

Abstract
BACKGROUND AND PURPOSE
Besides targeting the well-known oncogenic c-Met, crizotinib is the first oral tyrosine kinase inhibitor inhibiting anaplastic lymphoma kinase (ALK) in clinical trials for the treatment of non-small cell lung cancer. Here, we assessed the possible reversal of multidrug resistance (MDR) by crizotinib in vitro and in vivo.
EXPERIMENTAL APPROACH
1-(4,5-Dimethylthiazol-2-yl)-3,5- diphenylformazan was used in vitro and xenografts in nude mice were used in vivo to investigate reversal of MDR by crizotinib. To understand the mechanisms for MDR reversal, the alterations of intracellular doxorubicin or rhodamine 123 accumulation, doxorubicin efflux, ABCB1 expression level, ATPase activity of ABCB1 and crizotinib-induced c-Met, Akt and ERK1/2 phosphorylation were examined.
KEY RESULTS
Crizotinib significantly enhanced the cytotoxicity of chemotherapeutic agents which are also ABCB1 substrates, in MDR cells with no effect found on sensitive cells in vitro and in vivo. Additionally, crizotinib significantly increased intracellular accumulation of rhodamine 123 and doxorubicin and inhibited the drug efflux in ABCB1-overexpressing MDR cells. Further studies showed that crizotinib enhanced the ATPase activity of ABCB1 in a concentration-dependent manner. However, expression of ABCB1 was not affected, and reversal of MDR by crizotinib was not related to the phosphorylation of c-Met, Akt or ERK1/2. Importantly, crizotinib significantly enhanced the effect of paclitaxel against KBv200 cell xenografts in nude mice.
CONCLUSIONS AND IMPLICATIONS
Crizotinib reversed ABCB1-mediated MDR by inhibiting ABCB1 transport function without affecting ABCB1 expression or blocking the Akt or ERK1/2 pathways. These findings are useful for planning combination chemotherapy of crizotinib with conventional chemotherapeutic drugs.
Keywords: crizotinib, multidrug resistance, ATP-binding cassette transporters, P-glycoprotein, ABCG2, xenograft
Introduction
The ATP-binding cassette (ABC) transporters are a superfamily of transmembrane proteins that transport a wide variety of substrates across extracellular and intracellular membranes (Dean et al., 2001a). In the human genome, 48 different ABC transporters have been identified and are divided into seven subfamilies (A–G) based on sequence similarities (Dean et al., 2001a; 2001b). Some of them play a crucial role in the development of multidrug resistance (MDR) by pumping out substrate drugs out of the cells against a concentration gradient with the use of energy from ATP hydrolysis (Ambudkar et al., 1999; Perez-Tomas, 2006). In particular, the ABC transporters subfamily B member 1 (ABCB1/P-glycoprotein/P-gp), subfamily C member 1 (ABCC1/MRP1) and subfamily G member 2 (ABCG2/BCRP) are the most important transporters members mediating MDR (nomenclature follows Alexander et al., 2011). Overexpression of these transporters was commonly observed in drug-selected resistant cancer cell lines and has been suggested to cause failure of cancer chemotherapy in the clinic (Dean et al., 2001b).
These ABC transporters can extrude a wide range of structurally and mechanistically different anticancer drugs from the cells. For example, the spectrum of chemotherapeutic agents transported by ABCB1/P-gp include the frequently used chemotherapeutic agents, most of them are hydrophobic and either uncharged or slightly positively charged, such as anthracyclines, Vinca alkaloids, anthracyclines, epipodophyllotoxins and taxanes (Sauna et al., 2007). Drugs transported by ABCG2 include anthracyclines, mitoxantrone, camptothecin-derived and indolocarbazole topoisomerase inhibitors, methotrexate, and flavopiridol, as well as fluorescent dyes such as Hoechst 33342 (Doyle and Ross, 2003; Schinkel and Jonker, 2003). On the other hand, ABCC1 can transport a broad spectrum of substrate anticancer drugs mainly conjugated to glutathione, glucuronate and sulphate, including vincristine and doxorubicin (Bakos and Homolya, 2007). Therefore, compounds that fully or partly block ABC transporter activities may prevent the undesirable loss of intracellular substrate anticancer drugs and thus could be beneficial when used in combination chemotherapy. Enormous effort has been devoted to the development of inhibitors for ABC transporters in the hope of circumventing MDR. To date, three generations of MDR inhibitors have been developed, some of which are currently under clinical trials to evaluate their usefulness in circumventing anticancer drug resistance (Dean et al., 2005; Coley, 2010).
Tyrosine kinase inhibitors (TKIs) are an important new class of targeted chemotherapeutic agents, which work by reversible competition against ATP binding to the intracellular catalytic domain of oncogenic tyrosine kinases. Consequently, they can attenuate downstream signalling pathways involved in cancer proliferation, invasion, metastasis and angiogenesis, thereby representing a promising class of anticancer agents in the clinic (Shawver et al., 2002). Crizotinib (PF-02341066) is a novel oral multitargeted TKI that inhibit c-Met and ALK. It is also the first agent that can selectively target the echinoderm microtubule-associated protein–like 4 anaplastic lymphoma kinase (EML4-ALK) translocation commonly found in non–small cell lung cancer (NSCLC) patients. Currently, clinical development of crizotinib is focused primarily on its effect on ALK rearranged NSCLC. Besides displaying antitumour activity by directly inhibiting tumour cell proliferation and survival via c-Met and ALK inhibition, crizotinib was also suggest to suppress tumour angiogenesis via VEGFR inhibition (Zou et al., 2007).
Previously, it has been reported that several tyrosine kinase inhibitors including lapatinib (Dai et al., 2008), gefitinib (Kitazaki et al., 2005), erlotinib (Shi et al., 2007), cediranib (Tao et al., 2009), vandetanib (Zheng et al., 2009) and sunitinib (Dai et al., 2009) can inhibit functions of ABC transporters, thereby overcoming chemotherapy resistance in MDR cancer cells. Taken together, these reports suggest that TKIs may be promising MDR inhibitors.
Given that crizotinib may be used in combination chemotherapy to achieve its maximum clinical efficacy and to extend its coverage to tumour types that do not have the EML4-ALK translocation, it will be beneficial to have a detailed understanding about its interaction with various ABC transporters. In this study, we investigated the circumvention of MDR by crizotinib via its interactions with ABC transporters in MDR cancer cells in vitro and in a tumour xenograft model.
Methods
Cell lines and cell culture
The following cell lines were cultured in DMEM or RPMI 1640 supplemented with 10% FBS at 37°C in a humidified atmosphere of 5% CO2: the human breast carcinoma cell line MCF-7, its doxorubicin-selected ABCB1-overexpressing derivative MCF-7/adr (Fu et al., 2004); the human oral epidermoid carcinoma cell line KB and its vincristine-selected ABCB1-overexpressing derivative KBv200 (a kind gift from Dr Xu-Yi Liu, Cancer Hospital of Beijing, Beijing) (Zhang et al., 2007); the human leukaemia cell lines HL60 and its doxorubicin-selected ABCC1-overexpressing derivative HL60/adr (Tang et al., 2004); the human colon carcinoma cell line S1 and its mitoxantrone-selected ABCG2-overexpressing derivative S1-M1-80 (Honjo et al., 2001) and the human embryonic kidney cell line HEK293 and its stable pcDNA3.1 or ABCB1 transfectant HEK293/pcDNA3.1, HEK293/ABCB1, obtained from Dr Susan Bates (National Cancer Institute, NIH) (Robey et al., 2008). The transfected cells were cultured in medium containing 2 mg·mL−1 G418. All resistant cells were authenticated by comparing their fold resistance with that of the parental drug-sensitive cells and examining the expression levels of ABC transporters. All cells were grown in drug-free culture medium for more than 2 weeks before assay.
