obtained in human aortic SMC stimulated by MCP-1 (Supporting Information Figure S1). In combination, these results suggest that PI3Ks are directly activated downstream CCR2 activation in vascular SMC.
Figure 1.
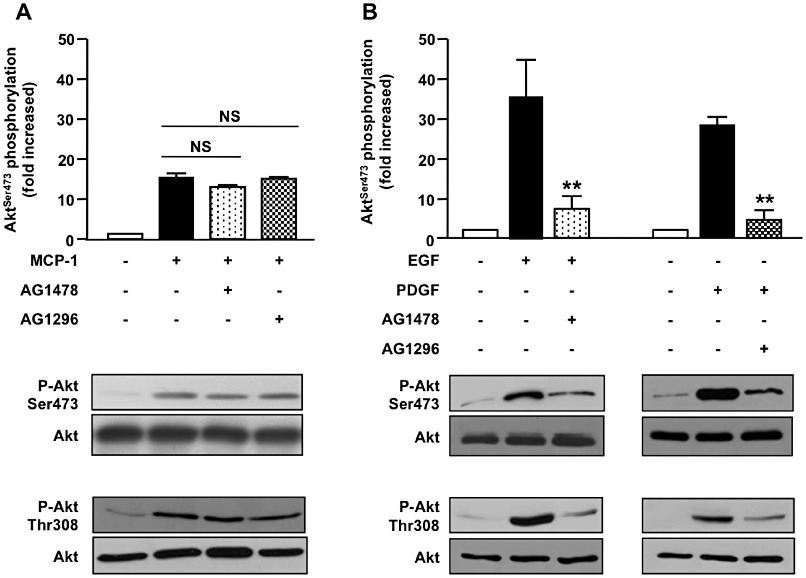
PKB (Akt) phosphorylation induced by MCP-1 in aortic SMCs does not require tyrosine kinase receptor transactivation. (A, B) Primary pig aortic SMCs were pretreated for 30 min with tyrosine kinase activity inhibitors (AG1478: 300 nM, AG1296: 10 µM) and stimulated with MCP-1 (10 ng·mL−1, 5 min) (A), EGF (10 ng·mL−1, 2 min) or PDGF-BB (10 ng·mL−1, 2 min) (B). PKB phosphorylation and expression were analysed with antibodies against phosphorylated PKB (serine 473 and threonine 308) and total PKB. Immunoblots shown are representative of three independent experiments (n= 3). **P < 0.01 compared with EGF or PDGF stimulation alone.
PI3Kγ mediates MCP-1-induced PKB phosphorylation
To investigate the potential role of PI3Kγ activity in PKB phosphorylation induced by MCP-1, aortic SMCs were treated with a selective inhibitor of PI3Kγ (AS-252424) (Condliffe et al., 2005). PKB phosphorylation induced by MCP-1 was reduced by 85% by AS-252424 and eliminated by wortmannin, a pan PI3K inhibitor (Figure 2A). This indicates that PI3K activity is required and that PI3Kγ is the major isoform involved in this process although other class I PI3K (PI3Kα and β) is also expressed in these cells (Supporting Information Figure S2). To control AS-252424 specificity, we investigated PKB phosphorylation after treating cells with EGF and PDGF, which are known to recruit class IA PI3K. We did not observe any modification of PKB phosphorylation by either treatment (Figure 2B), demonstrating the selectivity of the inhibitor against class IB PI3K. Similarly, infection of aortic SMC with an adenovirus encoding an inactive form of PI3Kγ (PI3Kγ KR) completely blocked PKB phosphorylation induced by MCP-1 (Figure 2C) whereas adenovirus encoding a GFP (Ad-GFP) as a control did not significantly modify the level of PKB phosphorylation, confirming the involvement of PI3Kγ in MCP-1-induced PKB phosphorylation in aortic SMC.
Figure 2.
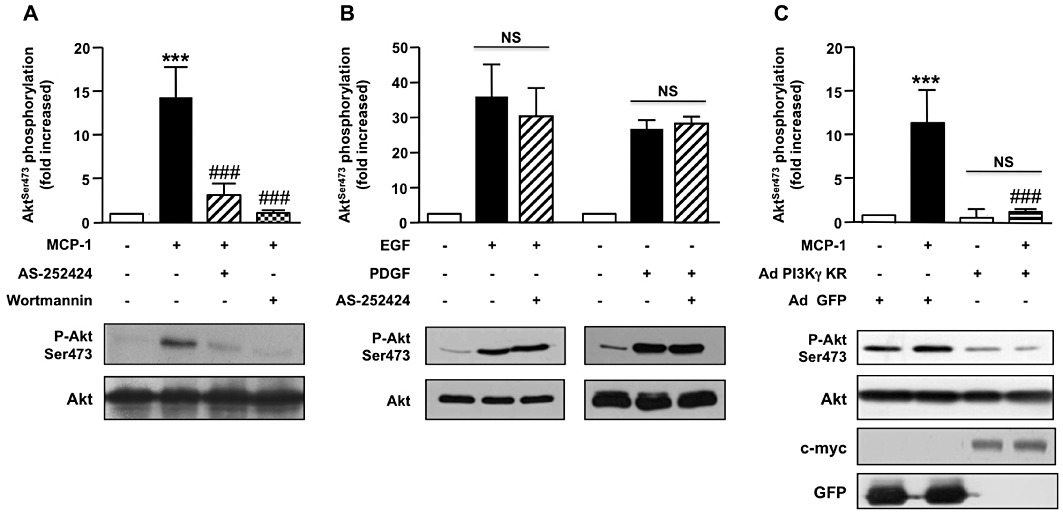
PI3Kγ is required for PKB (Akt) phosphorylation in MCP-1-activated aortic SMC. (A, B) Pig aortic SMCs were pre-incubated (30 min) (A) with wortmannin (100 nM) or AS-252424 (100 nM) and stimulated with MCP-1 (10 ng·mL−1, 5 min) or (B) with AS-252424 and stimulated with EGF (10 ng·mL−1, 5 min) or PDGF (10 ng·mL−1, 5 min). Lysates were analysed by Western blot using antibodies against phosphorylated PKB (serine 473) or total PKB. Immunoblots from representative experiments and densitometry analyses are shown (means ± SEM, n= 4). (C) Pig aortic SMCs were infected with PI3Kγ KR-myc adenovirus (Ad PI3Kγ KR) or a GFP encoding adenovirus (Ad-GFP) as a control and stimulated with MCP-1 (10 ng·mL−1, 5 min). PKB phosphorylation and expression were analysed with antibodies against phosphorylated PKB (serine 473) or total PKB. Expression of PI3Kγ KR was analysed with anti-c-myc antibody and control adenovirus with anti-GFP antibody. Immunoblots from representative experiments are shown (n= 3). ###P < 0.001 versus MCP-1 alone and ***P < 0.001 compared with unstimulated cells.
