

Abstract
BACKGROUND AND PURPOSE
JNJ-26070109 [(R)4-bromo-N-[1-(2,4-difluoro-phenyl)-ethyl]-2-(quinoxaline-5-sulfonylamino)-benzamide] is a novel antagonist at cholecystokinin CCK2 receptors with good pharmacokinetic properties and represents a novel mechanism for the treatment of gastro-oesophageal reflux disease (GORD). The purpose of the present study was to determine whether chronic treatment with JNJ-26070109 could prevent, as well as treat, acid rebound in rats.
EXPERIMENTAL APPROACH
A chronic fistula was surgically inserted into the stomach of rats to enable the measurement of acid secretion under basal, pentagastrin and histamine-stimulated conditions. JNJ-26070109 and omeprazole were administered separately and in combination.
KEY RESULTS
Sustained administration of omeprazole alone and in combination with JNJ-26070109 inhibited gastric acid secretion by >90%. However, 3 days after withdrawing treatment, there was a rebound hypersecretion by ∼1.5-fold in omeprazole-treated animals. No such acid rebound was observed with JNJ-26070109 alone or with co-administration of JNJ-26070109 and omeprazole. The anti-trophic effects of JNJ-26070109 in the gastric mucosal paralleled the effects on acid rebound. Administration of JNJ-26070109 for 3 days after cessation of omeprazole prevented the occurrence of acid rebound. Interestingly, chronic, but not acute, treatment with JNJ-26070109 also inhibited histamine-stimulated acid secretion.
CONCLUSIONS AND IMPLICATIONS
Chronic administration of JNJ-26070109 effectively inhibited gastric acid secretion and suppressed proton pump inhibitor (PPI)-induced acid rebound in the rat. This work advances the field by demonstrating that modest doses of a competitive CCK2 receptor antagonist have significant and functionally important anti-trophic actions in the gastric mucosa. These properties make JNJ-26070109 a suitable candidate for clinical investigation for the treatment of GORD.
Keywords: JNJ-26070109, omeprazole, gastro-oesophageal reflux disease, GORD, gastric acid secretion, acid rebound, CCK receptor, gastrin, ECL cell, parietal cell
Introduction
Gastrin and cholecystokinin (CCK) are gut–brain hormones that exert their actions through seven-transmembrane receptors now called CCK1 and CCK2 receptors (Noble et al., 1999; Cawston and Miller, 2010; Alexander et al., 2011). While CCK is an agonist at both receptors, gastrin is highly selective for the CCK2 receptor. In the periphery, gastrin is a major hormonal stimulant of gastric acid secretion and growth of the gastric mucosa. Accordingly, antagonism of the CCK2 receptor could be an effective means to treat gastro-oesophageal reflux disease (GORD; Wettstein et al., 1994). The potential value of this approach becomes apparent upon examination of the control of gastric acid secretion (Black and Shankley, 1987; Morton et al., 2011a). Acid secretion is regulated in a complex, non-hierarchical fashion with the two most important determinants being central input via the vagus nerve and meal-stimulated gastrin release from the antrum of the stomach (Dockray et al., 2001). The dominant effect of gastrin is to activate CCK2 receptors on the enterochromaffin-like (ECL) cell to stimulate the local release of histamine. Histamine subsequently acts on histamine H2 receptors on parietal cells and stimulates the hydrogen-potassium ATPase, the site of action of the proton pump inhibitors (PPIs), to secrete acid into the stomach. Input from the vagus nerve, and to a lesser degree gastrin activation of CCK2 receptors, on the parietal cells has also been shown to be required for maximum acid secretion. Acetylcholine released from the vagus nerve acts via muscarinic M3 receptors on the parietal cell to facilitate gastric acid secretion but also on M2 and M4 sub-types on somatostatin releasing D cells. Inhibition of D cell release of somatostatin releases a tonic restraint on ECL and parietal cells to facilitate gastric acid secretion. Gastrin release from the antrum of the stomach is facilitated by input from the vagus. Given the important role of gastrin in the regulation of acid secretion, acute antagonism of the CCK2 receptor is expected to provide effective and rapid inhibition of gastric acid secretion.
