

Abstract
BACKGROUND AND PURPOSE
The µ-conopeptide family is defined by its ability to block voltage-gated sodium channels (VGSCs), a property that can be used for the development of myorelaxants and analgesics. We characterized the pharmacology of a new µ-conopeptide (µ-CnIIIC) on a range of preparations and molecular targets to assess its potential as a myorelaxant.
EXPERIMENTAL APPROACH
µ-CnIIIC was sequenced, synthesized and characterized by its direct block of elicited twitch tension in mouse skeletal muscle and action potentials in mouse sciatic and pike olfactory nerves. µ-CnIIIC was also studied on HEK-293 cells expressing various rodent VGSCs and also on voltage-gated potassium channels and nicotinic acetylcholine receptors (nAChRs) to assess cross-interactions. Nuclear magnetic resonance (NMR) experiments were carried out for structural data.
KEY RESULTS
Synthetic µ-CnIIIC decreased twitch tension in mouse hemidiaphragms (IC50= 150 nM), and displayed a higher blocking effect in mouse extensor digitorum longus muscles (IC = 46 nM), compared with µ-SIIIA, µ-SmIIIA and µ-PIIIA. µ-CnIIIC blocked NaV1.4 (IC50= 1.3 nM) and NaV1.2 channels in a long-lasting manner. Cardiac NaV1.5 and DRG-specific NaV1.8 channels were not blocked at 1 µM. µ-CnIIIC also blocked the α3β2 nAChR subtype (IC50= 450 nM) and, to a lesser extent, on the α7 and α4β2 subtypes. Structure determination of µ-CnIIIC revealed some similarities to α-conotoxins acting on nAChRs.
CONCLUSION AND IMPLICATIONS
µ-CnIIIC potently blocked VGSCs in skeletal muscle and nerve, and hence is applicable to myorelaxation. Its atypical pharmacological profile suggests some common structural features between VGSCs and nAChR channels.
Keywords: cone snail venom, µ-conotoxin, voltage-gated sodium channel, nicotinic acetylcholine receptor, myorelaxant, twitch tension, NMR structure
Introduction
Venoms of the marine snails belonging to the Conoidean superfamily provide a diverse source of bioactive components that are useful as pharmacological tools and for drug discovery. With 800 described species and hundreds of bioactive ingredients in each venom, the venom of Conus species appears as one of the richest sources of naturally occurring peptides, exhibiting a wide array of biological activities (Olivera and Teichert, 2007; Halai and Craik, 2009). Conopeptides (or conotoxins) target numerous and diverse molecular entities with high affinity and specificity, including voltage- and ligand-gated ion channels, and G-protein-coupled receptors (McIntosh et al., 1999; Favreau and Stöcklin, 2009). Biomedical applications for conopeptides have attracted attention, such as the clinical use of ω-conotoxin MVIIA (Prialt®, Elan Corporation, Dublin, Ireland) for the treatment of severe chronic morphine-resistant pain (Miljanich, 2004). Human clinical trials of conopeptides have been reviewed by Terlau and Olivera (2004).
The µ-conopeptide family features a conserved cysteine pattern that causes a constrained tertiary structure, and leads to interaction with sodium channels as pore blockers (Ott et al., 1991; Wakamatsu et al., 1992; Hill et al., 1996; Nielsen et al., 2002). These bioactive peptides were mainly isolated from fish-hunting cone snail species. Previous studies have demonstrated that most µ-conopeptides preferentially target the muscle-type voltage-gated sodium channels (VGSCs) with less selectivity towards neuronal VGSCs (Zhang et al., 2006). The resulting pharmacological effect consists in a block of conductance, leading to loss of function of the neuromuscular system, and to the rapid paralysis of the prey during envenomation. This pharmacological activity is regarded as potentially useful for the development of new analgesics (Norton, 2010) and, more directly, as myorelaxants. A number of reviews have focused on the particular pharmacology, structures and possible uses of this family of cone snail peptides (Li and Tomaselli, 2004; Ekberg et al., 2008).
In the present work, we report the structure and pharmacological profile of the novel µ-conotoxin CnIIIC (from Conus consors). Following chemical synthesis of a peptide identical to the native product, the synthetic µ-conotoxin CnIIIC was tested on a number of preparations and molecular targets to assess its potential as a myorelaxant. Further structural study by nuclear magnetic resonance (NMR) also indicated interesting features of this new µ-conotoxin.
Methods
Bioassays
All animal care and experimental procedures were performed in accordance with the European legislation on animal experimentation. We used adult male Swiss-Webster mice (20–25 g body weight), wild male and female European pike (Esox lucius, 0.8–1.2 kg body weight), and adult female Xenopus laevis. Isolated tissues were obtained as previously described for mice (Schlumberger et al., 2010), and pike (Luzzati et al., 2000). Stage V–VI oocytes were isolated from X. laevis as previously mentioned (Liman et al., 1992). Fifteen specimens of C. consors were collected in Chesterfield Island (New Caledonia) and immediately frozen at −80°C.
Conus consors venom extraction and fractionation
The C. consors venom was obtained from 15 dissected venom ducts, extracted with 0.08% trifluoroacetic acid (TFA) in water and stored at −80°C until required. Fractionation of the crude lyophilized venom was performed using a high-pressure liquid chromatography system equipped with an ultraviolet detector. Elution solvents used for reverse-phase chromatography were the following: solvent A, H2O/0.1% TFA; and solvent B, H2O/CH3CN 40/60 0.1% TFA. Semi-preparative runs on the crude venom were performed with a C18 Vydac 218TP510 column using the gradient 0–8% B/5 min, 8–80% B/70 min, 80–100% B/10 min, followed by 100% B/10 min (flow rate, 2 mL/min). Further purification steps using an analytical C18 Vydac 218TP54 column were carried out with the following gradient 0–10% B/5 min, 10–20% B/10 min, 20–40% B/40 min. The effluent was monitored at 214 nm.
Electrospray ionization mass spectrometry (ESI-MS) and sequencing
Molecular mass measurements were performed on a quadrupole time-of-flight I instrument (Micromass/Waters, Manchester, UK) equipped with an electrospray ion source. Sample analysis was carried out in positive ionization mode using a carrier infusion solvent of H2O/CH3CN/HCOOH (49.9/49.9/0.2). Tandem mass spectrometry was carried out for structural investigations. In this configuration, collision-induced dissociation was performed by manually adjusting the collision energy. Samples were previously reduced using 100 mM dithiothreitol in an ammonium bicarbonate buffer (pH 7.8) at 56°C for 3 h. The reduced peptide was then desalted using a ZipTip (Millipore, USA), according to the manufacturer's protocol. Data acquisition and data analysis were performed with the MassLynx software (Micromass/Waters). Multiply-charged mass spectra were transformed into singly charged data using the MaxEnt3 option of MassLynx. Manual and semi-automatic data treatment was then operated for sequence characterization.
Chemical synthesis
The peptide was assembled using a ABI 433A peptide synthesizer adapted to Boc chemistry. Classical Boc protected amino acids were used during the assembly and deprotection. Cleavage from the resin was performed with HF. After purification by reverse-phase HPLC, the linear peptide purity and integrity were controlled by ESI-MS. Refolding was carried out using Tris 100 mM, guanidinium chloride 0.5 M and reduced/oxidized glutathione 0.5/0.1 mM. This mixture was stirred overnight, at room temperature, and was then acidified using acetic acid, and concentrated using a C18 SepPak (Micromass/Waters) cartridge following the manufacturer's protocols. The folded peptide was purified by reverse-phase chromatography at a semi-preparative scale. The final product was found to be homogeneous upon standard HPLC and MS squality control analyses.
Twitch tension recordings on isolated skeletal muscle
For isometric twitch tension measurements, mouse hemidiaphragms with their respective associated phrenic nerves, or the mouse extensor digitorum longus (EDL) muscles were carefully removed, and mounted in silicone-lined organ baths (4 mL volume) containing Krebs-Ringer solution of the following composition (in mM): 154 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES buffer and 11 glucose (pH 7.4), gassed with pure O2. For direct muscle stimulation an electrode assembly was placed along the length of the fibres and 20 µM d-tubocurarine (Sigma-Aldrich, Saint Quentin Fallavier, France) added to the medium to block nicotinic acetylcholine receptors (nAChRs) and thereby neuromuscular transmission. Muscle twitch tension recordings were evoked by current pulses of 0.15 ms duration and supramaximal intensity supplied at 0.1 Hz, as previously described (Schlumberger et al., 2010).
Electrophysiological recordings from isolated mouse and pike nerves
The sciatic nerves (left and right) were dissected from mice and kept in oxygenated Krebs-Ringer solution at room temperature for 30 min prior to recordings. Left and right olfactory nerves were collected and rinsed, for at least 30 min before recordings, with an oxygenated pike Ringer's solution containing (in mM) 82.5 NaCl, 2.5 KCl, 1 CaCl2, 1 Na2HPO4 buffer, 5 HEPES buffer and 1 MgCl2 (pH 7.3), at room temperature for a 30 min period prior to use. Electrophysiological recordings of the global nerve action potential (GAP) were obtained as described (Luzzati et al., 2000).