MCP-1/CCR2 signalling induces aortic SMC migration via a PI3Kγ-dependent pathway
PKB, and especially PKB1, has been demonstrated to be a key event in SMC migration (Fernandez-Hernando et al., 2009). Because MCP-1 is a chemoattractant for several cell types including SMC, we evaluated the possible involvement of PI3Kγ/PKB pathway in MCP-1-induced aortic SMC migration. Using a wound-healing cell migration assay, we showed that MCP-1 induced aortic SMC migration at the dose of 5 ng·mL−1 with a maximum efficiency at 10 ng·mL−1 (Supporting Information Figure S3). This effect was totally abolished by a PKB inhibitor demonstrating the essential function of PKB in MCP-1-induced cell migration (Figure 3A). RS-102895, an inhibitor of the chemokine receptor CCR2 (Figure 3B), prevented MCP-1-induced migration indicating that the effect of MCP-1 on aortic SMC migration is mediated by CCR2 and PKB. We next investigated the involvement of PI3Kγ in this process. Cells were pretreated with the PI3Kγ inhibitor AS-252424 or infected by an adenovirus encoding an inactive form of PI3Kγ. Basal migration was significantly decreased in both conditions indicating that a weak activation of PI3Kγ persisted in the basal condition. Nevertheless, the PI3Kγ inhibitor as well as an adenovirus encoding inactive PI3Kγ totally prevented MCP-1-induced migration (Figure 3C,D). To verify the involvement of PI3Kγ in migration processes induced by MCP-1, the chemotaxis of SMC induced by MCP-1 was performed using a Boyden chamber assay. AS-25224 totally abolished the migration in this condition (Supporting Information Figure S4A). Moreover, BrdU staining showed that MCP-1 was not able to induce aortic pig SMC proliferation (Supporting Information Figure S4B). These data clearly indicate that PI3Kγ activation downstream of MCP-1/CCR2 signalling plays a crucial role in aortic SMC migration.
Figure 3.
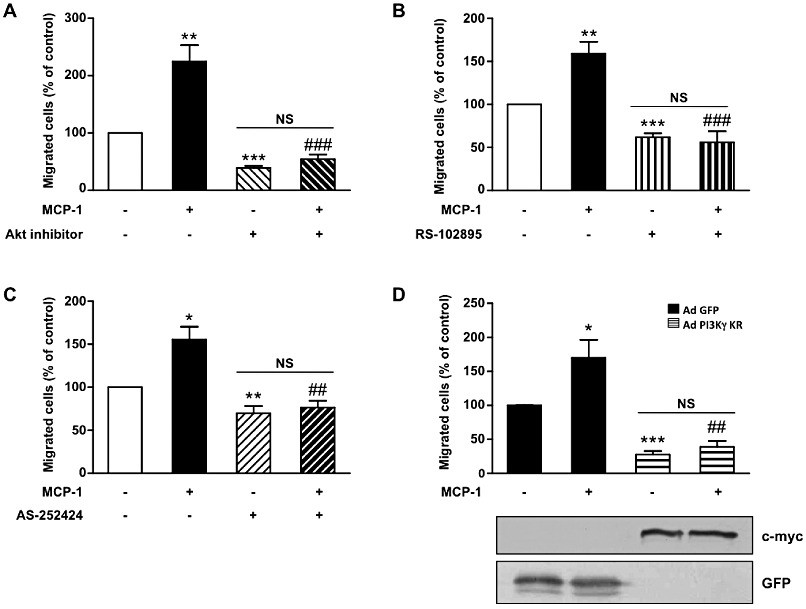
PI3Kγ is essential for MCP-1-induced aortic SMC migration. Pig aortic SMC migration was quantified with a wound-healing assay using the Oris cell migration kit. Confluent SMCs were treated with MCP-1 (10 ng·mL−1) in serum-free medium and allowed to migrate in the wound surface for 48 h. Migrated cells were stained with DAPI and counted under a fluorescence microscope. Results are expressed as a percentage of control. (A) SMCs were treated with MCP-1 (10 ng·mL−1) only or together with PKB inhibitor (20 µM) in serum-free medium. (B) SMCs were treated with MCP-1 (10 ng·mL−1) alone or together with MCP-1 receptor inhibitor (RS-102895: 5 µg·mL−1). (C) SMCs were treated with MCP-1 (10 ng·mL−1) alone or together with a specific PI3Kγ inhibitor (AS-252424: 100 nM). (D) SMCs infected with GFP adenovirus (Ad-GFP) or with an adenovirus encoding a dominant negative form of PI3Kγ (Ad PI3Kγ KR) for 72 h were treated with MCP-1 (10 ng·mL−1). Expression of PI3Kγ KR was analysed with anti-c-myc antibody and control adenovirus with anti-GFP antibody. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control, ##P < 0.01 compared with MCP-1 alone stimulated cells, ###P < 0.001 compared with MCP-1 alone stimulated cells.