Animals
All animal care and experimental procedures have been approved by the Ethics Committee for Animal Experimentation and were carried out in accordance with the guidelines on animal care and experiments of laboratory animals (Center of Experimental Animals, Sun Yat-Sen University, China). As there are gender-related differences in the pharmacokinetics and toxicity of crizotinib in mice (Zhong et al., 2010), only female mice was used in these experiments. The KBv200 tumour xenografts were developed in athymic female nude mice (BALB/c-nu/nu), 6 to 7 weeks old and weighing 18 to 24 g, obtained from the Center of Experimental Animals, Sun Yat-Sen University (China). The experimental animals had free access to sterilized food and water.
Cell cytotoxicity assay
The assay using 1-(4,5-dimethylthiazol-2-yl)-3,5- diphenylformazan (MTT) was carried out, as described previously, to assess the sensitivity of cells to chemotherapeutic drugs (Chen et al., 2004b). Briefly, cells were plated in 96-well microtitre plates, and then various concentrations of crizotinib and/or a full range concentration of conventional chemotherapeutic drug were added to the wells. After 68 h of incubation, MTT (5 mg·mL−1, 20 µL per well) was added to the wells, and the cells were incubated for an additional 4 h (37°C). Subsequently, the medium was discarded, and 200 µL of DMSO was added to dissolve the formazan product from the metabolism of MTT. The optical density was measured at 540 nm with background subtraction at 670 nm using a Model 550 Microplate Reader (BIO-RAD, Hercules, CA). The concentration required to inhibit cell growth by 50% (IC50) was calculated from survival curves using the Bliss method (Shi et al., 2006). The degree of resistance was estimated by dividing the IC50 for the MDR cells by that of the parental sensitive cells; the fold-reversal factor of MDR was calculated by dividing the IC50 of the anticancer drug in the absence of crizotinib by that obtained in the presence of crizotinib. Besides using the ABCB1-overexpressing cell line models, two other ABCC1-overexpressing HL60/adr or ABCG2-overexpressing S1-M1-80 cell lines were also used in our study to assess if crizotinib was specific for ABCB1.
Nude mouse xenograft model
The KBv200-inoculated nude mice xenograft model previously established by Chen and colleagues was used in this study (Chen et al., 2004a). These xenografts were found to maintain the MDR phenotype in vivo and were extremely resistant to paclitaxel treatment (Chen et al., 2004a). Briefly, KBv200 cells grown in vitro were harvested and implanted s.c. under the shoulder in the nude mice. When the tumours reached a mean diameter of 0.5 cm, the mice were randomized into four groups and treated with various regimens: (a) saline (q3d × 4); (b) paclitaxel (18 mg·kg−1, i.p., q3d × 4); (c) crizotinib (25 mg·kg−1, p.o., q3d × 4); and (d) crizotinib (25 mg·kg−1, p.o., q3d × 4 given 1 h before injecting paclitaxel) + paclitaxel (18 mg·kg−1, i.p., q3d × 4). The body weights of the animals and the two perpendicular diameters (A and B) were recorded every 2 days, and tumour volume (V) was estimated according to the following formula (Chen et al., 2004a):
The curve of tumour growth was drawn according to tumour volume and time of implantation. The mice were anaesthetized and killed when the mean tumour weight was more than 1 g in the control group. Tumour tissues were excised from the mice, and their weights were measured. The ratio of growth inhibition (IR) was calculated according to the following formula (Chen et al., 2004a):
Doxorubicin and rhodamine 123 accumulation
The effect of crizotinib on the accumulation of doxorubicin and rhodamine 123 was measured by flow cytometry as previously described (Fu et al., 2004). Briefly, the cells were incubated with crizotinib at a range of concentrations or vehicle at 37°C for 3 h. 10 µM doxorubicin or 5 µM rhodamine 123 was added, and incubation was continued for additional 3 or 0.5 h respectively. The cells were then collected, washed three times with ice-cold PBS and analysed by flow cytometric analysis (Beckman Coulter, Cytomics FC500, Brea, CA, USA). Verapamil, a known ABCB1 inhibitor, was used as a positive control (Rabindran et al., 2000).
Studies of doxorubicin efflux
Doxorubicin efflux was assayed following a modification of methods described earlier (Dai et al., 2009). KB and KBv200 cells were treated with 10 µM doxorubicin for 3h at 37°C, the cells were washed then twice with ice-cold PBS and subsequently maintained at 37°C and without doxorubicin with culture media with or without 1.5 µM crizotinib. Subsequently, at 0, 15, 30, 60 and 120 min, cells were gathered and washed twice with ice-cold PBS. Finally, cells were resuspended in ice-cold PBS buffer for flow cytometric analysis immediately (Beckman Coulter), and the fluorescence intensity was determined.
ABCB1 ATPase activity assay
The changes of ATPase activity were estimated by Pgp-Glo™ assay systems (Promega Corp., Madison, WI, USA). The inhibitory effects of crizotinib were examined against a verapamil-stimulated ABCB1 ATPase activity. Sodium orthovanadate (Na3VO4) was used as an ABCB1 ATPase inhibitor. Various concentrations of crizotinib diluted with assay buffer were incubated in 0.1 mM verapamil, 5 mM MgATP and 25 µg recombinant human ABCB1 membranes at 37°C for 40 min. Luminescence was initiated by ATP detection buffer. After incubation at room temperature for 20 min to allow the luminescent signal to develop, the untreated white opaque 96-well plate (Corning, New York, NY, USA) was read on a luminometer (spectraMax M5, Molecular Devices, Sunyvale CA, USA). The changes of relative light units (ΔRLU) were determined by comparing Na3VO4-treated samples with crizotinib and verapamil combination-treated samples, and hence, the ATP consumed was obtained by comparing to a standard curve.
RT-PCR and real-time quantitative PCR
After drug treatment for 48 h, total cellular RNA was isolated by Trizol Reagent RNA extraction kit following the manufacturer's instruction (Molecular Research Center, Cincinnati, OH, USA). The first strand cDNA was synthesized by Oligo dT primers with reverse transcriptase (Promega Corp.). PCR primers were 5′-CCCATCATTGCAATAGCAGG-3′ (forward) and 5′-GTTCAAACTTCTGCTCCTGA-3′ (reverse) for ABCB1 and 5′-CTTTGGTATCGTGGAAGGA-3′ (forward) and 5′-CACCCTGTTGCTGTAGCC-3′ (reverse) for GAPDH respectively. Using the GeneAmp PCR system 9700 (PE Applied Biosystems, Foster City, CA, USA), reactions were carried out at 94°C for 2 min for initial denaturation, and then at 94°C for 30 s, 58°C for 30 s and 72°C for 1 min. After 35 cycles of amplification, additional extensions were carried out at 72°C for 10 min. Products were resolved and examined by 1.5% agarose gel electrophoresis. Expected PCR products were 157 bp for ABCB1 and 475 bp for GAPDH respectively.
Real-time PCR was performed with Real-time PCR Master Mix containing SYBR GREEN I and hotstart Taq DNA polymerase. GAPDH was amplified as control. The primers are 5′-GTGGGGCAAGTCAGTTCATT-3′ (forward) and 5′-TCTTCACCTCCAGGCTCAGT-3′ (reverse) for ABCB1 and 5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-GATCTCGCTCCTGGAAGATG-3′ (reverse) for GAPDH respectively. Real-time detection of the emission intensity of SYBR GREEN bound to double-stranded DNAs was performed using the Icycler Instrument (Bio-Rad, Hercules, CA, USA). At the endpoint of PCR cycles, melting curves were examined to check for product purity. The level of ABCB1 mRNA was expressed as a ratio relative to the GAPDH mRNA in each sample. The slopes of Ct and dCt [(target gene) – (reference gene)] and R2 values of each sample were calculated by the Bio-Rad Chromo4 real-time PCR system and Microsoft Excel 2007 for Windows. Relative quantification of ABCB1 was performed using the 2−ΔΔCt method (Livak and Schmittgen, 2001). The results were obtained from three reactions in each sample and analysed by the SPSS software (Version 11.0) (SPSS Inc., Chicago, IL, USA).