Electrophysiological recordings from HEK cells expressing recombinant VGSCs
Whole-cell patch-clamp recordings (Hamill et al., 1981) were performed at room temperature (20–22°C) on HEK 293 cells transiently expressing rat skeletal muscle NaV1.4, rat brain NaV1.2, mouse cardiac NaV1.5, neuronal mouse NaV1.6 and NaV1.7 VGSC, as described earlier (Chen et al., 2000). Rat TTX-resistant NaV1.8 channels were expressed in Neuro-2A cells and currents were measured in the presence of 1 µM TTX to block endogenous NaV channels as described by Schirmeyer et al. (2010). Channel and receptor nomenclature follows Alexander et al., (2011).
Electrophysiological recordings from Xenopus oocytes expressing nAChRs
Two-microelectrode voltage-clamp experiments were carried out using Xenopus oocytes prepared and injected as described previously (Hogg et al., 2008). Briefly, cDNA encoding for the human α3, α4, α7 and β2 subunits of nAChRs were cloned into pCR 3.1 or pRc/CMV expression vectors (Invitrogen, Carlsbad, CA, USA) and injected into the nucleus of the Xenopus oocytes (Hogg et al., 2008). Measurements were performed 2–3 days after injection. Currents were recorded using an automated process, as previously described (Hogg et al., 2008).
1H-NMR spectroscopy
NMR experiments were carried out on Bruker AVANCE 600 and 750 MHz spectrometers equipped with 5 mm triple-resonance z-gradient probes. Samples of µ-CnIIIC (1.3 mM) were examined at 278–298 K, pH 4, in 95/5% H2O/D2O and in 100% D2O. The following NMR experiments were acquired: 2-D total correlation spectroscopy (TOCSY) (Bax and Davis, 1985) with a spin-lock time of 80 ms; 2-D nuclear Overhauser effect spectroscopy (NOESY) (Jeener et al., 1979; Kumar et al., 1980) with mixing times of 250 ms in 100% D2O, and 75 and 300 ms in 95/5% H2O/D2O; double quantum-filtered correlation spectroscopy (COSY) (Rance et al., 1983) and exclusive COSY (Griesinger et al., 1987) in 100% D2O. Complex data matrices of 4096 × 600 points for TOCSY, for NOESY, and 8192 × 600 points for DQF-COSY and 40, 48, and 96 scans per t1 increment in the indirect detected dimension were used.
Structure calculations
Volumes of 529 cross-peaks were collected from 2-D NOESY spectra recorded at 5°C with 300 ms and 75 ms mixing times; at 20°C with a 250 ms mixing time and from 2-D NOESY spectra recorded in 100% D2O at 27°C with a 250 ms mixing time. All NMR spectra were processed using TopSpin 1.3 (Bruker) and analysed using SPARKY (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Scrutiny of NOESY spectra provided no evidence of non-transient self-association. Backbone ϕ angle restraints were derived from 3JHN-Hα coupling constraints (Pardi et al., 1984) measured from the DQF-COSY spectrum (3JHN-Hα > 8 Hz, ϕ=−120 ± 30°; 3JHN-Hα < 6 Hz, ϕ=−60 ± 30°). All other residues were restricted to a negative ϕ-value except for Gly2, Gly6, and Gly9, which were not restrained. The overall quality of the final structure was validated with WHATIF (Vriend, 1990) and PROCHECK (Morris et al., 1992). Eight χ-angles were derived from 3JHα-Hβ coupling constraints provided by the exclusive COSY spectrum in 100% D2O. Structures were generated using the torsion angle dynamics program CYANA 2.1 (Herrmann et al., 2002), and refined in explicit water solvent in the programme Cristallography and NMR System (CNS) (Brünger et al., 1998) using standard distance geometry and simulation annealing scripts. Residue pyroGlu was added into the CNS and CYANA topology libraries based on data available at http://xray.bmc.uu.se/hicup/PCA/. Sequential Hδi-Hαi-1 NOEs indicated unique trans conformation for the sole Pro residue. Pseudo-distance restraints of 3.1 Å for Cβ-Sγ bonds and 2.1 Å for Sγ-Sγ bonds were added for all three disulfide bonds Cys3-Cys15, Cys4-Cys21 and Cys10-Cys22, based on the sequence alignment of µ-CnIIIC to closely related µ-conotoxins (Table 1) and the proximity and geometry of Cys sidechains in an inititial set of structure calculations using CYANA2.1 that did not incorporate disulfide restraints.
Table 1.
Amino acid sequence of µ-conotoxins, origin and known targets to date
| Name | Sequence | Clade/species | Target | References |
|---|---|---|---|---|
| Gastridium | ||||
| µ-GIIIA | RDCCTPP--KK-CKDRQCKPQ-RCCA* | Conus geographus | NaV1.4 | Cruz et al. (1985) |
| µ-GIIIB | RDCCTPP--RK-CKDRRCKPM-KCCA* | C. geographus | NaV1.4, NaV1.2, NaV1.3 | Cruz et al. (1985); Lewis et al. (2007) |
| µ-GIIIC | RDCCTPP--KK-CKDRRCKPL-KCCA* | C. geographus | NaV1.4 | Cruz et al. (1985) |
| µ-TIIIA | RHGCCKGP--KG-CSSRECRPQ-HCC* | C. tulipa | NaV1.4, NaV1.2 | Lewis et al. (2007) |
| Chelyconus | ||||
| µ-PIIIA | ZRLCCGFP--KS-CRSRQCKPH-RCC* | C. purpurascens | NaV1.4, NaV1.2, hNaV1.7 | Safo et al. (2000); Nielsen et al. (2002) |
| Pionoconus | ||||
| µ-CnIIIA | GRCCDVP--NA-CSGRWCRDHAQCC* | C. consors | fTTX-R | Zhang et al. (2006) |
| µ-CnIIIB | QGCCGEP--NL-CFTRWCRNNARCCRQQ | C. consors | fTTX-R | Zhang et al. (2006) |
| µ-CnIIIC | ZGCCNGP--KG-CSSKWCRDHARCC* | C. consors | NaV1.4, NaV1.2, α3β2 nAChRs | This work |
| µ-CIIIA | GRCCEGP--NG-CSSRWCKDHARCC* | C. catus | fTTX-R, fTTX-S | Zhang et al. (2006) |
| µ-MIIIA | QGCCNVP--NG-CSGRWCRDHAQCC* | C. magus | fTTX-R | Zhang et al. (2006) |
| µ-SIIIA | QNCCN----GG-CSSKWCRDHARCC* | C. striatus | NaV1.2, NaV1.4, fTTX-R | Bulaj et al. (2005); Schroeder et al. (2008) |
| µ-SIIIB | QNCCN----GG-CSSKWCKGHARCC* | C. striatus | NaV1.4, NaV1.2 | Schroeder et al. (2008) |
| Textilia | ||||
| µ-BuIIIA | VTDRCC--K-GKRECG-RWCRDHSRCC* | C. bullatus | NaV1.4 | Holford et al. (2009) |
| µ-BuIIIB | VGERCC--KNGKRGCG-RWCRDHSRCC* | C. bullatus | NaV1.4 | Holford et al. (2009) |
| µ-BuIIIC | IVDRCCNKGNGKRGCS-RWCRDHSRCC* | C. bullatus | NaV1.4 | Holford et al. (2009) |
| µ-SmIIIA | QRCCNG---RRGCSSRWCRDHSRCC* | C. stercusmuscarum | fTTX-R | West et al. (2002) |
| Asprella | ||||
| µ-KIIIA | CCN-------CSSKWCRDHSRCC* | C. kinoshitai | NaV1.2, NaV1.4, NaV1.1, NaV1.6, NaV1.7, fTTX-R | Bulaj et al. (2005); Zhang et al. (2007) |
Indicates C-terminal amidation. Sequence alignment was manually performed and cysteine residues are highlighted in blue.
All target channels are from rat except hNaV1.7 (human), fTTX-R (frog) and mNaV1.7 (mouse).
Data analysis
Responses are expressed as percentage of control, with data presented as the mean ± SEM, and unless otherwise noted, statistical comparisons were made using either one-way anova, or a two-tailed unpaired t-test for non-paired comparisons.
Other µ-conopeptides used
µ-TIIIA from Conus tulipa (Lewis et al., 2007), µ-SIIIA from C. striatus (Schroeder et al., 2008), µ-SmIIIA from C. stercusmuscarum and µ-PIIIA from C. purpurascens (Keizer et al., 2003), and µ-T3.1 from C. tulipa (Olivera et al., 2004) were synthesized according to the cited references.
Results
Isolation, purification, characterization and synthesis of a novel µ-conopeptide
The dried dissected venom of C. consors was subjected to reverse-phase HPLC and provided a complex chromatogram, analyzed by absorbance at 214 nm (Figure 1). A fraction eluting at approximately 28 min demonstrated a potent blocking activity of muscle contraction in mouse hemidiaphragm neuromuscular preparations. The fraction was further purified to homogeneity and displayed a single monoisotopic molecular mass of 2374.10 Da. The compound was unsuccessfully subjected to Edman degradation, indicating a probable blocked N-terminus. After disulfide bridge reduction, the conopeptide was analysed by tandem mass spectrometry (MS/MS). Selection and collision-induced dissociation of the ion at m/z 596 ([M+4H+]/4) allowed complete fragmentation of the peptide. Manual interpretation of the data led to the assignment of a highly probable sequence bearing 22 amino acids. The sequence shared homology with previously isolated µ-conopeptides and, based on preliminary pharmacological data, was named µ-CnIIIC (Table 1). Synthesis of the linear peptide was carried out by conventional Boc chemistry and folding was performed using a reduced/oxidized glutathione mixture. This led to a major folded peptide (35% yield from the linear form) that was shown to be identical to the native material by HPLC co-elution experiments and ESI-MS/MS data. The synthetic µ-CnIIIC was used for all biological assays and structural investigations.