PI3Kγ can amplify PDGF-induced aortic SMC migration via an MCP-1/CCR2 pathway
MCP-1 mRNA is rapidly increased in arterial tissue after arterial injury and might be involved in both monocyte recruitment and the SMC migration process (Taubman et al., 1992; Wysocki et al., 1996). While the mechanism regulating CCR2 expression in SMC is not completely understood, induction of MCP-1 is well characterized. PDGF has been shown to induce MCP-1 accumulation in rat SMC not only by activation of a specific region in the promoter (Poon et al., 1996; Bogdanov et al., 1998) but also by increasing MCP-1 mRNA stability (Liu et al., 2006). We therefore investigated whether PI3Kγ is not just restricted to a role in MCP-1 signalling but could also amplify migration induced by PDGF through an autocrine/paracrine loop. To investigate this mechanism in our aortic SMC model, we first evaluated MCP-1 mRNA and protein level accumulation in response to PDGF. We showed an increase in mRNA encoding MCP-1 after as little as 1 h of PDGF stimulation (Figure 4A) and a 10-fold accumulation of MCP-1 protein after 48 h (Figure 4B). To explore a possible paracrine/autocrine loop in PDGF-induced SMC migration, aortic SMCs were incubated with a CCR2 inhibitor or an MCP-1-neutralizing antibody before stimulation with PDGF. Under these conditions, PDGF-induced aortic SMC migration was reduced by about 50% indicating an important function of the MCP-1/CCR2 pathway in PDGF-mediated migration (Figure 4C). To determine the possible involvement of PI3Kγ in this process, the same experiments were performed in aortic SMC pre-incubated with AS-252424. Treatment with the PI3Kγ inhibitor reduced by, about 40%, the migration (Figure 4C) and chemotactic response of SMCs (Supporting Information Figure S4C) induced by PDGF. To investigate the possible involvement of PI3Kγ in the proliferation mechanism of PDGF in our wound healing experiments, we investigated SMC proliferation under the same conditions using BrdU staining. The results, presented in Supporting Information Figure S4D, showed that the PI3Kγ inhibitor did not significantly modulate SMC proliferation induced by PDGF indicating a specific role of PI3Kγ in PDGF-induced migration in aortic SMCs (Supporting Information Figure S4D). Infection of SMCs with an adenovirus encoding the inactive form of PI3Kγ confirmed these results (Figure 4D). In these experiments, PI3Kγ inhibition reduced basal aortic SMC migration as effectively as the CCR2 inhibitor or MCP-1-neutralizing antibody treatment indicating that a weak activation of MCP-1/CCR2/PI3Kγ pathway still occurred in our basal conditions. To confirm the involvement of PI3Kγ in the amplification of aortic SMC migration upon PDGF stimulation, we isolated primary SMCs from aortas of PI3Kγ+/+, PI3Kγ−/− mice or mice expressing a catalytically inactive form of PI3Kγ (PI3KγKD/KD). These cells expressed CCR2 and PI3Kγ as pig aortic SMCs (Supporting Information Figure S5). These cells stained positive for smooth muscle actin and did not show any difference in the number of positive cells in different genotypes (Figure 5A). In line with the results in SMCs treated with PI3Kγ inhibitors, murine SMC derived from PI3Kγ−/− or PI3KγKD/KD mice exhibited a reduced migratory response to MCP-1 and PDGF compared with PI3Kγ+/+ cells. This demonstrates the importance of PI3Kγ in both the MCP-1 and the PDGF-induced migration process (Figure 5B).
Figure 4.