Western blot analysis
To identify whether crizotinib affected ABCB1 protein expression, the cells were incubated with different concentrations of crizotinib for 48 h. To determine whether crizotinib is able to block c-Met, Akt or ERK1/2 phosphorylation, we incubated cells with different concentrations of crizotinib for 24 h and various hours for 1.5 µM. Then, whole cell lysates were harvested and washed twice with ice-cold PBS. Cell extracts were collected in cell lysis buffer (PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 µg·mL−1 PMSF, 10 µg·mL−1 aprotinin, 10 µg·mL−1 leupeptin). Equal amounts of cell lysate from various treatments were resolved by SDS-PAGE. After blocking in TBST (10 mM Tris–HCl, 150 mM NaCl and 0.1% Tween 20, pH 8.0) with 5% non-fat milk for 2 h at room temperature, the membranes were incubated with appropriately diluted primary antibodies overnight at 4°C. The membranes were then washed three times with TBST and incubated with HRP-conjugated secondary antibody at 1:5000 dilution for 2 h at room temperature. After three washes with TBST, the protein–antibody complexes were visualized by the enhanced Phototope TM-HRP Detection Kit (Cell Signaling) and exposed to Kodak medical X-ray processor (Carestream Health, Atlanta, GA, USA). GAPDH was used as a loading control.
Data analysis
Results are shown as means ± SD, unless otherwise stated. All experiments were repeated at least three times, and the differences were determined by using Student's t-test. The significance was determined at P < 0.05.
Materials
Crizotinib (PF-02341066) was purchased from Selleck Chemicals (Houston, TX, USA), with a molecular structure as shown in Figure 1A. Monoclonal antibodies against ABCB1 and total c-Met (C-28) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antiphospho-c-Met and Akt antibody was a product of Cell Signaling Technology, Inc. (Danvers, MA). Phosphorylated Akt, phosphorylated ERK, Mark/2 (ERK1/2) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Kangchen Co. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM) and RPMI-1640 were products of Gibco BRL (Grand Island, NY, USA). Platinum® SYBR® Green qPCR SuperMix-UDG with ROX was obtained from Invitrogen Co. (Grand Island, NY, USA) Rhodamine 123, 1-(4,5-dimethylthiazol-2-yl)-3,5- diphenylformazan (MTT), fumitremorgin C, paclitaxel, doxorubicin, vincristine, mitoxantrone, MK571 and other chemicals were purchased from Sigma Chemical Co (St. Louis, MO).
Figure 1.
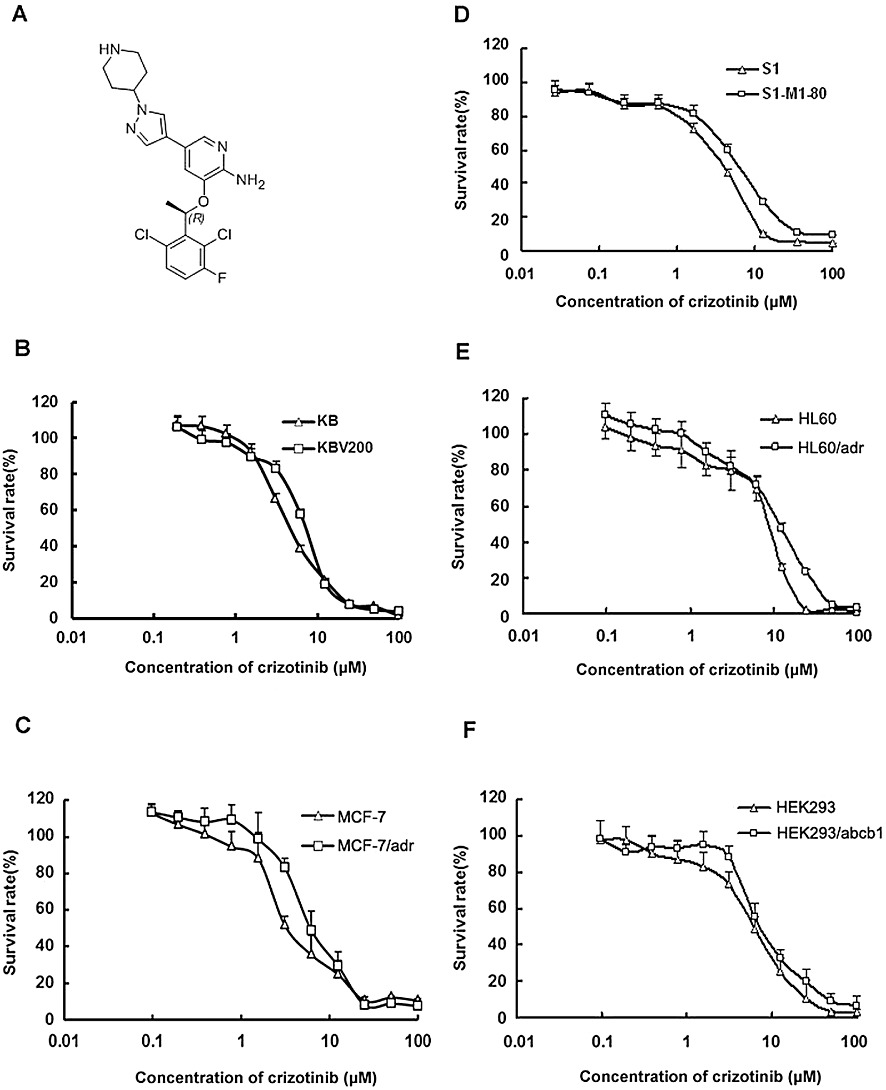
Cytotoxicity of crizotinib in the drug-resistant and parental, sensitive, cancer cells. A, the structure of crizotinib; MTT cytotoxicity assay was assessed in pairs of parental and transporter-overexpressing cells: B, ABCB1-negative KB and ABCB1-positive KBv200 cells. C, ABCB1-negative MCF-7 and ABCB1-positive MCF-7/adr cells. D, ABCG2-negative S1 and ABCG2-positive S1-M1-80 cells. E, ABCC1-negative HL60 and ABCC1-positive HL60/adr cells. F, ABCB1-negative HEK293 and HEK293/ABCB1 cells. Data shown are means ± SD for three determinations. Each experiment was performed in three replicate wells.
Results
Cytotoxicity effect of crizotinib on MCF-7/adr, KBv200, HL60/adr, S1-M1-80, HEK293/ABCB1 and their corresponding parental cells
The cytotoxicity of crizotinib in different cell lines was determined by the MTT assay. The IC50 values were 4.68 ± 0.67, 6.67 ± 0.87, 3.34 ± 0.52, 6.11 ± 0.78, 3.46 ± 0.54, 6.23 ± 0.31, 8.52 ± 0.93, 11.59 ± 1.06, 6.10 ± 0.79 and 6.79 ± 1.31 µM for KB, KBv200, MCF-7, MCF-7/adr, S1, S1-M1-80, HL60, HL60/adr, HEK293/pcDNA3.1 and HEK293/ABCB1 cells respectively (Figure 1 and Supporting Information Table S1). Based on the cytotoxicity curves, more than 85% of cells were viable at the concentrations of 1.5 µM crizotinib in KB, KBv200, MCF-7, MCF-7/adr, HL60, HL60/adr, HEK293 and HEK293/ABCB1 cells and 1.0 µM in S1 and S1-M1-80 cells. Therefore, crizotinib at a concentration of 1.5 µM (in KB, KBv200, MCF-7, MCF-7/adr, HL60, HL60/adr, HEK293 and HEK293/ABCB1 cells) or 1.0 µM (in S1 and S1-M1-80 cells) was chosen as a maximum concentration for combination treatment with known ABCB1 (doxorubicin and paclitaxel), ABCC1 (doxorubicin) or ABCG2 (topotecan) substrate anticancer drugs.