Under normal physiological conditions, the release of gastrin is under feedback control so that prolonged periods of hypergastrinaemia do not occur. Thus, gastrin is released phasically in response to a meal. When the acid buffering capacity of the meal is overcome by the secreted acid, the intragastric pH decreases. This decrease in pH inhibits the secretion of further gastrin from the gastric antrum. Consequently, any effective form of gastric acid secretion inhibition (e.g. histamine H2 receptor antagonism and PPIs) causes sustained hypergastrinaemia. Hypergastrinaemia increases the acid secretion capacity of the stomach as gastrin in addition to its acute actions also exerts a powerful trophic action on the gastric mucosa via CCK2 receptors. This trophic action has been implicated in occurrence of ‘acid rebound’ upon withdrawal of acid suppression therapy (Waldum et al., 1996; Gillen et al., 1999; Gillen and McColl, 2001). This effect has been investigated in detail in rats where it was shown that chronic acid suppression results in hypergastrinaemia, increased histamine content of the gastric mucosa, increased ECL cell density and thickness of the gastric mucosa as well as a sustained increase in pentagastrin-stimulated gastric acid secretion for up to 56 days after cessation of treatment (Nishida et al., 1995, also see Waldum et al., 1991). These effects were prevented by co-administration of the CCK2 receptor antagonist YM022 (Nishida et al., 1995). This work supported the role of hypergastrinaemia and ECL proliferation in acid rebound and also established an experimental model that facilitates the evaluation and development of CCK2 receptor antagonists for the treatment of GORD. The consequences of acid-suppression therapy induced hypergastrinaemia have also been investigated in man. Sustained treatment with PPIs or histamine H2 receptor antagonists increases the size and number of ECL and parietal cells in humans (Lamberts et al., 1988; Driman et al., 1996; Waldum et al., 1996; Weinstein et al., 1996). Acid rebound is correlated to the duration of treatment with acid inhibition therapy and is long lasting after discontinuing treatment (Larsson et al., 1988; Zhao et al., 1998; Gillen and McColl, 2001; Reimer et al., 2009). The hyperplasia within the acid secretory apparatus might account for the difficulties some patients have in terminating acid suppression therapy (e.g. >70% of patients resume PPI use within 1 year, Björnsson et al., 2006). Taken together these data suggest that CCK2 receptor antagonism could be an effective treatment for GORD in its own right and differentiated from existing therapies by preventing the consequences of hypergastrinaemia that occur with all known forms of acid suppression therapy.
The purpose of the current study was to evaluate the effects of a recently described low MW, orally bioavailable CCK2 receptor antagonist, (R)4-bromo-N-[1-(2,4-difluoro-phenyl)-ethyl]-2-(quinoxaline-5-sulfonylamino)-benzamide (JNJ-26070109; Morton et al., 2011b), in a rat model of acid hypersecretion (Nishida et al., 1995). Sustained treatment with JNJ-26070109 over 21–24 days suppressed basal, pentagastrin and histamine-stimulated gastric acid secretion. When administered concurrently with omeprazole, JNJ-26070109 prevented acid rebound and suppressed the ECL cell proliferation but did not cause hypoplasia when administered alone. JNJ-26070109 also reversed acid rebound when given for a short time (3 days) following omeprazole treatment. Overall, these studies demonstrated that chronic administration of JNJ-26070109 effectively inhibited basal and stimulated acid secretion and also suppressed PPI-stimulated acid rebound in the rat. These properties make JNJ-26070109 a suitable candidate for clinical investigation in the treatment of GORD.
Methods
All animal care and experimental procedures were performed according to the internationally accepted guidelines for the care and use of laboratory animals in research and were approved by the local Institutional Animal Care and Use Committee.
Implementation of gastric fistulas
Male Sprague Dawley rats (160–190 g), fasted for 18 h (water ad libitum), were anaesthetized with isoflurane (1–3%) mixed in clean dry air. A stainless steel gastric fistula was implanted and analgesia was provided for 3 days post-operatively (0.03 mg·g−1 buprenorphine subcutaneously every 12 h). A post-operative recovery period of 7–10 days was allowed before experimentation. During this recovery period, the rats were conditioned by having their stomachs rinsed with unbuffered gastric mucosal physiological solution (135 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2 and 31.6 mM glucose at pH 6.6) and by being placed for 3 h in the metabolic cages used to house the animals during gastric acid secretion collection. The rats adapted readily and rapidly to the fistula and operated animals were maintained for as long as 4 months without complications.