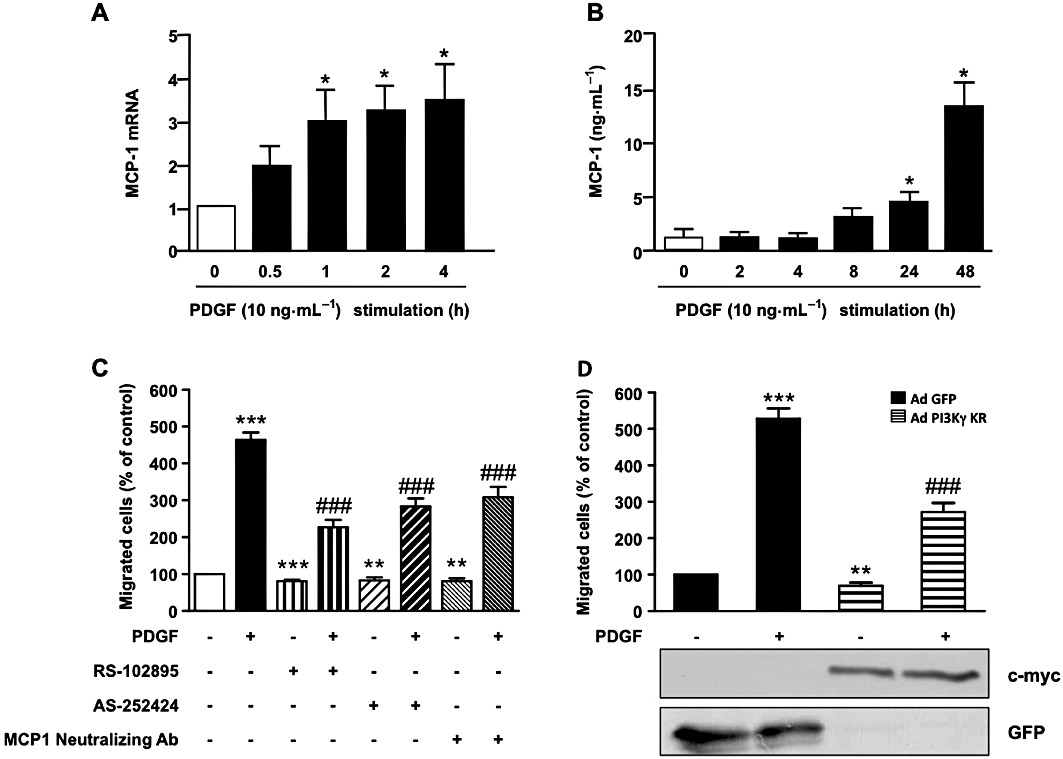
PI3Kγ amplifies PDGF-stimulated aortic SMC migration by an MCP-1/CCR2 pathway. (A) Pig aortic SMCs were stimulated with PDGF-BB (10 ng·mL−1) in serum-free medium for the indicated times. Total RNA was extracted and subjected to quantitative real-time PCR using specific primers for MCP-1. (B) Pig aortic SMCs were stimulated with PDGF-BB (10 ng·mL−1) in serum-free medium for the indicated times. Cell supernatants were analysed for MCP-1 protein content by a specific ELISA kit as described in Methods. (C) Confluent pig aortic SMCs were treated with PDGF-BB (10 ng·mL−1) alone or together with MCP-1 receptor inhibitor (RS-102895: 5 µg·mL−1) or a specific PI3Kγ inhibitor (AS-252424: 100 nM) or MCP-1-neutralizing antibody (5 µg·mL−1) in serum-free medium. Cells were allowed to migrate for 48 h and migrated cells were stained with DAPI and counted under a fluorescence microscope. Results are expressed as a percentage of the control. (D) Pig aortic SMCs infected with GFP adenovirus or with an adenovirus encoding a dominant negative form of PI3Kγ (PI3Kγ KR) for 72 h were treated with PDGF-BB (10 ng·mL−1) and analysed for migration as above. Expression of PI3Kγ KR was analysed with anti-c-myc antibody and control adenovirus with anti-GFP antibody. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control, ###P < 0.001 compared with PDGF alone stimulated cells.
Figure 5.
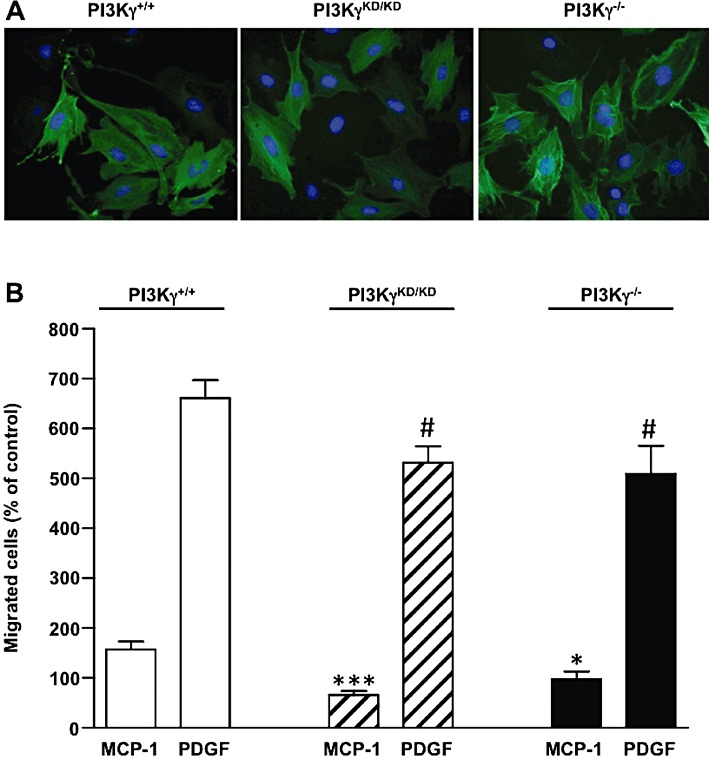
Effect of PI3Kγ deficiency in MCP-1 and PDGF-induced migration of mouse aortic SMCs. (A) Aortic SMCs from wild-type (PI3Kγ+/+), PI3Kγ-deficient mice (PI3Kγ−/−) or mice expressing a catalytically inactive PI3Kγ (PI3KγKD/KD) were fixed and stained with an anti-smooth muscle α-actin antibody. Photomicrographs from representative experiments are shown. (B) Aortic SMCs from PI3Kγ+/+, PI3Kγ−/− or PI3KγKD/KD mice were stimulated with MCP-1 (10 ng·mL−1) or PDGF-BB (10 ng·mL−1) in serum-free medium and allowed to migrate for 48 h. Migrated cells were stained with DAPI and counted under a fluorescence microscope. Results are expressed as a percentage of PI3Kγ+/+, PI3Kγ−/− or PI3KγKD/KD non-stimulated cells, respectively. *P < 0.05, ***P < 0.001 compared with MCP-1-stimulated PI3Kγ+/+ cells; #P < 0.05 compared with PDGF-stimulated PI3Kγ+/+ cells.