Modulation of MDR in MDR cell lines by crizotinib
The IC50 values of the anticancer drugs in sensitive and resistant cells in the absence or presence of crizotinib are shown in Table 1. Crizotinib produced a concentration-dependent decrease in the IC50 values of doxorubicin and paclitaxel in MCF-7/adr cells and KBv200 cells but did not alter the cytotoxicity of cisplatin, which is not an ABCB1 substrate. Furthermore, crizotinib significantly decreased the IC50 values of doxorubicin and paclitaxel in stably transfected HEK293/ABCB1 cells (Table 2). However, no enhancement effects of crizotinib were observed in the parental cells. In addition, crizotinib had no significant reversal effect on ABCC1-mediated drug resistance in HL60/adr cells or ABCG2-mediated drug resistance in S1-M1-80 cells. These results demonstrate that crizotinib significantly sensitized ABCB1-overexpressing cells to anticancer agents that are ABCB1 substrates.
Table 1.
Effect of crizotinib on reversing ABCB1-, ABCC1- and ABCG2-mediated MDR in pairs of sensitive and drug-resistant cell lines
| IC50± SD (µM) (fold-reversal) | ||||
|---|---|---|---|---|
| Compounds | MCF-7 | MCF-7/adr (ABCB1) | ||
| Doxorubicin | 0.341 ± 0.029 | (1.00) | 8.321 ± 0.602 | (1.00) |
| +0.375 µM Crizotinib | 0.339 ± 0.041 | (1.01) | 2.119 ± 0.151** | (4.13) |
| +0.75 µM Crizotinib | 0.326 ± 0.017 | (1.01) | 1.038 ± 0.062** | (8.50) |
| +1.5 µM Crizotinib | 0.303 ± 0.016 | (1.13) | 0.849 ± 0.051** | (10.2) |
| +10 µM Verapamil | 0.334 ± 0.020 | (1.02) | 0.496 ± 0.005** | (17.8) |
| Paclitaxel | 0.031 ± 0.025 | (1.00) | 0.877 ± 0.021 | (1.00) |
| +0.375 µM Crizotinib | 0.030 ± 0.014 | (1.03) | 0.381 ± 0.019** | (2.32) |
| +0.75 µM Crizotinib | 0.029 ± 0.028 | (1.07) | 0.303 ± 0.021** | (2.68) |
| +1.5 µM Crizotinib | 0.030 ± 0.098 | (1.03) | 0.217 ± 0.025** | (4.00) |
| +10 µM Verapamil | 0.027 ± 0.019 | (1.14) | 0.052 ± 0.065** | (16.9) |
| Cisplatin | 15.431 ± 1.571 | (1.00) | 18.672 ± 0.671 | (1.00) |
| +1.5 µM Crizotinib | 16.012 ± 0.468 | (1.18) | 17.956 ± 0.997 | (1.04) |
| +10 µM Verapamil | 15.285 ± 0.299 | (1.26) | 18.316 ± 0.346 | (1.02) |
| KB | KBv200 (ABCB1) | |||
| Doxorubicin | 0.190 ± 0.061 | (1.00) | 6.536 ± 0.219 | (1.00) |
| +0.375 µM Crizotinib | 0.205 ± 0.074 | (0.93) | 4.136 ± 0.137* | (1.58) |
| +0.75 µM Crizotinib | 0.181 ± 0.031 | (1.04) | 2.422 ± 0.149** | (2.70) |
| +1.5 µM Crizotinib | 0.169 ± 0.006 | (1.12) | 1.578 ± 0.341** | (4.14) |
| +10 µM Verapamil | 0.158 ± 0.053 | (1.20) | 0.408 ± 0.083** | (16.0) |
| Paclitaxel | 0.0010 ± 0.0012 | (1.00) | 0.388 ± 0.023 | (1.00) |
| +0.375 µM Crizotinib | 0.0011 ± 0.0007 | (0.90) | 0.201 ± 0.003** | (1.93) |
| +0.75 µM Crizotinib | 0.0010 ± 0.0006 | (1.00) | 0.145 ± 0.042** | (2.67) |
| +1.5 µM Crizotinib | 0.0009 ± 0.0003 | (1.11) | 0.105 ± 0.049** | (3.70) |
| +10 µM Verapamil | 0.0009 ± 0.0004 | (1.11) | 0.017 ± 0.002** | (22.8) |
| Cisplatin | 2.284 ± 0.219 | (1.00) | 3.631 ± 0.058 | (1.00) |
| +1.5 µM Crizotinib | 2.503 ± 0.061 | (0.91) | 3.393 ± 0.764 | (1.07) |
| +10 µM Verapamil | 2.461 ± 0.021 | (0.93) | 2.980 ± 0.023 | (1.22) |
| HL60 | HL60/adr (ABCC1) | |||
| Doxorubicin | 0.033 ± 0.002 | (1.00) | 5.950 ± 0.259 | (1.00) |
| +0.375 µM Crizotinib | 0.032 ± 0.002 | (1.03) | 4.605 ± 0.389 | (0.99) |
| +0.75 µM Crizotinib | 0.029 ± 0.004 | (1.14) | 4.656 ± 0.211 | (1.31) |
| +1.5 µM Crizotinib | 0.030 ± 0.002 | (1.10) | 4.889 ± 0.362 | (1.21) |
| +50 µM MK571 | 0.031 ± 0.002 | (1.06) | 0.801 ± 0.401** | (7.89) |
| S1 | S1-M1-80 (ABCG2) | |||
| Topotecan | 0.517 ± 0.017 | (1.00) | 18.671 ± 0.164 | (1.00) |
| +0.25 µM Crizotinib | 0.515 ± 0.022 | (1.00) | 17.515 ± 0.158 | (1.06) |
| +0.5 µM Crizotinib | 0.514 ± 0.024 | (1.01) | 17.839 ± 0.259 | (1.04) |
| +1.0 µM Crizotinib | 0.485 ± 0.065 | (1.08) | 14.746 ± 0.808 | (1.14) |
| +2.5 µM FTC | 0.499 ± 0.088 | (1.06) | 2.051 ± 0.032** | (9.13) |
Cell survival was determined by MTT. Data are the means ± SD of at least three independent experiments performed in triplicate. The fold reversal of MDR was calculated by dividing the IC50 value for cells with the anticancer drug in the absence of inhibitor by that obtained in the presence of inhibitor.
P < 0.05,
P < 0.01, significantly different from those obtained in the absence of inhibitor.
Table 2.