Figure 1.
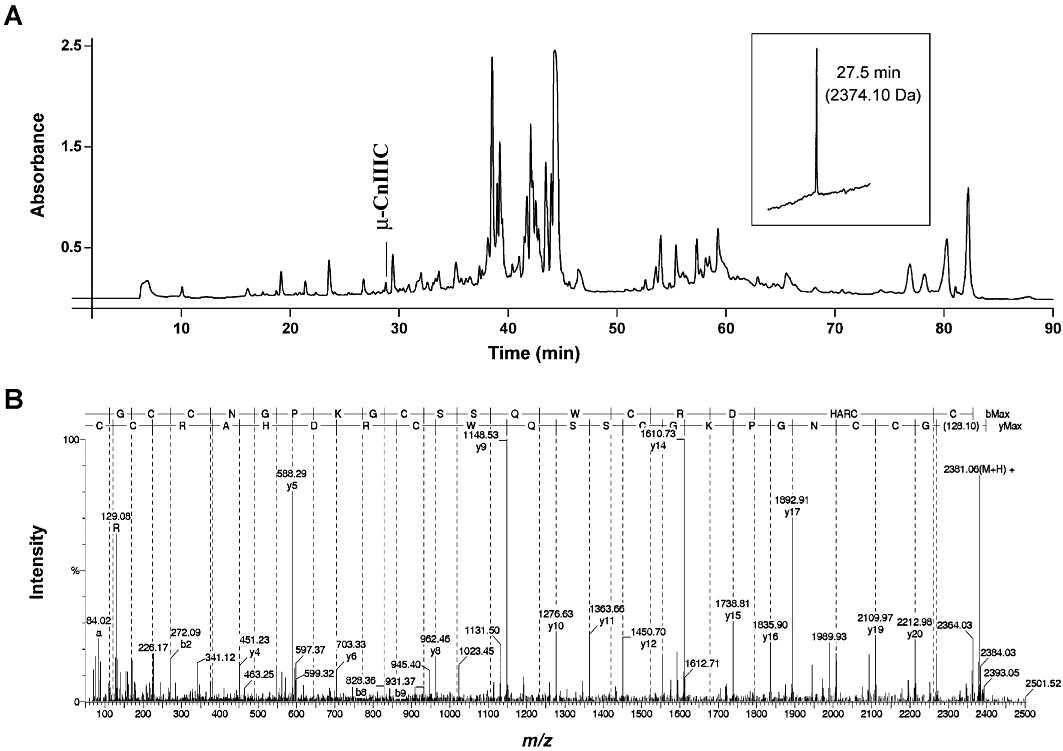
Isolation and characterization of µ-CnIIIC. (A) Reverse-phase HPLC chromatogram (monitored by absorbance at 214 nm) of the venom duct extract from Conus consors. The peak eluting at ∼28 min contained µ-CnIIIC that was purified to homogeneity at an analytical scale and showed a single monoisotopic molecular mass (inset). The peptide was then subjected to reduction and mass spectrometry analysis. (B) ESI-MS/MS spectrum of the the m/z 596 undergoing collision-induced dissociation. The good fragmentation coverage allowed complete amino acid sequence assignment with y and b ions.
Effect of µ-CnIIIC on skeletal muscle contraction
The activity of µ-CnIIIC was first assessed on isolated mouse hemidiaphragm muscles by recording isometric twitch tension in response to direct electrical stimulation. For each µ-CnIIIC concentration, muscle contraction recordings were carried out 2 h after peptide application and in the presence of 20 µM d-tubocurarine. Contraction was diminished in the presence of 0.1 µM µ-CnIIIC and was completely inhibited by 0.6 µM of the peptide (Supporting Information Figure S1). By comparison, a concentration of at least 2 µM of µ-conopeptide GIIIA or µ-conopeptide GIIIB was necessary for complete inhibition in the same preparation and under identical conditions. µ-CnIIIC thus appeared at least three times more potent than these two latter µ-conopeptides tested in this in vitro model. The concentration–response curve yielded a half-maximal inhibition concentration of 150 nM µ-CnIIIC (Supporting Information Figure S1).
The effect of µ-CnIIIC on twitch tension was also studied on the mouse EDL muscle, and was compared with that of other µ-conopeptides (µ-TIIIA, µ-T3.1, µ-PIIIA, µ-SmIIIA, µ-SIIIA) (Figure 2A). For this purpose, concentration–response curves were generated in individual muscles (the contraction measured in the presence of various concentrations of a given µ-conotoxin being expressed as percent of the control twitch response). Each µ-conopeptide concentration was applied by perfusion and allowed to equilibrate for at least 45–60 min. IC50 values for each µ-conopeptide (Figure 2B) showed the following decreasing order of potency: µ-CnIIIC ∼µ-TIIIA > µ-SIIIA > µ-SmIIIA > µ-PIIIA > µ-T3.1. Therefore, we concluded that the inhibitory potential of µ-CnIIIC was equal to, or greater than, that of the other µ-conopeptides in this preparation.
Figure 2.
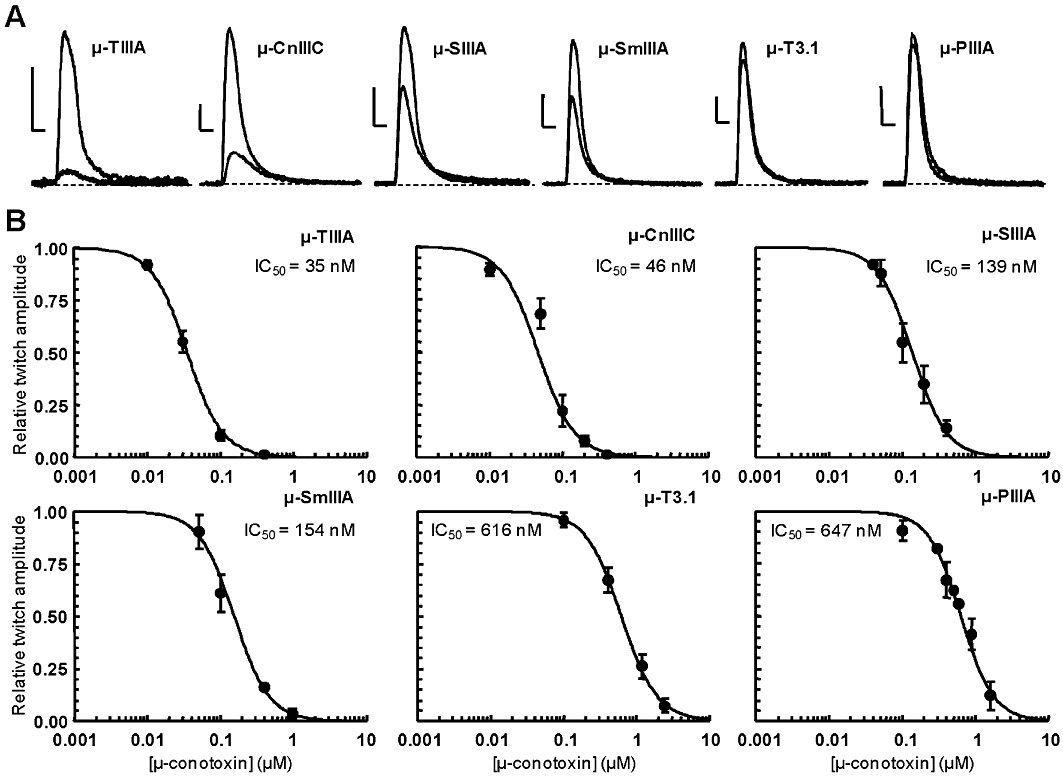
Effects of different µ-conotoxins on isometric twitch tension elicited by direct electrical stimulation of isolated mouse EDL muscles. (A) Effects of µ-conotoxins on skeletal muscle contraction. Contractions evoked by direct muscle stimulation and recorded in the absence and presence of 0.1 µM µ-conotoxins. Scale bars: 0.2 g (vertical) and 0.1 s (horizontal). (B) Concentration–response curves of peptide effects on directly elicited muscle contraction. For each µ-conopeptide concentration, the maximal twitch peak amplitude is expressed relative to its control value as the mean ± SEM of three to six experiments. The theoretical curves were calculated from typical sigmoidal nonlinear regression through data points (correlation coefficient r2≥ 0.948). The µ-conopeptide concentration that inhibited 50% of the twitch tension (IC50) is indicated for each peptide studied.
Effects of µ-CnIIIC on the GAP of mouse sciatic and pike olfactory nerves
In addition to the well-established effect of µ-conopeptides on skeletal muscle, we investigated the effect of µ-CnIIIC on myelinated and unmyelinated nerves from mouse and pike, respectively. Indeed, some µ-conotoxins have previously been shown to interact with neuronal sodium channels that are present in such preparations (Table 1). Increasing concentrations of µ-CnIIIC were found to decrease the GAP amplitude, which was almost nil when the sciatic and olfactory nerves were treated for 30–60 min with 50 and 10 µM of conotoxin, respectively (Figure 3A–D). The stimulation intensity necessary to reach 50% of maximum GAP amplitude was enhanced with increasing concentrations of µ-CnIIIC, while the propagation velocity of the GAP was not markedly modified. Altogether, these results conclusively show that µ-CnIIIC acts on mouse sciatic and pike olfactory nerves by decreasing individual fibre responses.