Study A: The effect of sustained treatment (21 days) with 10 µmol·kg−1 JNJ-26070109 and/or 400 µmol·kg−1 omeprazole on acid secretion, hypergastrinaemia, trophism and acid rebound
Rats with a gastric fistula (n= 7 per treatment group) were dosed orally with either 400 µmol·kg−1 omeprazole once a day, 10 µmol·kg−1 JNJ-26070109 twice a day (∼10 h apart), a combination of omeprazole and JNJ-26070109 or vehicle controls (see Figure 1 for study design). At peak plasma concentration this dose of JNJ-26070109 produced near maximal inhibition of pentagastrin-stimulated (30 nmol·kg−1 s.c.) acid secretion. The dose was administered 45 min before measurement of acid secretion and assessment of each end-point was 90 min in duration; see Morton et al., 2011b. The frequency of administration of JNJ-26070109 was selected to provide sufficient antagonism of CCK2 receptors to prevent the trophic actions of the evoked hypergastrinaemia in this model (half-life of JNJ-26070109 = 1.8 h in rats, Morton et al., 2011b). On day 21, gastric acid secretion was measured 12–14 h after the final administration of the compounds. These rats were maintained for a further 3 days with no drug administration and gastric acid secretion was measured again ∼84 h after the final dose of compounds. An additional group of omeprazole-treated rats was included in the study to investigate whether short-term treatment with a CCK2 receptor antagonist could reverse omeprazole-induced acid hypersecretion (acid rebound). In this group of rats, treatment was switched from omeprazole to JNJ-26070109 after 21 days and treatment with JNJ-26070109 (10 µmol·kg−1 b.i.d.) was continued for a further 3 days. The last dose of JNJ-26070109 was administered ∼14 h before measurement of gastric acid secretion with the expectation that JNJ-26070109 plasma levels would be below those required for significant CCK2 receptor occupancy at the time when gastric acid secretion was measured (for pharmacokinetic analysis of JNJ-26070109 see Morton et al., 2011b). Thus, any effect observed at this time could be attributed to the anti-trophic effects of CCK2 receptor antagonism rather than an acute effect that would require effective plasma concentrations of JNJ-26070109. A blood sample was also taken after collection of the final sample of gastric secretion to confirm this. An additional group of rats, which did not have gastric fistulas, were included in the study to evaluate the effects of omeprazole and JNJ-26070109 on gastrin plasma levels and markers of gastric mucosal proliferation. On day 21, these rats were killed using a rising concentration of CO2 and blood and tissue samples were taken as described later.
Figure 1.
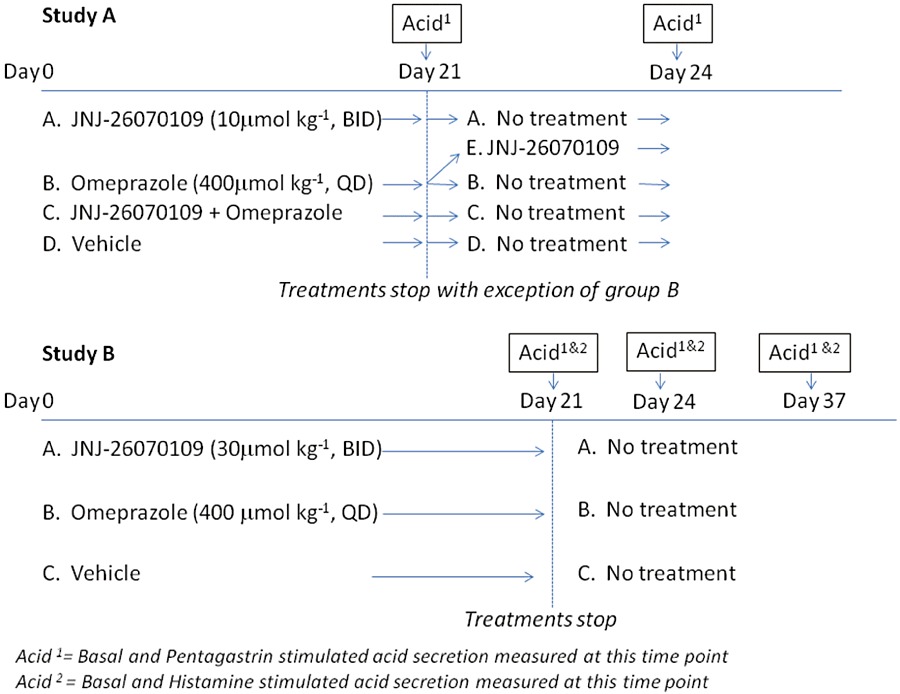
Experimental design for Studies A and B.
Study B: The effect of sustained treatment (21–24 days) with 30 µmol·kg−1 JNJ-26070109 or 400 µmol·kg−1 omeprazole on basal, pentagastrin and histamine-stimulated acid secretion and acid rebound in rats
This series of experiments were conducted to evaluate the effects of both a higher dose of JNJ-26070109 (30 µmol·kg−1 p.o.) and also the additional acid-secretagogue, histamine (45 µmol·kg−1 s.c.) on acid secretion in conscious rats (for study design see Figure 1, Study B). JNJ-26070109 (30 µmol·kg−1) and omeprazole (400 µmol·kg−1) were given for 21 days to rats implemented with gastric fistulas. Acid secretion was measured 12–14 h after the final dose of compounds. Basal and pentagastrin-stimulated acid secretion were measured on day 21, day 24 and day 37. Basal and histamine-stimulated acid secretion were measured within 2 days of measuring pentagastrin-stimulated acid secretion. There was no significant difference between basal acid secretion measured when pentagastrin and histamine responses were measured and therefore, only the basal secretion on the day that pentagastrin-stimulated acid secretion is presented for simplicity.