Discussion
MCP-1 has been shown to contribute to all steps of the arterial remodelling process by inducing monocyte infiltration and functional switching of SMCs to the synthetic phenotype, leading to increased proliferation and migration. Most of the effects mediated by MCP-1 appear to be mediated by its interaction with the GPCR CCR2 but downstream signalling pathways are not well characterized. The involvement of PI3K lipid products, especially PIP3, has been reported in restenosis processes such as SMC proliferation and migration (Goncharova et al., 2002; Irani et al., 2002; Mehrhof et al., 2005; Radhakrishnan et al., 2008; Zhou et al., 2009). Here, we have investigated whether PI3K isoform(s) are involved in MCP-1-mediated SMC migration. Early signalling events occurring downstream of GPCR activation have been subject to intense investigation. It has been shown that GPCR agonists induce tyrosine phosphorylation of multiple substrates in target cells (Force et al., 1991; Leeb-Lundberg and Song, 1991; Laffargue et al., 1999). In SMCs, GPCR activation leads to rapid activation of tyrosine kinase receptors such as EGF and PDGF receptors that might be involved in PI3K/PKB activation (Voisin et al., 2002; Baudhuin et al., 2004; Tanimoto et al., 2004; Hsieh et al., 2009). However, previous reports indicated that in SMCs, GPCR signalling can directly activate the PI3Kγ isoform (Vecchione et al., 2005). In this respect, our study demonstrated a direct link between MCP-1/CCR2 activation and the PI3K/PKB pathway. Furthermore, we have provided evidence that the γ isoform of PI3K is specifically involved in this MCP-1/CCR2 signalling pathway. Whereas PI3Kγ has been widely reported to be involved in the migration of haematopoietic cells (Hirsch et al., 2000; Hannigan et al., 2002; Jones et al., 2003; Del Prete et al., 2004), our study is the first to demonstrate its involvement in SMC migration. Only a few studies have reported a role for PI3Kγ in non-haematopoietic cell migration (Heller et al., 2008; Guzman-Hernandez et al., 2009). Heller et al. (2008) investigated the role of different isoforms of PI3K in sphingosine 1-phosphate-induced migration of endothelial cells and demonstrated that PI3Kβ and γ are equally involved in this process. Whereas the structure of PI3Kβ predicted activation downstream of the tyrosine kinase receptor, recent data obtained from mouse mutants expressing a catalytically inactive PI3Kβ showed that PI3Kβ was more effective at activating the GPCR than tyrosine kinases (Ciraolo et al., 2008; Guillermet-Guibert et al., 2008; Jia et al., 2008). Using PI3Kβ inhibitors, we also found that PI3Kβ is somewhat involved in MCP-1-induced aortic SMC migration (data not shown). This could explain the difference in PKB phosphorylation induced by MCP-1 in cells treated with a pan-PI3K inhibitor such as wortmannin compared with cells treated with a specific PI3Kγ inhibitor. Even if PI3Kγ is not the predominant isoform expressed in aortic SMCs, we clearly demonstrated that its involvement predominates in MCP-1-stimulated aortic SMC because PKB phosphorylation decreased by more than 85% in the absence of PI3Kγ activity and MCP-1-induced migration was totally abolished in the same conditions.
High levels of MCP-1 are expressed not only by inflammatory cells but also by cells of the arterial wall during the development of atherosclerosis and after balloon injury (Taubman et al., 1992; Wysocki et al., 1996; Sako et al., 2008). In vitro, PDGF could be responsible for part of this increase in MCP-1 secretion by SMCs (Bogdanov et al., 1998; Liu et al., 2006). Our data extended these observations and showed the involvement of the MCP-1/CCR2 pathway in PDGF-induced aortic SMC migration. In addition, our results demonstrated that PI3Kγ, by this autocrine pathway, could amplify the migration process activated by PDGF. Other agonists, such as TNF-α or thrombin, are able to induce MCP-1 secretion in SMC (Kranzhofer et al., 1996; Biswas et al., 1998). Moreover, it has been shown that other cell types found in the arterial wall, such as inflammatory cells or endothelial cells, secrete MCP-1 in response to interleukin-6 (Biswas et al., 1998; Rott et al., 2003). It would be interesting to investigate the possible involvement of PI3Kγ downstream of the effectors that are key players in intimal hyperplasia formation.
Although PI3Kγ activation mechanisms require GPCR activation, other agonists acting via the tyrosine kinase receptor have been shown to activate PI3Kγ. For example, migration induced by CSF-1, a tyrosine kinase receptor agonist, was altered in PI3Kγ−/− macrophages (Jones et al., 2003). It is also of interest that incubating cells with pertussis toxin to block Gi coupled receptors had a marked effect on cell morphology, indicating the involvement of a Gi-activated signalling pathway (Jones et al., 2003). In the same cells, MCP-1-induced migration was markedly reduced in the absence of PI3Kγ. One explanation for this defect in CSF-1-induced migration of these PI3Kγ−/− macrophages could be the presence of an autocrine/paracrine pathway involving secretion of chemokines. Hence, the pathway described in our study in aortic SMC could also be present in other cell types.