Effect of crizotinib on reversing ABCB1- mediated MDR in transfected cell lines
| IC50± SD (µM) (fold-reversal) | ||||
|---|---|---|---|---|
| Compounds | HEK293 | HEK293/ABCB1 | ||
| Doxorubicin | 0.058 ± 0.001 | (1.00) | 0.643 ± 0.013 | (1.00) |
| +0.375 µM Crizotinib | 0.063 ± 0.009 | (0.91) | 0.367 ± 0.021** | (1.75) |
| +0.75 µM Crizotinib | 0.056 ± 0.007 | (1.03) | 0.244 ± 0.059** | (2.64) |
| +1.5 µM Crizotinib | 0.046 ± 0.008 | (1.25) | 0.133 ± 0.030** | (3.87) |
| +10 µM Verapamil | 0.044 ± 0.006 | (1.32) | 0.049 ± 0.008** | (13.1) |
| Paclitaxel | 0.039 ± 0.002 | (1.00) | 0.8137 ± 0.098 | (1.00) |
| +0.375 µM Crizotinib | 0.035 ± 0.004 | (1.11) | 0.488 ± 0.160** | (1.67) |
| +0.75 µM Crizotinib | 0.034 ± 0.004 | (1.15) | 0.334 ± 0.079** | (2.43) |
| +1.5 µM Crizotinib | 0.033 ± 0.003 | (1.18) | 0.195 ± 0.035** | (4.16) |
| +10 µM Verapamil | 0.031 ± 0.003 | (1.26) | 0.055 ± 0.007** | (18.0) |
| Cisplatin | 2.014 ± 0.341 | (1.00) | 2.281 ± 0.046 | (1.00) |
| +1.5 µM Crizotinib | 1.989 ± 0.360 | (1.18) | 2.360 ± 0.058 | (0.96) |
| +10 µM Verapamil | 2.380 ± 0.081 | (1.26) | 2.032 ± 0.083 | (1.12) |
Cell survival was determined by MTT assays. Data are the means ± SD of at least three independent experiments performed in triplicate. The fold reversal of MDR (values given in parentheses) was calculated by dividing the IC50 for cells with the anticancer drugs in the absence of inhibitor by that obtained in the presence of inhibitor.
P < 0.01, significantly different from values obtained in the absence of inhibitor.
Crizotinib reversed ABCB1-mediated MDR in nude mouse xenografts
An established KBv200 cell xenograft model in female nude mice was used to evaluate the efficacy of crizotinib to reverse the resistance to paclitaxel in vivo. There was no significant difference in tumour size between animals treated separately with saline, crizotinib or paclitaxel, indicating the in vivo resistance to paclitaxel. However, the combination of crizotinib and paclitaxel produced a significant inhibition of tumour growth compared with animals treated with saline, paclitaxel, or crizotinib alone (P < 0.05; Figure 2B and C). The ratio of tumour growth inhibition by the combination was 46.1% (Supporting Information Table S2). Furthermore, at the doses tested, no mortality or apparent decrease in body weight was observed in the combination treatment groups, suggesting that the combination regimen did not increase the incidence of toxic side effects (Figure 2A).
Figure 2.

Potentiation of the antitumour effects of paclitaxel by crizotinib in a KBv200 xenograft model in athymic nude mice. A, changes of body weight after tumour cell inoculation. Data shown are means ± SD for each group of seven mice after implantation. B, changes in tumour volume with time after tumour cell inoculation. Data shown are means ± SD for each group of seven mice after implantation. C, tumour size. The photograph was taken on the 24th day after implantation. The various treatments were as follows: control (vehicle alone); paclitaxel (18 mg·kg−1, i.p., q3d × 4); crizotinib (25 mg·kg−1, p.o., q3d × 4) and paclitaxel (18 mg·kg−1, i.p., q3d × 4) plus crizotinib (25 mg·kg−1, p.o., q3d × 4, given 1 h before paclitaxel administration).
Crizotinib enhanced the accumulation of doxorubicin and rhodamine 123 in MDR cells overexpressing ABCB1
The results above indicated that crizotinib could enhance the sensitivity of MDR cancer cells to certain ABCB1 substrate anticancer drugs. To understand the underlying mechanisms, the intracellular accumulation of doxorubicin and rhodamine 123 in the presence or absence of crizotinib was examined by flow cytometric analysis. Upon incubation with the fluorescent substrates alone, intracellular fluorescence intensity of doxorubicin was significantly higher in the KB (3.46-fold) and MCF-7 cells (4.27-fold) than that in the KBv200 and MCF-7/adr cells, whereas that of rhodamine 123 was 18.3-fold higher in KB and 12.5-fold higher in MCF-7 cells, compared with KBv200 and MCF-7/adr cells respectively (Figure 3). When the KBv200 and MCF-7/adr cells were treated with crizotinib, the intracellular accumulation of doxorubicin was increased by 1.27-, 1.95-, 2.37-fold in KBv200 cells and 1.23-, 1.57-, 1.98-fold in MCF-7/adr cells, but no alteration in KB cells and MCF-7 cells was observed in the presence of 0.375, 0.75 and 1.5 µM of crizotinib respectively (Figure 3A and B). As shown in Figure 3C and D, crizotinib at 0.375, 0.75 and 1.5 µM increased the intracellular accumulation of rhodamine 123 by 2.07-, 3.21-, 4.90-fold in KBv200 cells and 2.40-, 3.87- and 5.32-fold in MCF-7/adr cells at the concentration of 0.375, 0.75 and 1.5 µM respectively. However, no significant change in the intracellular accumulation of rhodamine 123 was observed in the parental MCF-7 and KB cells upon combination treatment with crizotinib. Taken together, these results suggest that crizotinib is able to inhibit the transport activity of ABCB1 in MDR cells.
Figure 3.

Effect of crizotinib on the accumulation of doxorubicin and rhodamine 123. The accumulation of doxorubicin (Dox; A, B) and rhodamine 123 (C, D) were measured by flow cytometric analysis. The results are presented as fold change in fluorescence intensity relative to control MDR cells. Data shown are means ± SD of triplicate determinations. **P < 0.01 significantly different from control group.
Crizotinib inhibited the efflux of doxorubicin in MDR cells overexpressing ABCB1
Crizotinib increased intracellular accumulation of anticancer agents such as doxorubicin and of rhodamine 123 in ABCB1 MDR cells; we now determined if the increased accumulation of anticancer agents was due to inhibition of efflux. The time course of doxorubicin efflux during 2 h after accumulation is shown in Figure 4A. This Figure also shows that crizotinib inhibited drug efflux of ABCB1 in KBv200 cells but did not influence drug efflux in sensitive KB cells. For example, at 120 min, 49.7% of accumulated doxorubicin was pumped out of KBv200 cells in the presence of 1.5 µM crizotinib, while 70.3% of accumulated doxorubicin was lost from KBv200 cells in the absence of crizotinib (P < 0.05). In KB cells, 21.6% of accumulated doxorubicin was lost from KB cells at 120 min in the presence of 1.5 µM crizotinib, while 23.8% of accumulated doxorubicin was lost in the absence of crizotinib (P > 0.05). These results indicated that crizotinib could effectively inhibit drug efflux of ABCB1.
Figure 4.
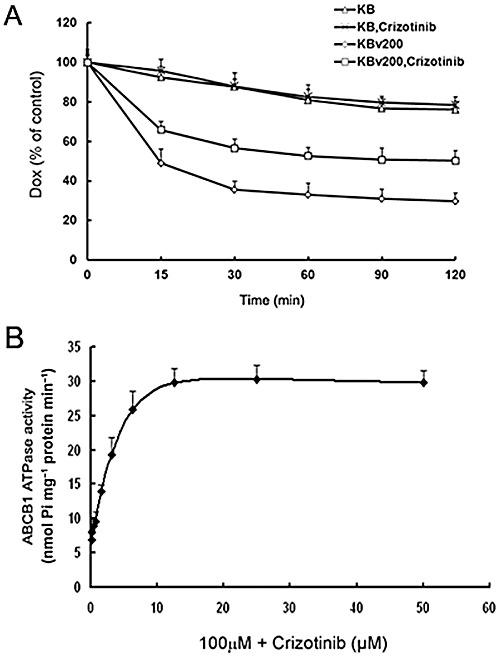
Effect of crizotinib on the efflux of doxorubicin and on ABCB1 ATPase activity. A, Time course of doxorubicin (Dox) efflux was measured in KB and KBv200 cells, with or without 1.5 µM crizotinib. B, ABCB1 ATPase assays were performed according to the instruction of Pgp-Glo™ Assay Systems. All these experiments were repeated at least three times. Data shown are means ± SD for independent determinations in triplicate.