Study C: Acute actions of JNJ-26070109 on basal and histamine-stimulated acid secretion
The effect of a single intravenous dose of JNJ-26070109 (60 µmol·kg−1) on basal gastric acid secretion was evaluated. In addition, the effect of a single oral administration of JNJ-26070109 (30 µmol·kg−1) on histamine-stimulated gastric acid secretion was measured. These experiments were conducted identically to the chronic studies with the exception that JNJ-2607109 was given 45 min before administration of histamine.
Measurement of gastric acid secretion in conscious rats
Test compounds were administered orally 45 min before the stomach was flushed with 50–60 mL of warm mucosal solution to remove any residual gastric contents. A tube was then inserted in the fistula and the gastric secretions were collected under gravity into a volumetric cylinder. For these studies, animals were housed in modified metabolic cages and were unrestrained for the duration of the experiment. A single subcutaneous injection of 5 mL of normal saline was given to offset dehydration due to collection of the gastric secretions. Basal acid secretion was determined over a 90 min period and secretagogue-stimulated secretion was assessed for a further collection period of 90 min following subcutaneous administration of 30 nmol·kg−1 pentagastrin or 45 µmol·kg−1 histamine. Aliquots were collected every 30 min and the amount of acid secreted was determined by titration with 0.01 N NaOH to pH 7.0 using an automatic titration assembly (Metrohm Herisau, Switzerland). The values were pooled for the basal and secretagogue-stimulated periods (90 min each) and were expressed as total acid secretion (H+ moles).
Measurement of serum gastrin levels and markers of cell proliferation
Blood samples were collected from animals immediately after death and serum gastrin levels were determined by enzyme immunoassay according to manufacturer's instructions (Enzo Life Sciences, Plymouth Meeting, PA, USA). This assay is specific for the detection of gastrin-17 and, as such, may underestimate the total gastrin content, as the method does not detect other forms of gastrin as efficiently. The concentration of JNJ-26070109 in the serum was determined by high-pressure liquid chromatography/tandem mass spectrometry in the electrospray-positive mode by selected reaction monitoring (ACE C18 column, 2 × 50 mm, 3 or 5 µM particle size, Phenomenex, Torrance, CA, USA).
For histological analysis, the stomach was removed, washed clean with saline and weighed. The stomach was divided along its long axis and half of it was fixed in 40% neutral buffered formalin (Sigma Chemical, Poole, Dorset, UK) at room temperature for histological evaluation (performed by HistoGenex, Edegem, Belgium). A sample of the gastric mucosa was obtained by injecting saline between the mucosa and the smooth muscle layer (blistering technique) to allow easy dissection of mucosa. The density of histidine decarboxylase (HDC) and PCNA were determined by immunoflorescent staining of formalin fixed, paraffin embedded sections (6 µm thick) of the stomach. The number of cells staining positive for HDC was taken as an index of ECL cell number while the number of cells staining positive for PCNA was taken as an index of the rate of cell division. Sections were incubated overnight with an anti-HDC antibody (B-GP 265-1; Eurodiagnostica, Arnhem, Netherlands) diluted 1/1000 Antigen-antibody binding was detected with goat-anti guinea pig antibody (Jackson Immuno Research, West Grove, PA) conjugated to alkaline phosphatase, and visualized using Fast Blue BB. For determination of the state of proliferation sections were incubated with an anti-PCNA antibody (M0879; DakoCytomation, Glostrup, Denmark) diluted 1/1000 for 1 h. Bound antibody was visualized using the EnVision horseradish peroxidase (HRP) mouse system (DakoCytomation), 3-amino-9-ethylcarbazole and hydrogen peroxide. A double-staining protocol was used to identify ECL cells that were actively proliferating using the methods described earlier (i.e. those cells that stained positive for both HDC and PCNA). Controls to rule out the occurrence of non-specific signals during the setup of the double staining procedure included: single staining of (serial) sections with either the PCNA or HDC antibody and double staining procedures lacking one of the two primary antibodies and including both secondary antibodies. No cross reactivity of the antibodies was observed. For each staining run a negative control was included, that is, the primary antibodies were omitted. No specific staining was observed in the negative controls. An investigator who was unaware of the original numbering and classification of the specimens performed the counts of HDC and HDC-PCNA positive cells.
For quantitative reverse-transcription PCR (rt-PCR) studies, samples of tissue were preserved on ice (4°C) in RNALater (Ambion, Austin, TX, USA). The quality of the RNA in the gastric mucosa samples was assessed using the RNA 6000 Nano LabChip kit (Agilent Technologies, Palo Alto, CA, USA) and the relative expression of HDC, chromogranin A (CGA) and CCK2 receptor mRNA were determined using two-tube quantitative real-time PCR. Reverse transcription was conducted using TaqMan Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR was performed using an iCycler iQ detection system (Bio-Rad, Hercules, CA, USA). The following primer sequences were used: HDC: AGCACAAGCTGTCGTCCTTT (forward) and GGTGACTTCTGGGCACTCAT (reverse); CGA: GCAGGAGGAGGAAGAGGAAG (forward) and TCTCTGCTGTCAGCTCCTTG (reverse); glyceraldehyde 3-phosphate dehydrogenase (GAPDH): GAGGACCAGGTTGTCTCCTG (forward) and ATGTAGGCCATGAGGTCCAC (reverse), CCK2: CCATAGCCCTGGAGCGATAC (forward) and AGGTCCCACTGGCTGTACC (reverse). PCR was performed using 15 µL of the diluted RT reaction, 300 nM of each primer, 20 nM Fluorescein (Bio-Rad) and SYBR Green PCR Master Mix (Applied Biosystems) with a final volume of 50 µL. Reactions were incubated as directed for the SYBR Green PCR Master Mix. Levels of mRNA were normalized to GAPDH mRNA levels.