In summary, our study shows that PI3Kγ is the major PI3K involved in PKB phosphorylation and migration induced by the chemokine MCP-1. Moreover, we demonstrated that the involvement of PI3Kγ in aortic SMC migration was not restricted to MCP-1 because PI3Kγ could amplify PDGF-induced cell migration by an autocrine/paracrine pathway involving MCP-1 secretion and CCR2 activation (Figure 6). These results together with the anti-inflammatory properties of the PI3Kγ inhibitor in the arterial wall that we demonstrated previously (Fougerat et al., 2008) make PI3Kγ a promising target for the prevention of restenosis.
Figure 6.
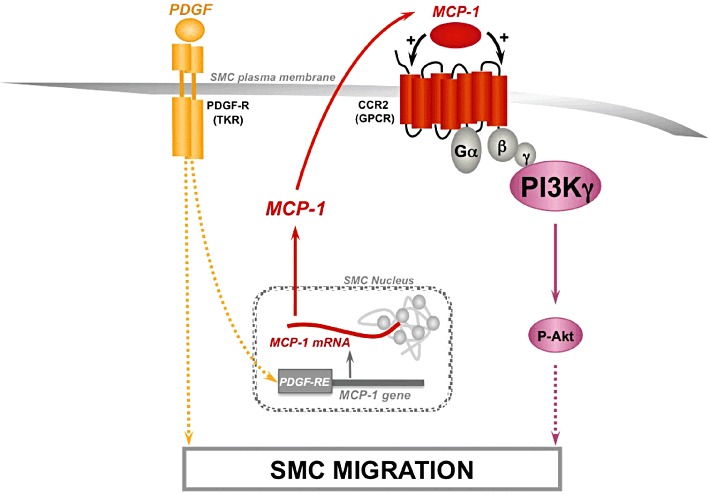
Proposed signalling model of the essential role of PI3Kγ in MCP-1-stimulated aortic smooth muscle cell as an amplifier of PDGF-induced cell migration. In aortic SMC migration, PI3Kγ is the major PI3K isoform implicated in MCP-1-induced PKB (Akt) phosphorylation. Moreover, PI3Kγ could play a role as an amplifier of PDGF-induced SMC migration by an autocrine/paracrine pathway. MCP-1 mRNA level is increased upon PDGF stimulation leading to MCP-1 secretion and CCR2 activation. Dashed lines indicate potential signalling mechanism not fully established at this time. Abbreviation: TKR, tyrosine kinase receptor; PDGF-RE, PDGF-responsive-element.
Acknowledgments
We thank the technical service of the animal facilities of the Bio-Medical Research Federative Institute of Toulouse (Genotoul Anexplo Platform IFR150).
This work was supported by the Fondation de France (No. Engt 2007001907) and the Agence Nationale de la Recherche (ANR-BMF#GENO 102 01). SG is supported by FRM (Fondation pour la Recherche Médicale) fellowship and AF by NSFA (Nouvelle Société Française d'Athérosclérose) fellowship.
Glossary
- AG-1478
N- (3- chlorophenyl)- 6, 7- dimethoxy- 4- quinazolinamine
- AG-1296
6, 7- dimethoxy- 2- phenyl- quinoxaline
- AS-252424
5- [5- (4- fluoro- 2- hydroxy- phenyl)- furan- 2- ylmethylene]- thiazolidine- 2, 4- dione
- CCR2
CC-chemokine receptor 2
- EGF
epidermal growth factor
- MCP-1
monocyte chemotactic protein-1
- PDGF
platelet-derived growth factor
- PI3K
phosphoinositide 3-kinase
- RS102895
1'-[2-[4-(trifluoromethyl)phenyl]ethyl]-spiro[4H-3,1-benzoxazine-4,4'-piperidin]-2(1H)-one hydrochloride
- SMC
smooth muscle cell
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 PKB phosphorylation induced by MCP-1 in human aortic SMC does not require tyrosine kinase receptor transactivation.
Figure S2 PI3K isoforms are expressed in pig aortic SMC. PI3K isoform expression was analysed with specific antibodies against p110α, p110β, p110γ in pig aortic SMC.
Figure S3 Dose–response of MCP-1-induced aortic SMC migration.
Figure S4 PI3Kγ is essential to MCP-1 and PDGF-induced aortic SMC migration and not proliferation.
Figure S5 PI3Kγ and CCR2 are expressed in mouse and pig aortic SMC.