Crizotinib stimulated the ATPase activity of ABCB1
Like all other ABC transporters, the drug efflux function of ABCB1 is driven by ATP hydrolysis. Therefore, ATP consumption has been generally used to reflect ATPase activity of the transporter. To assess the effect of crizotinib on the ATPase activity of ABCB1, ABCB1-mediated ATP hydrolysis at different concentrations of crizotinib was measured. We found that crizotinib was an activator of ABCB1 ATPase. As shown in Figure 4B, crizotinib increased verapamil-stimulated ATPase activity in a dose-dependent manner.
Crizotinib did not alter ABCB1 expression at both mRNA and protein levels
Apart from the inhibition of transport by ABCB1, reversal of ABC transporter-mediated MDR could also be achieved by decreased transporter expression. Therefore, we determined the effects of crizotinib on the expression of ABCB1. To assess the effect of crizotinib on ABCB1 expression at mRNA and protein levels, reverse transcription-PCR, real time PCR and Western blot analysis were performed. Our results showed that ABCB1 expression at mRNA or protein levels (Figure 5) was not significantly altered. These results indicate that the modulation of ABCB1 expression was not involved in the reversal of ABCB1-mediated MDR by crizotinib.
Figure 5.

Effect of crizotinib on the expression of ABCB1 in MDR cells. KBv200 cells were treated with crizotinib at various concentrations for 48 h: A, equal amounts of total cell lysates were loaded and detected by Western blot; B, the mRNA level of ABCB1 was determined by RT-PCR. C, summary data from the mRNA level of ABCB1 determined by RT real-time-PCR. A representative result is shown from at least three independent experiments. All these experiments were repeated at least three times, and a representative experiment is shown in each panel.
MDR reversal by crizotinib did not involve the blockade of phosphorylation of c-Met, Akt and ERK1/2
The phosphorylation of Akt and ERK1/2, the downstream markers of crizotinib targets, can be utilized to test the targeted activity of crizotinib (Zillhardt et al., 2010). Previous studies have shown that the inhibition of the Akt and ERK1/2 pathways may enhance the efficacy of chemotherapeutic agents in cancer cells (Oh et al., 2006; Gagnon et al., 2008). We therefore tested phosphorylation of c-Met, Akt or ERK1/2 over a range of concentrations of crizotinib. 10 µM crizotinib was used as a positive control for blockade of c-Met phosphorylation. Another ABCB1-inhibiting TKI, lapatinib, was used as a positive control for blockade of Akt and ERK1/2 phosphorylation. As shown in Figure 6, after incubation with a range of concentrations of crizotinib and over 24 h, the phosphorylation of c-Met, Akt and ERK1/2 were not significantly affected. These results suggest that MDR reversal by crizotinib in the drug-resistant KBv200 cells did not involve inhibition of c-Met, Akt or ERK1/2 phosphorylation.
Figure 6.

Effect of crizotinib on phosphorylation of c-Met, Akt and ERK1/2. KB and KBv200 cells were treated with crizotinib at various concentrations for 24 h and for various times at 1.5 µM. 10 µM crizotinib was used as a positive control for blockade of c-Met phosphorylation. 10 µM lapatinib was used as a positive control for blockade of Akt and ERK1/2 phosphorylation. Equal amounts of protein were loaded for Western blot analysis. Independent experiments were performed at least three times and results from a representative experiment are shown.
Discussion and conclusions
The emerging paradigm of molecular targeted chemotherapy has attracted much basic science and clinical research on the novel inhibitors specific for oncogenic receptor tyrosine kinases (RTK) in various cancers (Schlessinger, 2000; Blume-Jensen and Hunter, 2001). Recent examples of successful therapeutic intervention with TKIs include imatinib in chronic myeloid leukaemia with oncoprotein BCR-ABL expression (Nimmanapalli et al., 2002; Azam et al., 2003), erlotinib in NSCLC with mutant and/or amplified epidermal growth factor receptor (Giaccone, 2005; Costa et al., 2008), trastuzumab in breast cancers with amplified/elevated HER-2 (Emens, 2005; Jackisch, 2006) and sunitinib targeting the von Hippel-Lindau (VHL)-dependent VEGF pathway in renal cell carcinoma (RCC)(Deprimo et al., 2007). Currently, a subset of NSCLC was found to carry a translocation, in which the echinoderm EML4 gene is fused to ALK, representing one of the newest molecular targets in NSCLC (Soda et al., 2007).
Crizotinib (PF-02341066) is the first agent in clinical use to selectively target the EML4-ALK translocation in NSCLC patients. Crizotinib inhibited both c-Met and ALK tyrosine kinases and their oncogenic variants, reduced c-Met and ALK phosphorylation in intact tumour cells, with IC50 values in the nM range and blocked cell cycle progression at the G1-S–phase checkpoint, inducing apoptosis (Christensen et al., 2007). Further studies demonstrated that crizotinib inhibited angiogenesis and progression of a number of xenograft and orthotropic nude mice models, including NSCLC, gastric carcinoma, glioblastoma, prostate carcinoma, breast carcinoma and colon carcinoma (Christensen et al., 2007; Zou et al., 2007). Phase I studies showed that crizotinib was generally well tolerated at dose up to 250 mg·day−1 with oral administration schedules (C. Li et al., 2011). More recently, crizotinib has entered phase II/III in its clinical development.
MDR-ABC transporters have recently been recognized as important determinants of the pharmacokinetic and toxicological properties of low MW TKIs, as well as key factors of resistance against targeted anticancer therapeutics (Brozik et al., 2011). Previous studies have shown that several TKIs can inhibit the functions of transporters, including ABCB1, ABCC1 and ABCG2, which are major factors in the development of MDR (Shi et al., 2007; Dai et al., 2009; Tao et al., 2009). Thus, it is possible that TKIs could be used, in combination with other anticancer drugs, to counteract or prevent MDR, thereby providing synergistic cytotoxic effects. The objectives of this study were to examine the reversal by crizotinib of ABC transporter-mediated drug resistance (MDR) and to understand the underlying mechanisms.
In the present study, we showed for the first time that crizotinib had potent reversing activity in ABCB1-expressing MDR cells in vitro. As demonstrated by MTT assay, the working concentrations of crizotinib chosen to study the MDR reversal effect was only weakly cytotoxic (inhibition rate < 15%). Crizotinib at 1.5 µM significantly increased the sensitivity of KBv200, MCF-7/adr and HEK293/ABCB1 cells to doxorubicin by 10.2, 4.1, 3.9-fold, and paclitaxel by 4.0, 3.7, 4.2 fold respectively (Table 1). However, crizotinib did not significantly sensitize the corresponding parental KB, MCF-7 or HEK293/pcDNA cells. Additionally, there were no additive or synergistic effects between crizotinib and non-ABCB1 substrates, such as cisplatin. Furthermore, crizotinib did not significantly alter cellular sensitivity to ABCG2 or ABCC1 substrates. These suggest that the sensitization of the resistant cells by crizotinib is probably due to its specific effect on ABCB1.