Data analysis and statistics
Values are represented as the mean ± SEM, n= 5–7. Statistical significance was determined using one-way anova (P < 0.05) followed by a Tukey test for multiple comparisons. Acid secretion was expressed as the total H+ (µmol) collected over the 60 or 90 min collection period, as described in the text. All data were analyzed using the software package GraphPad Prism®, version 3.01 or higher (GraphPad Software Inc., San Diego, CA, USA).
Materials
JNJ-26070109 was synthesized in house. Omeprazole was purchased from Sequoia Research (Oxford, UK). Both JNJ-26070109 and omeprazole were prepared as a sesame oil suspension for oral administration. For intravenous administration, JNJ-26070109 was prepared in a 5% (v/v) n-methyl-2-pyrrolidone in 20% hydroxypropyl-β-cyclodextrin solution.
Results
Study A. The effect of sustained administration (21 days) of JNJ-26070109 (10 µmol· kg−1) and omeprazole (400 µmol·kg−1) on basal and pentagastrin-stimulated acid secretion
Basal acid secretion in control rats was 438 ± 30 µmol H+, 12–14 h after final dose of vehicle. At this time point, there was no measurable basal acid secretion in rats treated with omeprazole alone or omeprazole in combination with 10 µmol·kg−1 JNJ-26070109 (Figure 2A). Basal acid secretion was also significantly reduced in rats treated with JNJ-26070109 alone (∼75% inhibition, P < 0.05). Pentagastrin (30 nmol·kg−1) stimulated gastric acid secretion in control animals (2606 ± 130 µmol H+, ∼sixfold over basal) and this response was inhibited in rats treated with omeprazole (98% inhibition) or omeprazole plus JNJ-26070109 for 21 days (99% inhibition). In rats treated with JNJ-26070109 alone, pentagastrin-stimulated acid secretion was reduced by 45%. The plasma concentration of JNJ-26070109 was assessed immediately after measurement of gastric acid secretion and found to be the same with and without treatment with omeprazole (0.2 ± 0.1 µM). This plasma concentration is ∼10-fold lower than that required for the acute pharmacological actions of JNJ-26070109 (Morton et al., 2011b).
Figure 2.
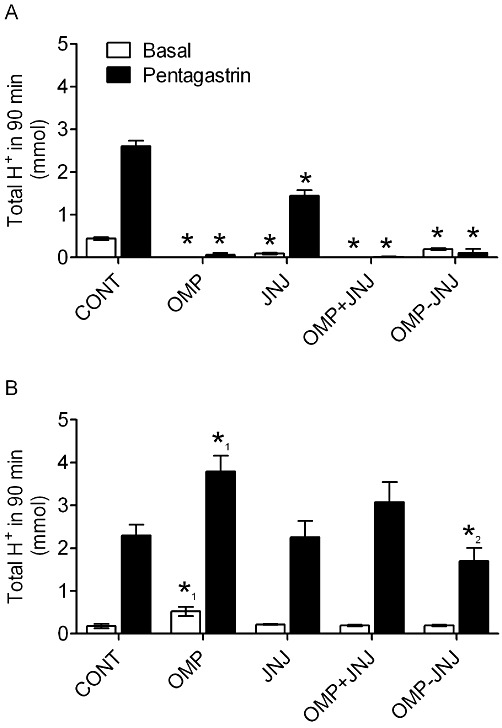
Inhibition of pentagastrin-stimulated gastric acid secretion while on sustained treatment with JNJ-26070109 with no acid rebound after withdrawal in rats. Study A; Basal and pentagastrin-stimulated acid secretion in conscious rats treated with vehicle (CONT), 400 µmol·kg−1 omeprazole (OMP), 10 µmol·kg−1 JNJ-26070109 (JNJ), 400 µmol·kg−1 omeprazole and 10 µmol·kg−1 JNJ-26070109 simultaneously for 21 days (OMP+JNJ) or 400 µmol·kg−1 omeprazole for 21 days followed by 10 µmol·kg−1 JNJ-26070109 for 3 days (OMP-JNJ). JNJ-26070109 was administered twice a day. Acid secretion was assessed while on treatment (day 21, 12 hours following final dose) (A) and 3 days after discontinuing treatment (day 24, 84 hours following final dose) (B). *=P < 0.01 versus corresponding control, *1=P < 0.05 for OMP versus all other groups and *2=P < 0.05 for JNJ-OMP versus OMP alone.