Figure 3.
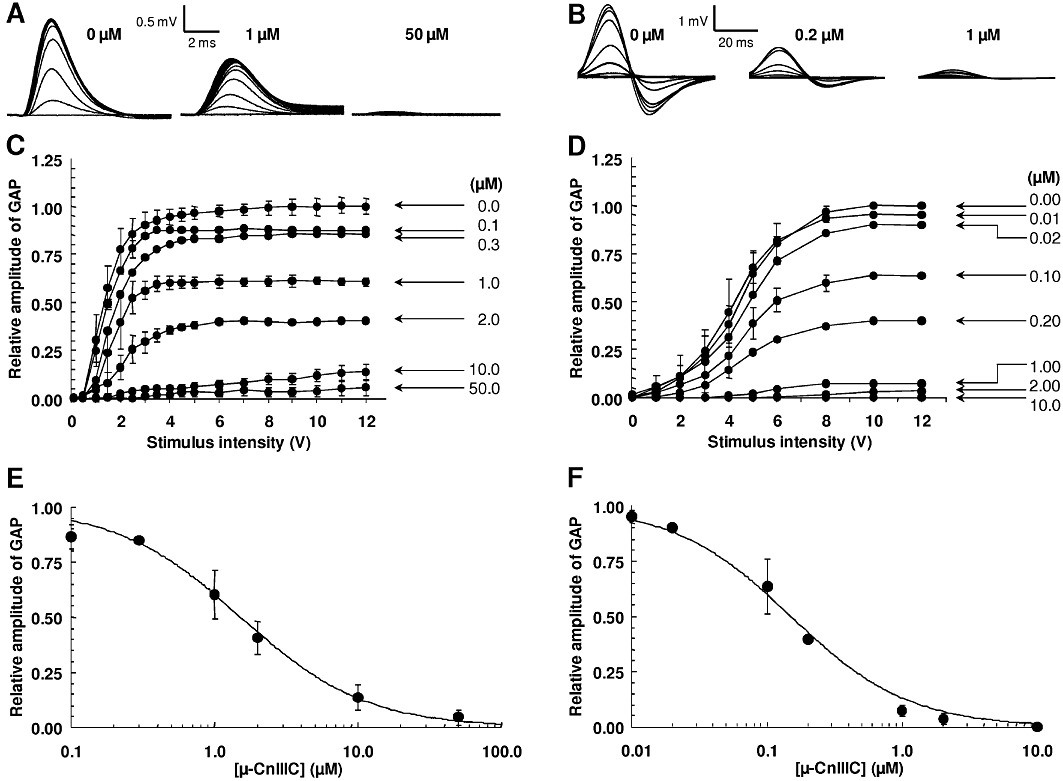
Effect of µ-CnIIIC on the GAP recorded on isolated mouse sciatic and pike olfactory nerves. (A–B) Traces of GAP recorded in response to 0.05 ms (mouse) and 8 ms (pike) and 0.1–15 V stimulations, under control conditions and after treatment with µ-CnIIIC at the indicated concentrations. (C–D) GAP amplitude in response to different intensities of 0.05 and 8 ms stimulations and to different concentrations of µ-CnIIIC. (E–F) GAP amplitude in the presence of various concentrations of µ-CnIIIC, and expressed relative to control values as mean ± SEM of three to six mouse sciatic and three to four pike olfactory nerves. The curves were calculated from typical sigmoidal nonlinear regression through data points (r2≥ 0.984). The µ-CnIIIC concentrations required to block 50% of the GAP amplitude (IC50) was 1.53 µM (mouse) and 0.15 µM (pike).
The concentration–response curves of the effects of µ-CnIIIC revealed that it reduced the GAP amplitude of sciatic and olfactory nerves by 50% at concentrations of 1.53 and 0.15 µM, respectively (Figure 3E, F), demonstrating a 10-fold higher potency on pike unmyelinated axons compared with mouse myelinated axons. Results also indicated that µ-CnIIIC was about 1000 times more potent than classical anaesthetics such as lidocaine on the mouse sciatic nerve, since millimolar concentrations of lidocaine were necessary to obtain a similar inhibitory effect (data not shown).
The reversibility of the effect of µ-CnIIIC was evaluated by recording the GAP amplitude of sciatic and olfactory nerves at various times from 2 to 24 h, following wash-out of physiological solutions with or without 1–50 µM µ-CnIIIC. When compared with controls, only a moderate increase in the GAP amplitude of the nerves treated with µ-CnIIIC was observed upon washing, even after 24 h. To test if the absence of reversibility was not due to a spontaneous decrease of the GAP amplitude over time, the nerves were bathed in conotoxin-free fresh solution for 2–24 h. No significant modification of the GAP amplitude was observed up to 24 h. Overall, these results strongly suggested that µ-CnIIIC was bound strongly to a receptor on the mouse sciatic and pike olfactory nerves, and that the dissociation of the conopeptide/receptor complex occurred slowly.
Effect of µ-CnIIIC on VGSCs
In order to identify the molecular target of µ-CnIIIC, the peptide was tested on rat skeletal muscle NaV1.4 channels, which were heterologously expressed in HEK 293 cells. Current responses at −20 mV were repeatedly recorded before and during local toxin application. µ-CnIIIC (1 µM) blocked NaV1.4 currents to leave 5.5 ± 0.5% (n= 23) of the current remaining; the remaining current exhibited the same kinetics as the control current (Figure 4A). Current inhibition did not reverse even after extensive washing with control saline (Figure 4B). A prominent characteristic feature of µ-CnIIIC was the slow onset of block (Figure 4B) at 1 µM, which followed a single-exponential time constant of 238 ± 22 s (n= 23). The inverse of this time constant and the remaining currents are shown for various µ-CnIIIC concentrations in Figure 4C, indicating that the on-rate becomes prohibitively slower at low toxin concentrations, which still would efficiently block the channels. Assuming a monomolecular reaction, we estimated an apparent IC50 value of 1.3 ± 0.4 nM and a non-blockable channel fraction of 5.0 ± 0.1% (data fit in Figure 4C). µ-CnIIIC at 1 µM also almost completely blocked rat brain NaV1.2 channels without affecting the kinetics of the remaining current (Figure 4D; remaining current: 9.9 ± 1.2%, n= 5). The rate of onset of block, however, was eight times slower than for NaV1.4 (1770 ± 171 s, n= 5). Similarly, µ-CnIIIC (500 nM) also blocked rat NaV1.4 channels expressed in Xenopus oocytes and sodium currents recorded from mouse neuroblastoma cells (data not shown).
Figure 4.
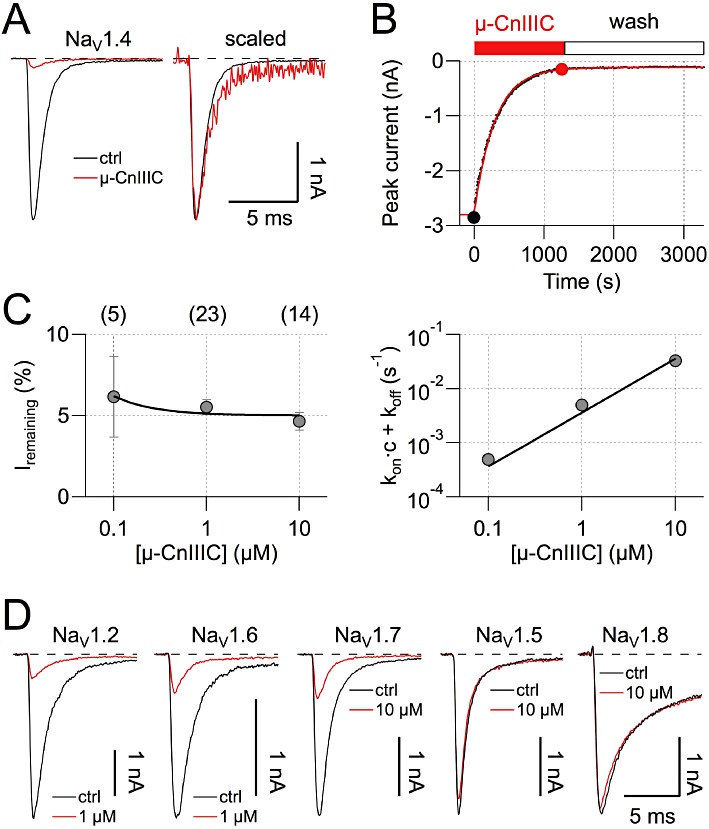
Effect of µ-CnIIIC on voltage-gated sodium channels. (A) Sodium currents at −20 mV recorded from HEK 293 cells expressing rat NaV1.4 channels, under control conditions and in the presence of 1 µM µ-CnIIIC (red). Holding potential was −120 mV, leak was corrected with a p/6 method. The panel on the right shows currents in the presence of toxin scaled and superimposed on the control current, indicating a lack of kinetic changes. (B) NaV1.4 peak current inhibition by µ-CnIIIC and wash-out with a conotoxin-free medium shown as a function of time. The continuous red curve is a single exponential; filled circles indicate data points corresponding to traces shown in (A). (C) The remaining current after toxin equilibration (left) as a function of µ-CnIIIC concentration and the inverse of the time constant characterizing the onset of block (right) as a function of µ-CnIIIC concentration. The number of experiments is indicated in parentheses. The continuous lines are global data fits assuming a first-order reaction yielding an apparent IC50 value of 1.3 ± 0.4 nM and a negligible off-rate. Error bars indicate SEM. (D) Current traces as in A for the indicated channel types before (black) and after application of µ-CnIIIIC at the indicated concentration (red). Traces for rat NaV1.8 channels were recorded from Neuro-2A cells at 10 mV in the presence of 1 µM TTX.