Appendix S1 Supplementary Methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Baudhuin LM, Jiang Y, Zaslavsky A, Ishii I, Chun J, Xu Y. S1P3-mediated Akt activation and cross-talk with platelet-derived growth factor receptor (PDGFR) FASEB J. 2004;18:341–343. doi: 10.1096/fj.03-0302fje. [DOI] [PubMed] [Google Scholar]
- Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, et al. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood. 1998;91:258–265. [PubMed] [Google Scholar]
- Bogdanov VY, Poon M, Taubman MB. Platelet-derived growth factor-specific regulation of the JE promoter in rat aortic smooth muscle cells. J Biol Chem. 1998;273:24932–24938. doi: 10.1074/jbc.273.38.24932. [DOI] [PubMed] [Google Scholar]
- Bondeva T, Pirola L, Bulgarelli-Leva G, Rubio I, Wetzker R, Wymann MP. Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science. 1998;282:293–296. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3. doi: 10.1126/scisignal.1161577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- Del Prete A, Vermi W, Dander E, Otero K, Barberis L, Luini W, et al. Defective dendritic cell migration and activation of adaptive immunity in PI3Kgamma-deficient mice. EMBO J. 2004;23:3505–3515. doi: 10.1038/sj.emboj.7600361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denger S, Jahn L, Wende P, Watson L, Gerber SH, Kubler W, et al. Expression of monocyte chemoattractant protein-1 cDNA in vascular smooth muscle cells: induction of the synthetic phenotype: a possible clue to SMC differentiation in the process of atherogenesis. Atherosclerosis. 1999;144:15–23. doi: 10.1016/s0021-9150(99)00033-7. [DOI] [PubMed] [Google Scholar]
- Egashira K, Zhao Q, Kataoka C, Ohtani K, Usui M, Charo IF, et al. Importance of monocyte chemoattractant protein-1 pathway in neointimal hyperplasia after periarterial injury in mice and monkeys. Circ Res. 2002;90:1167–1172. doi: 10.1161/01.res.0000020561.03244.7e. [DOI] [PubMed] [Google Scholar]
- Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, Sessa WC. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:2033–2040. doi: 10.1161/ATVBAHA.109.196394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force T, Kyriakis JM, Avruch J, Bonventre JV. Endothelin, vasopressin, and angiotensin II enhance tyrosine phosphorylation by protein kinase C-dependent and -independent pathways in glomerular mesangial cells. J Biol Chem. 1991;266:6650–6656. [PubMed] [Google Scholar]
- Fougerat A, Gayral S, Gourdy P, Schambourg A, Ruckle T, Schwarz MK, et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. 2008;117:1310–1317. doi: 10.1161/CIRCULATIONAHA.107.720466. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Ammit AJ, Irani C, Carroll RG, Eszterhas AJ, Panettieri RA, et al. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L354–L363. doi: 10.1152/ajplung.00010.2002. [DOI] [PubMed] [Google Scholar]
- Grassia G, Maddaluno M, Guglielmotti A, Mangano G, Biondi G, Maffia P, et al. The anti-inflammatory agent bindarit inhibits neointima formation in both rats and hyperlipidaemic mice. Cardiovasc Res. 2009;84:485–493. doi: 10.1093/cvr/cvp238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A. 2008;105:8292–8297. doi: 10.1073/pnas.0707761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Hernandez ML, Vazquez-Macias A, Carretero-Ortega J, Hernandez-Garcia R, Garcia-Regalado A, Hernandez-Negrete I, et al. Differential inhibitor of Gbetagamma signaling to AKT and ERK derived from phosducin-like protein: effect on sphingosine 1-phosphate-induced endothelial cell migration and in vitro angiogenesis. J Biol Chem. 2009;284:18334–18346. doi: 10.1074/jbc.M109.008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan M, Zhan L, Li Z, Ai Y, Wu D, Huang CK. Neutrophils lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl Acad Sci U S A. 2002;99:3603–3608. doi: 10.1073/pnas.052010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes IM, Jordan NJ, Towers S, Smith G, Paterson JR, Earnshaw JJ, et al. Human vascular smooth muscle cells express receptors for CC chemokines. Arterioscler Thromb Vasc Biol. 1998;18:397–403. doi: 10.1161/01.atv.18.3.397. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller R, Chang Q, Ehrlich G, Hsieh SN, Schoenwaelder SM, Kuhlencordt PJ, et al. Overlapping and distinct roles for PI3Kbeta and gamma isoforms in S1P-induced migration of human and mouse endothelial cells. Cardiovasc Res. 2008;80:96–105. doi: 10.1093/cvr/cvn159. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Horvath C, Welt FG, Nedelman M, Rao P, Rogers C. Targeting CCR2 or CD18 inhibits experimental in-stent restenosis in primates: inhibitory potential depends on type of injury and leukocytes targeted. Circ Res. 2002;90:488–494. doi: 10.1161/hh0402.105956. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Tung WH, Wu CY, Wang HH, Lin CC, Wang TS, et al. Thrombin induces EGF receptor expression and cell proliferation via a PKC(delta)/c-Src-dependent pathway in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2009;29:1594–1601. doi: 10.1161/ATVBAHA.109.185801. [DOI] [PubMed] [Google Scholar]
- Irani C, Goncharova EA, Hunter DS, Walker CL, Panettieri RA, Krymskaya VP. Phosphatidylinositol 3-kinase but not tuberin is required for PDGF-induced cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L854–L862. doi: 10.1152/ajplung.00291.2001. [DOI] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GE, Prigmore E, Calvez R, Hogan C, Dunn GA, Hirsch E, et al. Requirement for PI 3-kinase gamma in macrophage migration to MCP-1 and CSF-1. Exp Cell Res. 2003;290:120–131. doi: 10.1016/s0014-4827(03)00318-5. [DOI] [PubMed] [Google Scholar]