In human pharmacokinetic studies, the highest peak plasma crizotinib level was roughly 0.6 µM, the half-life was approximately 50 h and steady-state concentrations were achieved after 15 days after repeated dosing at 250 mg b.i.d. (Tan et al., 2010; Li et al., 2011). These data suggest that the lowest concentration of crizotinib used in our in vitro experiments could be attained in patients, while the highest and medium concentrations may exceed the plasma concentration after therapeutic treatment. However, higher concentrations of drugs may be detected in tumour tissues than in normal tissues and plasma, because of various functions of impaired tumour vasculature (Dreher et al., 2006). Therefore, it is possible the in vitro concentrations of crizotinib used in our reversal experiments could be obtained in tumour tissues after therapeutic treatment. In order to determine whether the in vitro effects of crizotinib can be translated to the in vivo setting, we examined the effect of crizotinib on the antitumour activity of paclitaxel in ABCB1-overexpressing KBv200-inoculated xenograft model. As gender affects the pharmacokinetics and toxicity of crizotinib in mice (Zhong et al., 2010), female mice were used in our experiments. Agreeing with the in vitro findings, our results indicated that the combination of crizotinib with paclitaxel resulted in markedly enhanced antitumour activity of paclitaxel in the KBv200 tumour xenograft model (Figure 2B and C). Additionally, we tested crizotinib in the KB tumour xenografts to exclude the effect of modulation of drug exposure. The results showed there was no significant difference in tumour size between paclitaxel and the combination of crizotinib with paclitaxel groups in the KB tumour xenograft model (Supporting Information Figure S1B, C and Table S3). Moreover, there was no substantially increased loss of body weight in mice treated with the drug combination compared with the individual drug treatment alone (Figure 2A and Supporting Information Figure S1A). Indeed, our results indicated that the combination of crizotinib with paclitaxel resulted in markedly enhanced antitumor activity of paclitaxel in the ABCB1-overexpressing tumour xenograft model.
The overexpression of ABCB1 was generally known to mediate MDR by actively pumping its substrate anticancer drugs out of the cells (Perez-Tomas, 2006). Therefore, to investigate the mechanism of ABCB1-mediated MDR reversal by crizotinib, ABCB1 transport activity was examined. Consistent with cytotoxicity data, crizotinib was found to significantly increase the intracellular accumulation of doxorubicin and rhodamine 123 in ABCB1-overexpressing MDR cells in a dose-dependent manner (P < 0.05) (Figure 3), without any observable effect in the corresponding parental KB and MCF-7 cells. Besides, crizotinb effectively inhibited drug efflux via ABCB1 (Figure 4A). Therefore, crizotinib may counteract MDR by increasing the intracellular concentration of its substrate anticancer drugs via inhibition of their efflux.
Because energy derived from ATP hydrolysis is required for ABC transporters to pump their substrate drugs out of cells, the profile of drug-stimulated ATPase activity in the ABCB1-expressing membrane is thought to reflect the nature of interaction of transporter pumps with drug substrates (Brozik et al., 2011). Based on their effect on ATPase activity of ABC transporters, a variety of transporter modulators can be categorized into three distinct classes. The first class of compounds stimulates ATPase activity at low concentrations but inhibits the activity at high concentrations, the second class of compounds enhances ATPase activity in a dose-dependent manner without any inhibition, whereas the third class of compounds inhibits both basal and stimulated ATPase activity (Ambudkar et al., 1999).We previously reported that some TKIs such as lapatinib, sunitinib and erlotinib can stimulate ATPase activities of the MDR transporters at low concentrations but inhibit the ATPase activities at higher concentrations (Shi et al., 2007; Dai et al., 2008). In the present experiments, crizotinib was found to stimulate the ABCB1 ATPase activity assay in a dose-dependent manner (Figure 4B and Supporting Information Figure S2). These data suggest that crizotinib belongs to the second class of compounds to interact with ABC transporters and is likely to be a substrate and therefore a competitive inhibitor of ABCB1.
To investigate the mechanism of ABCB1-mediated MDR reversal by crizotinib, the possible regulation of expression of ABCB1 by crizotinib was also examined. ABCB1 expression at both mRNA and protein levels in the resistant cells were not affected by a maximum concentration of up to 3 µM of crizotinib (48 h incubation) (Figure 5). Therefore, it is unlikely that crizotinib reversed ABCB1-mediated MDR via the downregulation of ABCB1 expression.
Crizotinib is a selective low MW inhibitor of both c-Met/ HGF receptors and ALK tyrosine kinases, and preclinical studies demonstrated that crizotinib inhibited cell proliferation and induced apoptosis via blocking downstream signalling pathways such as phosphorylation of Akt and ERK1/2 (Zou et al., 2007; Takezawa et al., 2011). Moreover, activation of PI3K/Akt and/or ERK pathways is related to resistance to conventional chemotherapeutic agents (West et al., 2002; Knuefermann et al., 2003; McCubrey et al., 2007). To determine whether these pathways were involved in the observed reversal of ABCB1-mediated MDR by crizotinib, activation of c-Met, Akt and ERK1/2 was examined. However, crizotinib (up to 1.5 µM) did not block the phosphorylation of c-Met, Akt or ERK1/2 in the tested cell lines (Figure 6), suggesting that inhibition of c-Met, Akt or ERK1/2 was not involved in the reversal of ABCB1-mediated MDR by crizotinib.
In conclusion, this study provides the first evidence that crizotinib significantly enhanced the efficacy of chemotherapeutic drugs in ABCB1-overexpressing MDR cells, which is likely to be attributable to the competitive inhibition of the transport function of ABCB1. Moreover, MDR reversal seems to be independent of the blockade of tyrosine kinases. Importantly, confirmation of MDR reversal by crizotinib in tumour xenograft model further supports the potential usefulness of combining crizotinib with other conventional anticancer drugs in combating MDR in cancer chemotherapy.
Acknowledgments
We like to thank Drs SE Bates and RW Robey (National Cancer Institute, NIH) for the ABCG2 expressing cell lines, ABCB1 transfectant cell lines (NIH, MD.). This work was funded by grants from China National Natural Sciences Foundation no. 81072669, 81061160 (L-W Fu) and no. 507/N_CUHK443/10 (K-K To).
Glossary
- ABC
ATP-binding cassette
- ABCB1
ABC transporter-subfamily B member 1
- ABCC1
ABC transporter-subfamily C member 1
- ABCG2
ABC transporter-subfamily G member 2
- ALK
anaplastic lymphoma kinase
- EML4
echinoderm microtubule-associated protein–like 4
- HER
human epidermal receptor
- MDR
multidrug resistance
- NSCLC
non-small cell lung cancer
- P-gp
(ABCB1) P-glycoprotein
- RCC
renal cell carcinoma
- RTK
receptor tyrosine kinases
- TKI
tyrosine kinase inhibitor
Conflicts of interest
There are no potential conflicts of interest to be disclosed.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Potentiation of the antitumour effects of paclitaxel by crizotinib in a KB xenograft model in athymic female nude mice. A, changes of body weight after tumour cell inoculation. Data shown are means ± SD for each group of nine mice after implantation. B, changes in tumour volume with time after tumour cell inoculation. Data shown are means ± SD for each group of nine mice after implantation. C, tumour size. The picture was taken on the 24th day after implantation. The various treatments were as follows: control (vehicle alone); paclitaxel (18 mg·kg−1, i.p., q3d × 4); crizotinib (25 mg·kg−1, p.o., q3d × 4) and paclitaxel (18 mg·kg−1, i.p., q3d × 4) plus crizotinib (25 mg·kg−1, p.o., q3d × 4, given 1 h before paclitaxel administration).