In contrast, mouse cardiac NaV1.5 and TTX-resistant rat NaV1.8 channels were practically resistant to µ-CnIIIC (Figure 4D, remaining current in 10 µM µ-CnIIIC: 78 ± 5.7%, n= 4 and 88.9 ± 1.3%, n= 6, respectively), while neuronal mouse NaV1.6 and NaV1.7 channels were of intermediate sensitivity (Figure 4D, remaining current in 10 µM µ-CnIIIC: 15.5 ± 1.9%, n= 6 and 16.1 ± 4.1%, n= 6, respectively; the respective onsets of block were 119 ± 30 s and 1460 ± 144 s).
µ-CnIIIC blocks neuronal nAChRs
In order to extend the pharmacological profile of µ-CnIIIC, its activity on voltage-gated potassium channels and nAChRs was investigated. These targets were selected as they are directly involved in signal transduction, as are VGSCs. Results from voltage-gated potassium channel subtypes (rKV1.1, rKV1.2, hKV1.3, rKV1.4, rKV1.5, rKV1.6, Shaker, hERG1), expressed in Xenopus oocytes, demonstrated no activity of µ-CnIIIC at concentrations up to 5 µM. Assays were also carried out in humans α7, α4β2 and α3β2 nAChR subunits expressed in Xenopus oocytes. Measurements were performed at −100 mV and channels were activated by 200 µM ACh. Application of several µ-CnIIIC concentrations, ranging from 1 nM to 10 µM, resulted in a weak inhibition of nAChR α7 channels (IC50= 22 µM, nH= 0.8; n= 7) (Figure 5A1–B1), whereas nAChR α4β2 channels were inhibited more strongly (IC50= 2.9 µM, nH= 0.6; n= 17) (Figure 5A2–B2). In both cases, inhibition was completely reversible. Interestingly, similar experiments performed on nAChR α3β2 showed a strong inhibition in the high nanomolar range (IC50= 450 nM, nH= 1; n= 8) (Figure 5A3–B3), which was only partially reversible.
Figure 5.
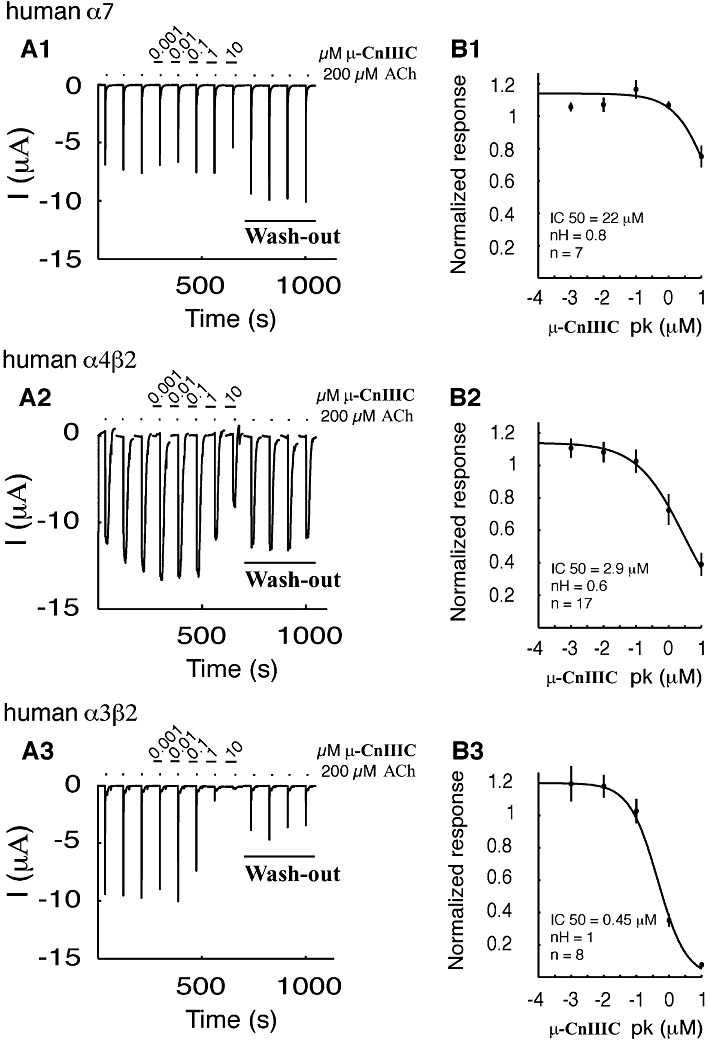
Effect of µ-CnIIIC on nAChRs expressed in Xenopus oocytes. (A1-A3) presents the inhibition of acetylcholine-induced currents on human α7 (A1), α4β2 (A2) and α3β2 (A3) nAChRs. All recordings were carried out at −100 mV and the first three traces are controls followed by concentration-dependent response to 2 min exposure to different concentrations of µ-CnIIIC, ranging from 1 nM to 10 µM. Each experiment was terminated by wash-out steps for a time period of 8 min. Complete wash-out was observed for α7 and α4β2, while only partial recovery occurred for α3β2. (B1–3) Inhibition curves of the fitted data. IC50, Hill coefficient (nH) and experimental numbers are presented under the fitted curve for α7 (B1), α4β2 (B2) and α3β2 (B3). Error bars indicate SEM.
3-D structure of µ-CnIIIC compared with other µ-conopeptides
The solution structure of µ-CnIIIC was characterized using NMR spectroscopy and molecular modelling. One- and two-dimensional 1H NMR spectra recorded on µ-CnIIIC showed good signal dispersion and indicated the presence of a single homogenic conformer. Secondary Hα chemical shifts (Wishart et al., 1991) and inter-residue NOE patterns were used to examine the secondary structure of µ-CnIIIC in solution.
A C-terminal α-helix is predicted in the third consensus loop (Supporting Information Figure S2), between residues 13–18, a feature that is commonly found in the solution structures of conotoxins from the M-5 branch of the M-superfamily. An additional short α-helix is predicted between residues 9–11, a feature also found in the solution structure of µ-PIIIA. The appearance of medium-range NOEs observed over residues 4–9 and 19–22 suggested the presence of β- or γ-turns. A backbone superimposition of the 20 lowest energy structures, for which no NOE violations greater than 0.3 Å nor dihedral violations greater than 5° were observed, revealed a well-defined NMR ensemble over the entire length of the polypeptide, with the exception of N-terminal residues Z1 and G2 (Figure 6A) and the N-terminal segment G6-K8. The root mean square deviation (RMSD) calculated from the mean structure between C3 and C22 yielded 0.47 Å for the backbone-heavy atoms and 1.37 Å for the side chains heavy atoms. Furthermore, 90% of the ϕ and ψ torsion angle values lie in the most favoured regions of the Ramachandran plot, whereas no values are in the disallowed regions. The predicted C-terminal α-helix and the shorter α-helix preceding it are observable in the lowest energy structure of the ensemble (Figure 6B).
Figure 6.
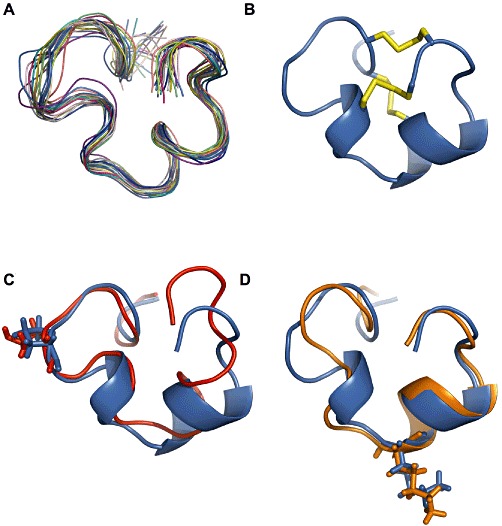
Structure of µ-CnIIIC. (A) Backbone superimposition over residues 3–22 for the 20 lowest energy structures (ribbon). (B) Lowest energy structure with the disulfide bridge shown in yellow. (C) Backbone superimposition of N-terminal region (residues 3–11) of µ-CnIIIC (blue) with N-terminal region (residues 4–12) of µ-TIIIA (red); Pro7 and Hyp8 of µ-CnIIIC and µ-TIIIA respectively are highlighted (sticks). (D) Backbone superimposition of C-terminal region (residues 12–22) of µ-CnIIIC (blue) with C-terminal region (residues 10–20) of µ-SIIIA (orange); Lys13 and Lys11 of µ-CnIIIC and µ-SIIIA respectively are highlighted (sticks). Structural data were used from biological magnetic resonance bank (BMRB) accession codes 20024 (TIIIA) and 20025 (SIIIA). µ-CnIIIC structural data can be accessed with protein databank (PDB) code 2YEN and BMRB accession code 17581.