Prevention and reversal of omeprazole-induced acid hypersecretion with JNJ-26070109
At day 21, all acid suppression therapies ceased (see Figure 1 for study design). Three days after discontinuing treatment (day 24, 3 days after the final dose), basal and pentagastrin-stimulated acid secretion in the omeprazole-treated rats was ∼2- and ∼1.5-fold greater than control animals respectively (Figure 2B). Conversely, no significant increase in basal or pentagastrin-stimulated acid secretion was observed in rats treated with either JNJ-26070109 alone or in combination with omeprazole. Furthermore, in animals administered omeprazole alone for 21 days and then given JNJ-26070109 for 3 days following cessation of omeprazole administration, no increase in basal or pentagastrin-stimulated acid secretion was observed. Once again the plasma concentration of JNJ-26070109 was measured and confirmed to be sub-threshold for acute pharmacological effects at the time when acid secretion was measured (0.2 ± 0.1 µM), which was consistent with the pharmacokinetic profile of this compound (see Morton et al., 2011b).
Effect of chronic administration of omeprazole and JNJ-26070109 on serum gastrin levels and markers of cellular proliferation
Sustained administration of omeprazole (21 days, 400 µmol·kg−1, QD) resulted in an approximately sevenfold increase in serum gastrin concentration (gastrin concentration in control animals and omeprazole-treated rats = 61 ± 11 pM and 428 ± 14 pM, respectively, P < 0.05). JNJ-26070109 (10 µmol·kg−1, b.i.d. for 21 days) produced an approximately threefold increase in serum gastrin (179 ± 26 pM, P < 0.05). Concurrent treatment with omeprazole and JNJ-26070109 for 21 days increased the concentration of gastrin to a similar degree to that achieved with omeprazole alone (363 ± 19 pM).
Omeprazole increased the expression of CCK2 receptor mRNA in the gastric mucosa. CCK2 receptor expression was reduced in the gastric mucosa of rats given JNJ-26070109 alone or JNJ-26070109 concomitantly with omeprazole. The expression of HDC and CGA mRNA in the gastric mucosa was elevated in omeprazole-treated rats when compared with control animals (Figure 3A). No effect on HDC and CGA mRNA expression was observed when JNJ-26070109 was given alone although both showed a trend towards a reduction. Co-administration of JNJ-26070109 and omeprazole resulted in a smaller increase in HDC and CGA mRNA compared with the animals that were given omeprazole alone.
Figure 3.
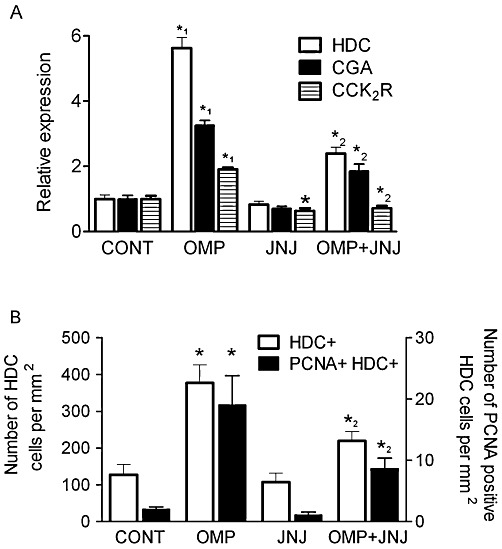
Effect of sustained treatment with omeprazole and/or JNJ-26070109 on markers of cellular proliferation in the gastric mucosa. In (A), Relative expression of HDC, CGA and CCK2 receptors (CCK2R) determined using quantitative PCR. In (B), measurement of the number of HDC-positive cells and PCNA-positive, HDC-positive cells using histological methods. Studies were conducted on the gastric mucosa of rats administered vehicle (CONT), 400 µmol·kg−1 omeprazole (OMP), 10 µmol·kg−1 JNJ-26070109 (JNJ), 400 µmol·kg−1 omeprazole and 10 µmol·kg−1 JNJ-26070109 simultaneously for 21 days (OMP+JNJ). *=P < 0.05 versus corresponding control, *1=P < 0.05 for OMP versus all other groups and *2=P < 0.05 for JNJ+OMP versus OMP alone.
The density of HDC and PCNA was determined from paraffin embedded sections of the body of the stomach to provide an index of the density of ECL cells and the proliferative state of these cells respectively. The number of ECL cells was increased ∼threefold in omeprazole-treated rats (Figure 3B). JNJ-26070109 had no significant effect on the number of HDC positive cells; however, rats that had concomitant administration of JNJ-26070109 and omeprazole had a lower number of HDC-positive cells than the omeprazole group alone (JNJ-26070109 and omeprazole 219 ± 25 HDC positive cells per mm2). The same trend was observed for the proliferation index of ECL cells, that is, cells that stained positive for both HDC and PCNA (Figure 3B).