Figure S2 Effect of crizotinib on ABCB1 ATPase at low concentrations. ABCB1 ATPase assays were performed according to the instruction of Pgp-Glo™ Assay Systems. All these experiments were repeated at least three times. Each point represents the mean ± SD for independent determinations in triplicate. *P < 0.05, **P < 0.01, significantly different from control.
Table S1 Effect of crizotinib in ABCB1-, ABCC1- and ABCG2-mediated sensitive and resistant cell lines. Cell survival was determined by MTT assay. Data are the means ± SD of at least three independent experiments performed in triplicate. **P < 0.01 significantly different from IC50 values of parental, sensitive cells
Table S2 Effect of crizotinib on the reversal of MDR in KBv200 cell xenografts. Changes of body weight and weight of tumour after KBv200 cell inoculation. The experiments were conducted as described in Methods. The values presented are the means ± SD for each group. *P < 0.05, significantly different from control values
Table S3 Effect of crizotinib on the reversal of MDR in KB cell xenografts. Changes of body weight and weight of tumour after KB cell inoculation. The experiments were conducted as described in Methods. The values presented are the means ± SD for each group; *P < 0.05, significantly different from control values
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- Bakos E, Homolya L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1) Pflugers Arch. 2007;453:621–641. doi: 10.1007/s00424-006-0160-8. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Brozik A, Hegedus C, Erdei Z, Hegedus T, Ozvegy-Laczka C, Szakacs G, et al. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol. 2011;7:623–642. doi: 10.1517/17425255.2011.562892. [DOI] [PubMed] [Google Scholar]
- Chen LM, Liang YJ, Ruan JW, Ding Y, Wang XW, Shi Z, et al. Reversal of P-gp mediated multidrug resistance in-vitro and in-vivo by FG020318. J Pharm Pharmacol. 2004a;56:1061–1066. doi: 10.1211/0022357043879. [DOI] [PubMed] [Google Scholar]
- Chen LM, Wu XP, Ruan JW, Liang YJ, Ding Y, Shi Z, et al. Screening novel, potent multidrug-resistant modulators from imidazole derivatives. Oncol Res. 2004b;14:355–362. doi: 10.3727/0965040041292378. [DOI] [PubMed] [Google Scholar]
- Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6((Pt 1)):3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- Coley HM. Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol Biol. 2010;596:341–358. doi: 10.1007/978-1-60761-416-6_15. [DOI] [PubMed] [Google Scholar]
- Costa DB, Nguyen KS, Cho BC, Sequist LV, Jackman DM, Riely GJ, et al. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin Cancer Res. 2008;14:7060–7067. doi: 10.1158/1078-0432.CCR-08-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Liang YJ, Wang YS, Tiwari AK, Yan YY, Wang F, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009;279:74–83. doi: 10.1016/j.canlet.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001a;42:1007–1017. [PubMed] [Google Scholar]
- Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001b;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Deprimo SE, Bello CL, Smeraglia J, Baum CM, Spinella D, Rini BI, et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32. doi: 10.1186/1479-5876-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- Emens LA. Trastuzumab: targeted therapy for the management of HER-2/neu-overexpressing metastatic breast cancer. Am J Ther. 2005;12:243–253. [PubMed] [Google Scholar]
- Fu L, Liang Y, Deng L, Ding Y, Chen L, Ye Y, et al. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol. 2004;53:349–356. doi: 10.1007/s00280-003-0742-5. [DOI] [PubMed] [Google Scholar]
- Gagnon V, Van Themsche C, Turner S, Leblanc V, Asselin E. Akt and XIAP regulate the sensitivity of human uterine cancer cells to cisplatin, doxorubicin and taxol. Apoptosis. 2008;13:259–271. doi: 10.1007/s10495-007-0165-6. [DOI] [PubMed] [Google Scholar]
- Giaccone G. Targeting HER1/EGFR in cancer therapy: experience with erlotinib. Future Oncol. 2005;1:449–460. doi: 10.2217/14796694.1.4.449. [DOI] [PubMed] [Google Scholar]
- Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–6639. [PubMed] [Google Scholar]
- Jackisch C. HER-2-positive metastatic breast cancer: optimizing trastuzumab-based therapy. Oncologist. 2006;11(Suppl. 1):34–41. doi: 10.1634/theoncologist.11-90001-34. [DOI] [PubMed] [Google Scholar]
- Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49:337–343. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- Li C, Alvey C, Bello A, Wilner KD, Tan W. Pharmacokinetics (PK) of crizotinib (PF-02341066) in patients with advanced non-small cell lung cancer (NSCLC) and other solid tumors. J Clin Oncol. 2011;29(Suppl) abstr e13065. [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmanapalli R, O’Bryan E, Huang M, Bali P, Burnette PK, Loughran T, et al. Molecular characterization and sensitivity of STI-571 (imatinib mesylate, Gleevec)-resistant, Bcr-Abl-positive, human acute leukemia cells to SRC kinase inhibitor PD180970 and 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2002;62:5761–5769. [PubMed] [Google Scholar]
- Oh SY, Song JH, Gil JE, Kim JH, Yeom YI, Moon EY. ERK activation by thymosin-beta-4 (TB4) overexpression induces paclitaxel-resistance. Exp Cell Res. 2006;312:1651–1657. doi: 10.1016/j.yexcr.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47–50. [PubMed] [Google Scholar]
- Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV, et al. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R) Biochem Pharmacol. 2008;75:1302–1312. doi: 10.1016/j.bcp.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1) J Bioenerg Biomembr. 2007;39:481–487. doi: 10.1007/s10863-007-9115-9. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–123. doi: 10.1016/s1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Shi Z, Liang YJ, Chen ZS, Wang XW, Wang XH, Ding Y, et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther. 2006;5:39–47. doi: 10.4161/cbt.5.1.2236. [DOI] [PubMed] [Google Scholar]
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17:2140–2148. doi: 10.1158/1078-0432.CCR-10-2798. [DOI] [PubMed] [Google Scholar]
- Tan W, Wilner KD, Bang Y, Kwak EL, Maki RG, Camidge DR, et al. Pharmacokinetics (PK) of PF-02341066, a dual ALK/MET inhibitor after multiple oral doses to advanced cancer patients. J Clin Oncol. 2010;28(15) Suppl. abstr 2596. [Google Scholar]
- Tang R, Faussat AM, Majdak P, Perrot JY, Chaoui D, Legrand O, et al. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia. 2004;18:1246–1251. doi: 10.1038/sj.leu.2403390. [DOI] [PubMed] [Google Scholar]
- Tao LY, Liang YJ, Wang F, Chen LM, Yan YY, Dai CL, et al. Cediranib (recentin, AZD2171) reverses ABCB1- and ABCC1-mediated multidrug resistance by inhibition of their transport function. Cancer Chemother Pharmacol. 2009;64:961–969. doi: 10.1007/s00280-009-0949-1. [DOI] [PubMed] [Google Scholar]
- West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat. 2002;5:234–248. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Wu HY, Xia XK, Liang YJ, Yan YY, She ZG, et al. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol Ther. 2007;6:1413–1421. doi: 10.4161/cbt.6.9.4543. [DOI] [PubMed] [Google Scholar]
- Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS ONE. 2009;4:e5172. doi: 10.1371/journal.pone.0005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong WZ, Zhan J, Kang P, Yamazaki S. Gender specific drug metabolism of PF-02341066 in rats–role of sulfoconjugation. Curr Drug Metab. 2010;11:296–306. doi: 10.2174/138920010791514207. [DOI] [PubMed] [Google Scholar]
- Zillhardt M, Christensen JG, Lengyel E. An orally available small-molecule inhibitor of c-Met, PF-2341066, reduces tumor burden and metastasis in a preclinical model of ovarian cancer metastasis. Neoplasia. 2010;12:1–10. doi: 10.1593/neo.09948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.