Structural homology with α-conotoxins
A striking similarity between the backbone trace of α-conotoxins and µ-conotoxins was revealed when the Cα atomic coordinates of the second, third and fourth cysteines of α-conotoxins were aligned with the third, fourth and sixth cysteines of µ-conotoxins respectively. It appears that the second disulfide bridge of α-conotoxins fulfills the same structural role as the second disulfide bridge of µ-conotoxins. However, while the Cα atomic coordinates of the third cysteine in α-conotoxins and the fourth cysteine in µ-conotoxins overlap, their disulfide-forming cysteine partners are not at equivalent positions. Of all α-conotoxin structures available to date, α-AuIB provides the closest match to µ-CnIIIC (RMSD 0.98 Å), followed by α-Vc1.1 (RMSD 1.21 Å) and α-PnIA (RMSD 1.33 Å) (Figure 7). The first α-conotoxin belongs to the 4/6 subclass, in which the second inter-cysteine loop contains six amino acids, while the last two α-conotoxins belong to the 4/7 subclass where seven amino acids are present in the equivalent loop. The structure of the C-terminal region of this shorter loop in α-AuIB, but not that of α-PnIA nor α-Vc1.1, closely resembles that of the C-terminal loop in µ-CnIIIC.
Figure 7.
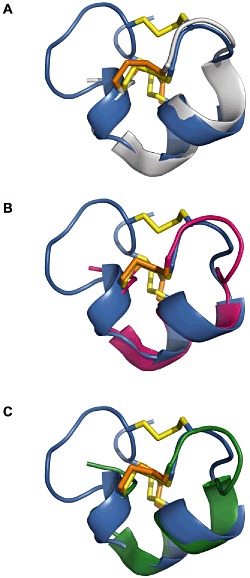
Structure comparison of µ-CnIIIC with α-conotoxins. (A–C) Backbone superimposition over Cα atoms of µ-CnIIIC (blue) with α-AuIB (grey), α-PnIA (magenta) and α-Vc1.1 (green). Disulfide bonds are shown in yellow for µ-CnIIIC and orange for the α-conotoxins. Protein databank (PDB) codes are 1DG2 (AuIB), 1PEN (PnIA) and 2H8S (Vc1.1).
Discussion and conclusions
Venom conopeptide sequences are usually derived from Edman sequencing and from DNA information, or a combination thereof. In this study, the fully validated µ-CnIIIC sequence was solely based on tandem mass spectrometry and de novo sequencing, supporting such approaches as a convenient route for natural peptide discovery (Quinton et al., 2006; Menin et al., 2008; Ueberheide et al., 2009). Along with minimal material use, this approach also added valuable information regarding post-translational modifications often found in cone snail venoms (Buczek et al., 2005).
While sharing between 50 and 90% sequence homology with other well-known µ-conotoxins, µ-CnIIIC displays an original pharmacological profile compared with previously described conotoxins in this family. µ-CnIIIC was found to strongly inhibit mouse muscle contraction, and was among the most potent of µ-conotoxins in this in vitro model. In addition, µ-CnIIIC demonstrated a 10-fold higher potency on pike unmyelinated axons compared with mouse myelinated axons. This difference in potency may be due to the distinct VGSC subtypes present in such axons (NaV1.2 in the pike olfactory nerve and NaV1.6 in the nodes of Ranvier of the mouse sciatic nerve) (Caldwell et al., 2000).
At the molecular level, these potencies are further reflected by the low apparent IC50 (1.3 nM) towards the rat muscle NaV1.4 channels. The complete block of the neuronal NaV1.2 channels at 1 µM was another original feature, whereas the cardiac NaV1.5 and DRG-specific NaV1.8 channels remained unaffected at this concentration. TTX-sensitive VGSCs expressed in the peripheral nervous system, mouse NaV1.6 and NaV1.7, displayed intermediate sensitivity. The extremely long-lasting activity supported by the virtually irreversible effect on VGSCs was another essential characteristic of µ-CnIIIC. This particular pharmacological profile and unprecedented long-lasting mode of action opens opportunities for new biomedical applications by acting atypically and almost irreversibly on the skeletal muscle VGSCs, as well as on unmyelinated and myelinated nerve fibres.
Unexpectedly, µ-CnIIIC also demonstrated some inhibitory activity at particular neuronal nAChR subtypes. The highest potency measured for nAChR inhibition in response to acetylcholine was in the IC50 range of ∼450 nM for α3β2 nicotinic receptors, which is relatively modest in comparison with α-conotoxin MII (Cartier et al., 1996) or α-conotoxin AnIB (Loughnan et al., 2004), which exhibit IC50 values of 0.5 and 0.3 nM respectively. In addition, the effective concentration for blocking nAChRs was more than two orders of magnitude greater than for NaV1.4 channels. This unexpected result led us to investigate the structural features of µ-CnIIIC in more detail, in particular, the similarities with nAChR-specific conopeptides.
The weak activity of µ-CnIIIC on nAChRs is reminiscent of the ψ-conotoxins, which share the same cysteine pattern and which are non-competitive inhibitors of mouse skeletal muscle and Torpedo electric organ nAChRs (Shon et al., 1997; Lluisma et al., 2008). The structure of ψ-PIIIE was shown to correlate well with typical µ-conotoxins, such as µ-GIIIA or µ-GIIIB (Mitchell et al., 1998), and it was suggested that the disulfide bond pattern was sufficient for keeping a CSα (Cysteine-Stabilized alpha-helical) motif, regardless of the inter-cysteine amino acid sequence. However, it should be noted that µ-CnIIIC does not show any primary sequence overlap with ψ-PIIIE or ψ-PrIIIE and does not inhibit the muscle-type nAChRs, as nAChR sensitivity was not altered at the mouse hemidiaphragm neuromuscular junction, at least up to 600 nM.
A comparison of the 3-D backbone trace of µ-CnIIIC with that of other µ-conotoxins revealed common features that may provide clues as to their common modes of action. The most obvious conserved feature is the positioning of the three disulfide bridges. A recent study on µ-KIIIA demonstrated that the removal of the disulfide bridge Cys1-Cys9 (equivalent to Cys3-Cys15 in µ-CnIIIC) did not significantly alter the biological activity of µ-KIIIA on its target VGSC, while the reverse was true for the removal of any of the other disulfide bridges (Han et al., 2009). Another common feature is the position of a basic residue (Lys13 in µ-CnIIIC) at the base of the well-conserved C-terminal α-helix in the penultimate intercysteine loop between the third and fourth cysteine (Figure 6D). This basic residue has been demonstrated to be critical to the activity of several µ-conotoxins (Sato et al., 1991; Keizer et al., 2003; Lewis et al., 2007; Zhang et al., 2007), and has been suggested to have a pore-blocking role in the interaction of µ-conotoxins with the affected VGSC. As well as having the highest sequence identity with µ-CnIIIC (Table 1), µ-SIIIA is also the closest structural homolog to µ-CnIIIC over the entire Cα backbone (RMSD 1.14 Å) and over the C-terminal half (Lys11 to Cys20) of the Cα backbone (RMSD 0.63 Å) (Figure 6C). A study on the dynamic properties of µ-SIIIA highlighted the rigidity of the C-terminal region (including at the putatively critical lysine), but also revealed flexibility on subnanosecond time scale for the N-terminal residues (pyroGlu1 and Asn2), as well as for Ser9, immediately following the third cysteine (Yao et al., 2008). While these observations cannot be directly extrapolated to µ-CnIIIC based on structural homology alone, the low level of convergence of the structural ensemble of µ-CnIIIC over the same regions is consistent with the dynamics analysis of µ-SIIIA (except over residues 7–8 in µ-CnIIIC, which are absent in µ-SIIIA). In a comparison of the selectivity profiles of the closest structural homologues to µ-SIIIA (i.e.µ-KIIIA and µ-SmIIIA), the same study suggested that the C-terminal region of these µ-conotoxins plays a critical role in blocking neuronal subtypes of VGSCs. Excluding the unstructured two N-terminal residues, the µ-conotoxin TIIIA is the closest structural homolog (RMSD 0.78 Å) to µ-CnIIIC over the Cα backbone of N-terminal residues – Gly3 to Lys9– (Figure 6C). Neither µ-CnIIIC, µ-TIIIA nor µ-PIIIA show evidence of conformational exchange over this loop region and prolines or hydroxyprolines for all three µ-conotoxins are in the trans configuration. The positioning of a highly conserved hydroxyproline or proline (Pro7 in µ-CnIIIC) in the first Cys loop (between the second and third cysteine) is conserved between µ-CnIIIC, µ-TIIIA and µ-PIIIA, and could be critical in constraining the loop orientation for this region. However, the substitution of Hyp7 to Pro in µ-GIIIA did not significantly alter the folding properties nor the activity of this conopeptide towards NaV1.4 channels, while a Hyp17 to Pro substitution in the C-terminal helix reduced activity 10-fold (Lopez-Vera et al., 2008).