- Kalmes A, Daum G, Clowes AW. EGFR transactivation in the regulation of SMC function. Ann N Y Acad Sci. 2001;947:42–54. doi: 10.1111/j.1749-6632.2001.tb03929.x. discussion 54–45. [DOI] [PubMed] [Google Scholar]
- Kranzhofer R, Clinton SK, Ishii K, Coughlin SR, Fenton JW, 2nd, Libby P. Thrombin potently stimulates cytokine production in human vascular smooth muscle cells but not in mononuclear phagocytes. Circ Res. 1996;79:286–294. doi: 10.1161/01.res.79.2.286. [DOI] [PubMed] [Google Scholar]
- Laffargue M, Raynal P, Yart A, Peres C, Wetzker R, Roche S, et al. An epidermal growth factor receptor/Gab1 signaling pathway is required for activation of phosphoinositide 3-kinase by lysophosphatidic acid. J Biol Chem. 1999;274:32835–32841. doi: 10.1074/jbc.274.46.32835. [DOI] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Song XH. Bradykinin and bombesin rapidly stimulate tyrosine phosphorylation of a 120-kDa group of proteins in Swiss 3T3 cells. J Biol Chem. 1991;266:7746–7749. [PubMed] [Google Scholar]
- Liu B, Poon M, Taubman MB. PDGF-BB enhances monocyte chemoattractant protein-1 mRNA stability in smooth muscle cells by downregulating ribonuclease activity. J Mol Cell Cardiol. 2006;41:160–169. doi: 10.1016/j.yjmcc.2006.03.426. [DOI] [PubMed] [Google Scholar]
- Ma J, Wang Q, Fei T, Han JD, Chen YG. MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood. 2007;109:987–994. doi: 10.1182/blood-2006-07-036400. [DOI] [PubMed] [Google Scholar]
- Mehrhof FB, Schmidt-Ullrich R, Dietz R, Scheidereit C. Regulation of vascular smooth muscle cell proliferation: role of NF-kappaB revisited. Circ Res. 2005;96:958–964. doi: 10.1161/01.RES.0000166924.31219.49. [DOI] [PubMed] [Google Scholar]
- Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, et al. Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits. Circulation. 2002;105:2905–2910. doi: 10.1161/01.cir.0000018603.67989.71. [DOI] [PubMed] [Google Scholar]
- Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2009;47:168–180. doi: 10.1159/000250095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Poon M, Hsu WC, Bogdanov VY, Taubman MB. Secretion of monocyte chemotactic activity by cultured rat aortic smooth muscle cells in response to PDGF is due predominantly to the induction of JE/MCP-1. Am J Pathol. 1996;149:307–317. [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan Y, Maile LA, Ling Y, Graves LM, Clemmons DR. Insulin-like growth factor-I stimulates Shc-dependent phosphatidylinositol 3-kinase activation via Grb2-associated p85 in vascular smooth muscle cells. J Biol Chem. 2008;283:16320–16331. doi: 10.1074/jbc.M801687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott D, Zhu J, Zhou YF, Burnett MS, Zalles-Ganley A, Epstein SE. IL-6 is produced by splenocytes derived from CMV-infected mice in response to CMV antigens, and induces MCP-1 production by endothelial cells: a new mechanistic paradigm for infection-induced atherogenesis. Atherosclerosis. 2003;170:223–228. doi: 10.1016/s0021-9150(03)00295-8. [DOI] [PubMed] [Google Scholar]
- Sako H, Miura S, Iwata A, Nishikawa H, Kawamura A, Matsuo K, et al. Changes in CCR2 chemokine receptor expression and plasma MCP-1 concentration after the implantation of bare metal stents versus sirolimus-eluting stents in patients with stable angina. Intern Med. 2008;47:7–13. doi: 10.2169/internalmedicine.47.0315. [DOI] [PubMed] [Google Scholar]
- Spinetti G, Wang M, Monticone R, Zhang J, Zhao D, Lakatta EG. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2004;24:1397–1402. doi: 10.1161/01.ATV.0000134529.65173.08. [DOI] [PubMed] [Google Scholar]
- Tanimoto T, Lungu AO, Berk BC. Sphingosine 1-phosphate transactivates the platelet-derived growth factor beta receptor and epidermal growth factor receptor in vascular smooth muscle cells. Circ Res. 2004;94:1050–1058. doi: 10.1161/01.RES.0000126404.41421.BE. [DOI] [PubMed] [Google Scholar]
- Taubman MB, Rollins BJ, Poon M, Marmur J, Green RS, Berk BC, et al. JE mRNA accumulates rapidly in aortic injury and in platelet-derived growth factor-stimulated vascular smooth muscle cells. Circ Res. 1992;70:314–325. doi: 10.1161/01.res.70.2.314. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Vecchione C, Patrucco E, Marino G, Barberis L, Poulet R, Aretini A, et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kgamma. J Exp Med. 2005;201:1217–1228. doi: 10.1084/jem.20040995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin L, Foisy S, Giasson E, Lambert C, Moreau P, Meloche S. EGF receptor transactivation is obligatory for protein synthesis stimulation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2002;283:C446–C455. doi: 10.1152/ajpcell.00261.2001. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling – which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- Wysocki SJ, Zheng MH, Smith A, Lamawansa MD, Iacopetta BJ, Robertson TA, et al. Monocyte chemoattractant protein-1 gene expression in injured pig artery coincides with early appearance of infiltrating monocyte/macrophages. J Cell Biochem. 1996;62:303–313. doi: 10.1002/(sici)1097-4644(199609)62:3<303::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci (Lond) 2008;114:257–264. doi: 10.1042/CS20070228. [DOI] [PubMed] [Google Scholar]
- Zhong L, Chen WQ, Ji XP, Zhang M, Zhao YX, Yao GH, et al. Dominant-negative mutation of monocyte chemoattractant protein-1 prevents vulnerable plaques from rupture in rabbits independent of serum lipid levels. J Cell Mol Med. 2008;12:2362–2371. doi: 10.1111/j.1582-4934.2008.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Takayama Y, Boucher P, Tallquist MD, Herz J. LRP1 regulates architecture of the vascular wall by controlling PDGFRbeta-dependent phosphatidylinositol 3-kinase activation. PLoS One. 2009;4:e6922. doi: 10.1371/journal.pone.0006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.