Study B. Effect of sustained administration (24 days) of JNJ-26070109 (30 µmol·kg−1) and omeprazole (400 µmol·kg−1) on basal, pentagastrin and histamine stimulated acid secretion
Consistent with the previous study, 21 day treatment with 400 µmol kg−1 omeprazole suppressed basal (79% inhibition) and pentagastrin-stimulated (78% inhibition) acid secretion during the treatment period (Figures 4A). In addition, omeprazole also inhibited acid secretion stimulated by subcutaneous administration of 45 µmol·kg−1 histamine (95% inhibition). JNJ-26070109 (21 days b.i.d.) at a dose of 30 µmol·kg−1 reduced basal acid secretion by ∼66% and pentagastrin stimulated acid secretion by ∼70%. The increased inhibition of pentagastrin stimulated acid secretion compared with the previous study is consistent with the increased dose and resulting increase in plasma concentration of JNJ-26070109 in this study (2.7 ± 0.04 µM). Interestingly, JNJ-26070109 produced a ∼25% reduction (P < 0.05) in histamine stimulated gastric acid secretion after 21 days of treatment.
Figure 4.
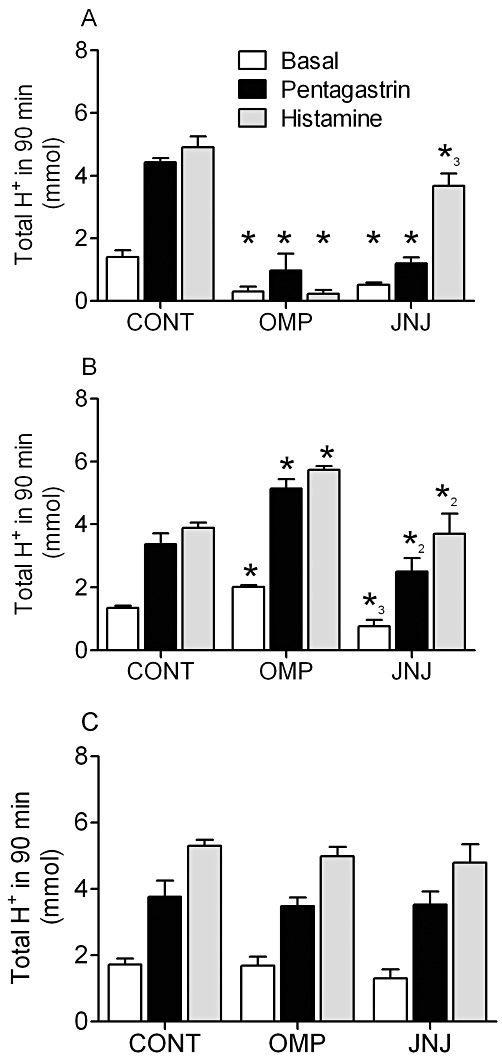
Inhibition of pentagastrin- and histamine-stimulated gastric acid secretion while on sustained treatment with JNJ-26070109 without acid rebound after withdrawal in rats. Study B: Basal, pentagastrin- and histamine-stimulated acid secretion in conscious rats dosed with vehicle (CONT), 400 µmol·kg−1 omeprazole (OMP) or 30 µmol·kg−1 JNJ-26070109 (JNJ). Gastric acid secretion after 21 days of sustained treatment (A), on day 24, 3 days after the final dose (B) and again on day 37, 13 days after the last dose (C) are shown. *=P < 0.01 versus the corresponding control, *2=P < 0.05 versus OMP-treated group and *3=P < 0.05 for JNJ versus both vehicle control and OMP-treated groups.
Three days after withdrawal of all drugs, basal and secretagogue-stimulated acid secretion was elevated by 1.5- to 1.75-fold in the omeprazole-treated group (Figure 4B). Conversely, animals treated with JNJ-26070109 showed no acid hypersecretion 3 days after termination of administration. The acid rebound in the omeprazole group was not present 13 days after discontinuing treatment (Figure 4C).
Acute actions of JNJ-260701209
A single 60 µmol·kg−1 intravenous dose of JNJ-26070109 suppressed basal acid secretion from 1517 ± 299 to 262 ± 190 µmol H+ in 90 min (>80% inhibition, P < 0.01). In contrast, acute oral administration of JNJ-26070109 (30 µmol·kg−1) had no effect on histamine-stimulated gastric acid secretion (3729 ± 362 in controls and 3264 ± 309 µmol in 90 min, in rats pre-treated with JNJ-26070109.