Based on its functional similarity with some neuronal α-conotoxins, it is tempting to speculate that the CSα region and the adjacent residues of µ-CnIIIC support the interaction site with nAChRs. On the one hand, the recent work of Schroeder et al., (2008) with µ-SIIIA demonstrated that critical residues for the interaction with NaV1.2 and NaV1.4 channels were located in a similar α-helical region, spanning from residues 11–16. On the other, the interaction site for α-conotoxins is provided by this unique structural platform constrained by the two disulfide bridges. Recently, an original α-conotoxin isolated from C. litteratus was found to lack some of the residues previously considered essential for the interaction with the α3β2 nicotinic receptors (Luo et al., 2010). This revealed a potential new interaction microsite between the toxin and its receptor. These results point to the potential diversity in residues supported by the α-conotoxin framework for its activity, which could also be exhibited by a similar region of µ-CnIIIC. More data are needed to support this hypothesis, but the present results undoubtedly provide a rationale for mutational studies and chimers between the α- and µ-conotoxin structures and provide new insights into their interaction with nAChRs and VGSCs. This might also be an important aspect when considering the molecular target underlying some pharmacological activity such as the analgesic activity observed for µ-KIIIA that acts primarily on NaV1.2 channels (Khoo et al., 2009). Interestingly, preliminary data indicated that the ability of µ-CnIIIC to block nAChRs is not unique, but also shared by several other µ-conopeptide members.
The fact that a conotoxin targets multiple pharmacologically distinct ion channels- or ligand-gated receptors remains atypical. However, in vivo pharmacological studies of a compound often show unexpected cross-interactions with different targets and this aspect should not be ignored. For instance, the N/P/Q-type Ca2+ channel blocker, ω-conotoxin MVIIC, inhibited rat α3β4 nAChR currents with an IC50 of 1.3 µM; the blockade was non-competitive and reversible. The ω-conotoxin GVIA (N-type Ca2+ channel blocker) at 1 µM, inhibited by 24 and 20% α3β4 and α7 nAChR currents, respectively (Herrero et al., 1999). The µ-O-conotoxins have been reported to target molluscan VGSCs and calcium channels (Fainzilber et al., 1995) and more recently a conopeptide from C. planorbis, pl14a, was shown to inhibit both voltage-gated (Kv1.6) and ligand-gated (muscle and α3β4 nAChRs) ion channels (Imperial et al., 2006). Also, β/ω-theraphotoxin-Tp2a (ProTxII) from spider venom inhibits particular sodium and calcium channels (Sokolov et al., 2008; Edgerton et al., 2010) and the τ/κ-theraphotoxin-Pc1a (Vanillotoxin-1) activates mammalian TRPV1 and inhibits KV2.1 potassium channels (Siemens et al., 2006). More recently, α-Vc1.1 and α-AuIB were shown to target α9α10 and α3β4 nAChRs respectively, as well as both inhibiting calcium channels through a GABAB receptor pathway (Sandall et al., 2003; Klimis et al., 2011), thus demonstrating the vast complexity of pharmacological interactions deriving from some conotoxins. Given the structural similarity between α-AuIB and µ-CnIIIC presented in this work, one can also hypothesize an inhibition of N-type calcium channels mediated by GABAB receptors in dorsal root ganglion neurons by µ-CnIIIC.
These multiple activities combined in a single entity may also give some hints about common interaction sites between distantly related ion channels or receptors. Due to their particular shape and mode of action, such gating modifiers offer unprecedented tools to evaluate common structural and functional features across various ion channel families.
In conclusion, µ-CnIIIC exhibits a new pharmacological profile distinct from previously reported µ-conopeptides. Characteristic features include its interaction with some nAChRs subtypes and its ability to potently block VGSCs in skeletal muscle and in myelinated and unmyelinated nerves through a long-lasting action. Such properties may offer promising perspectives for its local use as a myorelaxing drug candidate.
Acknowledgments
We would like to express our deepest gratitude to the Government of New Caledonia, the French Navy, the IRD-Nouméa ‘Institut de Recherche pour le Développement’ (Fabrice Colin, Jean-Louis Menou, Claude Payri, Napoléon Colombani) as well as to the Toxinomics Foundation office in Nouméa (Alain Gerbault and Jacques Pusset) and Paris (Renée Ménez), and to Robin Offord (Mintaka Foundation for Medical Research) for their efforts and constant support. We are most grateful to the European Commission for financial and strategic support. This study has been performed as part of the CONCO cone snail genome project for health (http://www.conco.eu) within the 6th Framework Program (LIFESCIHEALTH-6 Intergrated Project LSHB-CT-2007, contract number 037592.
Glossary
- BMRB
biological magnetic resonance bank
- COSY
correlation spectroscopy
- EDL
extensor digitorum longus
- ESI-MS
electrospray ionization mass spectrometry
- GAP
global action potential
- nAChRs
nicotinic acetylcholine receptors
- NOESY
nuclear Overhauser effect spectroscopy
- PDB
protein databank
- TFA
trifluoroacetic acid
- TOCSY
total correlation spectroscopy
- VGSCs
voltage-gated sodium channels
Conflicts of interest
None to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of µ-CnIIIC on directly elicited isometric twitch tension on mouse hemidiaphragms. (A) Representative twitch tension recordings obtained before and after treatment with 0.1 and 0.6 µM µ-CnIIIC. (B) Concentration–response curve for µ-CnIIIC generated indifferent muscles, and expressed relative to its control value. Points are mean values ± SEM obtained for each studied concentration of the conopeptide, with n = 3–6. When error bars are not apparent, this means that they are within the size of the symbols. The µ-CnIIIC concentration that inhibited 50% twitch tension was 150 nM.
Figure S2 Secondary structural indicators for µ-CnIIIC. (A) Short- and medium-range NOE patterns observed in the NOESY spectra. Consensus α-helices (red cylinder) and loops (straight line) shown above amino acid sequence. (B) Secondary Hα shifts calculated per residue using BMRB random shift values.
Please note: Wiley–Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A, Davis DG. MLEV-17 based two-dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson. 1985;65:355–360. [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Buczek O, Bulaj G, Olivera BM. Conotoxins and the posttranslational modification of secreted gene products. Cell Mol Life Sci. 2005;62:3067–3079. doi: 10.1007/s00018-005-5283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, et al. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry. 2005;44:7259–7265. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel NaV1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- Chen H, Gordon D, Heinemann SH. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and Lqh alphaIT. Pflugers Arch. 2000;439:423–432. doi: 10.1007/s004249900181. [DOI] [PubMed] [Google Scholar]
- Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, et al. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem. 1985;260:9280–9288. [PubMed] [Google Scholar]
- Edgerton GB, Blumenthal KM, Hanck DA. Inhibition of the activation pathway of the T-type calcium channel Ca(V)3.1 by ProTxII. Toxicon. 2010;56:624–636. doi: 10.1016/j.toxicon.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg J, Craik DJ, Adams DJ. Conotoxin modulation of voltage-gated sodium channels. Int J Biochem Cell Biol. 2008;40:2363–2368. doi: 10.1016/j.biocel.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Fainzilber M, van der Schors R, Lodder JC, Li KW, Geraerts WP, Kits KS. New sodium channel-blocking conotoxins also affect calcium currents in Lymnaea neurons. Biochemistry. 1995;34:5364–5371. doi: 10.1021/bi00016a007. [DOI] [PubMed] [Google Scholar]
- Favreau P, Stöcklin R. Marine snail venoms: use and trends in receptor and channel neuropharmacology. Curr Opin Pharmacol. 2009;9:594–601. doi: 10.1016/j.coph.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Griesinger C, Sørensen OW, Ernst RR. Practical aspects of the E.COSY technique. Measurement of scalar spin-spin coupling constants in peptides. J Magn Reson. 1987;75:474–492. [Google Scholar]
- Halai R, Craik DJ. Conotoxins: natural product drug leads. Nat Prod Rep. 2009;26:526–536. doi: 10.1039/b819311h. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han TS, Zhang MM, Walewska A, Gruszczynski P, Robertson CR, Cheatham TE, et al. Structurally minimized mu-conotoxin analogues as sodium channel blockers: implications for designing conopeptide-based therapeutics. ChemMedChem. 2009;4:406–414. doi: 10.1002/cmdc.200800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero CJ, García-Palomero E, Pintado AJ, García AG, Montiel C. Differential blockade of rat alpha3beta4 and alpha7 neuronal nicotinic receptors by omega-conotoxin MVIIC, omega-conotoxin GVIA and diltiazem. Br J Pharmacol. 1999;127:1375–1387. doi: 10.1038/sj.bjp.0702692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Hill JM, Alewood PF, Craik DJ. Three-dimensional solution structure of mu-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry. 1996;35:8824–8835. doi: 10.1021/bi960073o. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bandelier F, Benoit A, Dosch R, Bertrand D. An automated system for intracellular and intranuclear injection. J Neurosci Methods. 2008;169:65–75. doi: 10.1016/j.jneumeth.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Holford RC, Zhang MM, Gowd KH, Azam L, Green BR, Watkins M, et al. Pruning nature: biodiversity-derived discovery of novel sodium channel blocking conotoxins from Conus bullatus. Toxicon. 2009;53:90–98. doi: 10.1016/j.toxicon.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperial JS, Bansal PS, Alewood PF, Daly NL, Craik DJ, Sporning A, et al. A novel conotoxin inhibitor of Kv1.6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry. 2006;45:8331–8340. doi: 10.1021/bi060263r. [DOI] [PubMed] [Google Scholar]
- Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- Keizer DW, West PJ, Lee EF, Yoshikami D, Olivera BM, Bulaj G, et al. Structural basis for tetrodotoxin-resistant sodium channel binding by mu-conotoxin SmIIIA. J Biol Chem. 2003;278:46805–46813. doi: 10.1074/jbc.M309222200. [DOI] [PubMed] [Google Scholar]
- Khoo KK, Feng ZP, Smith BJ, Zhang MM, Yoshikami D, Olivera BM, et al. Structure of the analgesic mu-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry. 2009;48:1210–1219. doi: 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW, et al. A novel mechanism of inhibition of high-voltage activated calcium channels by alpha-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain. 2011;152:259–266. doi: 10.1016/j.pain.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Kumar A, Ernst RR, Wüthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- Lewis RJ, Schroeder CI, Ekberg J, Nielsen KJ, Loughnan M, Thomas L, et al. Isolation and structure-activity of mu-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol Pharmacol. 2007;71:676–685. doi: 10.1124/mol.106.028225. [DOI] [PubMed] [Google Scholar]
- Li RA, Tomaselli GF. Using the deadly mu-conotoxins as probes of voltage-gated sodium channels. Toxicon. 2004;44:117–122. doi: 10.1016/j.toxicon.2004.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Lluisma AO, Lopez-Vera E, Bulaj G, Watkins M, Olivera BM. Characterization of a novel psi-conotoxin from Conus parius Reeve. Toxicon. 2008;51:174–180. doi: 10.1016/j.toxicon.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vera E, Walewska A, Skalicky JJ, Olivera BM, Bulaj G. Role of hydroxyprolines in the in vitro oxidative folding and biological activity of conotoxins. Biochemistry. 2008;47:1741–1751. doi: 10.1021/bi701934m. [DOI] [PubMed] [Google Scholar]
- Loughnan ML, Nicke A, Jones A, Adams DJ, Alewood PF, Lewis RJ. Chemical and functional identification and characterization of novel sulfated alpha-conotoxins from the cone snail Conus anemone. J Med Chem. 2004;47:1234–1241. doi: 10.1021/jm031010o. [DOI] [PubMed] [Google Scholar]
- Luo S, Akondi KB, Zhangsun D, Wu Y, Zhu X, Hu Y, et al. Atypical alpha-Conotoxin LtIA from Conus litteratus targets a novel microsite of the alpha3beta2 nicotinic receptor. J Biol Chem. 2010;285:12355–12366. doi: 10.1074/jbc.M109.079012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzati V, Mateu L, Vachette P, Benoit E, Charpentier G, Kado R. Physical structure of the excitable membrane of unmyelinated axons: X-ray scattering study and electrophysiological properties of pike olfactory nerve. J Mol Biol. 2000;304:69–80. doi: 10.1006/jmbi.2000.4176. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Olivera BM, Cruz LJ. Conus peptides as probes for ion channels. Methods Enzymol. 1999;294:605–624. doi: 10.1016/s0076-6879(99)94034-x. [DOI] [PubMed] [Google Scholar]
- Menin L, Perchuć A, Favreau P, Perret F, Michalet S, Schöni R, et al. High throughput screening of bradykinin-potentiating peptides in Bothrops moojeni snake venom using precursor ion mass spectrometry. Toxicon. 2008;51:1288–1302. doi: 10.1016/j.toxicon.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- Mitchell SS, Shon KJ, Foster MP, Davis DR, Olivera BM, Ireland CM. Three-dimensional solution structure of conotoxin psi-PIIIE, an acetylcholine gated ion channel antagonist. Biochemistry. 1998;37:1215–1220. doi: 10.1021/bi972186t. [DOI] [PubMed] [Google Scholar]
- Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- Nielsen KJ, Watson M, Adams DJ, Hammarström AK, Gage PW, Hill JM, et al. Solution structure of mu-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J Biol Chem. 2002;277:27247–27255. doi: 10.1074/jbc.M201611200. [DOI] [PubMed] [Google Scholar]
- Norton RS. Mu-conotoxins as leads in the development of new analgesics. Molecules. 2010;15:2825–2844. doi: 10.3390/molecules15042825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera BM, Teichert RW. Diversity of the neurotoxic Conus peptides: a model for concerted pharmacological discovery. Mol Interv. 2007;7:251–260. doi: 10.1124/mi.7.5.7. [DOI] [PubMed] [Google Scholar]
- Olivera BM, MacIntosh JM, Garrett JE, Cruz LJ, Jones RM, Cartier GE, et al. 2004. Mu-conopeptides. US Patent 6,727,226.
- Ott KH, Becker S, Gordon RD, Ruterjans H. Solution structure of mu-conotoxin GIIIA analysed by 2D-NMR and distance geometry calculations. FEBS Lett. 1991;278:160–166. doi: 10.1016/0014-5793(91)80107-e. [DOI] [PubMed] [Google Scholar]
- Pardi A, Billeter M, Wüthrich K. Calibration of the angular dependence of the amide proton-C[alpha] proton coupling constants, 3JHN[alpha], in a globular protein : use of 3JHN[alpha] for identification of helical secondary structure. J Mol Biol. 1984;180:741–751. doi: 10.1016/0022-2836(84)90035-4. [DOI] [PubMed] [Google Scholar]
- Quinton L, Le Caer JP, Vinh J, Gilles N, Chamot-Rooke J. Fourier transform mass spectrometry: a powerful tool for toxin analysis. Toxicon. 2006;47:715–726. doi: 10.1016/j.toxicon.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Rance M, Sørensen OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- Safo P, Rosenbaum T, Shcherbatko A, Choi DY, Han E, Toledo-Aral JJ, et al. Distinction among neuronal subtypes of voltage-activated sodium channels by mu-conotoxin PIIIA. J Neurosci. 2000;20:76–80. doi: 10.1523/JNEUROSCI.20-01-00076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandall DW, Satkunanathan N, Keays DA, Polidano MA, Liping X, Pham V, et al. A novel alpha-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry. 2003;42:6904–6911. doi: 10.1021/bi034043e. [DOI] [PubMed] [Google Scholar]
- Sato K, Ishida Y, Wakamatsu K, Kato R, Honda H, Ohizumi Y, et al. Active site of mu-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J Biol Chem. 1991;266:16989–16991. [PubMed] [Google Scholar]
- Schirmeyer J, Szafranski K, Leipold E, Mawrin C, Platzer M, Heinemann SH. A subtle alternative splicing event of the Na(V)1.8 voltage-gated sodium channel is conserved in human, rat, and mouse. J Mol Neurosci. 2010;41:310–314. doi: 10.1007/s12031-009-9315-3. [DOI] [PubMed] [Google Scholar]
- Schlumberger S, Ouanounou G, Girard E, Sasaki M, Fuwa H, Louzao MC, et al. The marine polyether gambierol enhances muscle contraction and blocks a transient K(+) current in skeletal muscle cells. Toxicon. 2010;56:785–791. doi: 10.1016/j.toxicon.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Ekberg J, Nielsen KJ, Adams D, Loughnan ML, Thomas L, et al. Neuronally micro-conotoxins from Conus striatus utilize an alpha-helical motif to target mammalian sodium channels. J Biol Chem. 2008;283:21621–21628. doi: 10.1074/jbc.M802852200. [DOI] [PubMed] [Google Scholar]
- Shon KJ, Grilley M, Jacobsen R, Cartier GE, Hopkins C, Gray WR, et al. A noncompetitive peptide inhibitor of the nicotinic acetylcholine receptor from Conus purpurascens venom. Biochemistry. 1997;36:9581–9587. doi: 10.1021/bi970235w. [DOI] [PubMed] [Google Scholar]
- Siemens J, Zhou S, Piskorowski R, Nikai T, Lumpkin EA, Basbaum AI, et al. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature. 2006;444:208–212. doi: 10.1038/nature05285. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Kraus RL, Scheuer T, Catterall WA. Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol Pharmacol. 2008;73:1020–1028. doi: 10.1124/mol.107.041046. [DOI] [PubMed] [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Ueberheide BM, Fenyo D, Alewood PF, Chait BT. Rapid sensitive analysis of cysteine rich peptide venom components. Proc Natl Acad Sci USA. 2009;106:6910–6915. doi: 10.1073/pnas.0900745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K, Kohda D, Hatanaka H, Lancelin JM, Ishida Y, Oya M, et al. Structure-activity relationships of mu-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry. 1992;31:12577–12584. doi: 10.1021/bi00165a006. [DOI] [PubMed] [Google Scholar]
- West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. Mu-conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry. 2002;41:15388–15393. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- Yao S, Zhang MM, Yoshikami D, Azam L, Olivera BM, Bulaj G, et al. Structure, dynamics, and selectivity of the sodium channel blocker conotoxin SIIIA. Biochemistry. 2008;47:10940–10949. doi: 10.1021/bi801010u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, Fiedler B, Green BR, Catlin P, Watkins M, Garrett JE, et al. Structural and functional diversities among mu-conotoxins targeting TTX-resistant sodium channels. Biochemistry. 2006;45:3723–3732. doi: 10.1021/bi052162j. [DOI] [PubMed] [Google Scholar]
- Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, et al. Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J Biol Chem. 2007;282:30699–30706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.