Discussion
Gastrin, acting through the peripheral CCK2 receptors, is a major hormonal regulator of gastric acid secretion. Inhibition of acid secretion produces a increase in serum gastrin concentration, which results in acid hypersecretion for many weeks following the cessation of acid suppression therapy in animal models (Larsson et al., 1988; Nishida et al., 1995; Zhao et al., 1998) and in humans (Gillen and McColl, 2001; Reimer et al., 2009). It follows that inhibition of acid secretion using a CCK2 receptor antagonist may provide a novel therapy for acid secretory disorders whereby the consequences of hypergastrinaemia would be prevented. We recently described the identification of JNJ-26070109 (Morton et al., 2011b) a novel, high-affinity CCK2 receptor antagonist with good bioavailability and a robust relationship between plasma concentration and pharmacological effect. In this study, the effects of sustained administration of JNJ-26070109, on acid secretion, biomarkers of cellular proliferation and omeprazole-induced acid rebound, were evaluated in rats.
Chronic oral administration of JNJ-26070109 at 10 and 30 µmol·kg−1 for 21 days, resulted in an inhibition of basal, pentagastrin and histamine stimulated acid secretion in rats. The experimental design of Study A (using the 10 µmol·kg−1 dose of JNJ-26070109) was developed to assess whether chronic administration of JNJ-26070109 at doses maximally effective only at peak plasma concentrations in an acute model would result in sufficient functional and/or structural modification of the stomach to inhibit acid secretion. In order to achieve this, acid secretion was measured 12 h after the final dose when the serum concentration of JNJ-26070109 was below that required for significant CCK2 receptor antagonism. Histamine was included in study B to assess the potential of JNJ-26070109 to down-regulate parietal cell function as histamine acts directly on parietal cell histamine H2 receptors. After chronic administration, but not after acute administration, histamine-induced acid secretion was reduced. It is unlikely that the effect of JNJ-26070109 on histamine stimulated acid secretion was due to antagonism of the histamine receptor because a single dose of JNJ-26070109 had no effect on histamine-stimulated acid secretion and JNJ-26070109 had no demonstrable affinity for histamine H2 receptors in an in vitro assay (Morton et al., 2011b). These data demonstrate that chronic treatment with a CCK2 receptor antagonist alters the function of ECL and parietal cells in a fashion that reduces the acid secretory capacity of the stomach. A similar story has emerged from studies in CCK2 receptor knockout mice where a reduction in histamine-induced acid secretion was observed (Kanai et al., 2009).
Markers of cellular proliferation were also evaluated in this study. Unlike omeprazole, sustained administration of JNJ-26070109 did not result in an increase in any of the markers of ECL cell growth (CGA and HDC). Moreover, at doses that maximally suppressed gastric acid secretion at peak plasma concentrations, JNJ-26070109 did not result in a reduction in markers of ECL cell growth. These findings demonstrate that a dose of JNJ-26070109 can be selected that prevents the effects of evoked hypergastrinaemia without inducing a hypoplastic state with thinning of the gastric mucosa. CCK2 receptor density was reduced by treatment with JNJ-26070109 although the data as collected and expressed (whole gastric mucosal mRNA normalized to GAPDH expression) do not allow differentiation between the number of ECL or parietal cells or receptor expression per cell. Co-administration of JNJ-26070109 and omeprazole prevented the growth stimulatory effects elicited by treatment with omeprazole alone. These data are consistent with previous studies demonstrating that the proliferative effects of omeprazole are due to hypergastrinaemia secondary to inhibition of gastric acid secretion (Nishida et al., 1995; Kitano et al., 2000). An increase in serum gastrin concentration was also observed after JNJ-26070109 administration; however, because JNJ-26070109 antagonizes the cognate receptor for gastrin, the downstream consequences of CCK2 receptor activation were not observed. Sustained CCK2 receptor blockade using JNJ-26070109 resulted in a functional and/or structural modification of the gastric acid secretory machinery of the stomach. These anti-proliferative effects of JNJ-26070109 are consistent with previous reports using another CCK2 receptor antagonist, YF476, in the rat (Nishida et al., 1995; Kitano et al., 2000) and confirm the role of gastrin in mediating these effects. It is important to note that while the rat is particularly predisposed to the trophic effects of gastrin, similar effects of gastrin on the ECL and parietal cells have been observed in humans (Driman et al., 1996; Waldum et al., 1996; Weinstein et al., 1996; Gillen et al., 1999). If similar effects were produced in man, then JNJ-26070109 may offer advantages over current therapies by preventing acid rebound when administered chronically in addition to acutely reducing gastric acid secretion.
The effectiveness of JNJ-26070109 for prevention and treatment of acid rebound was also investigated. These studies confirmed that sustained administration of omeprazole resulted in an increase in basal, pentagastrin and histamine stimulated gastric acid secretion after administration of the PPI had ceased. Conversely, 3 days after discontinuing sustained treatment with JNJ-26070109, basal, pentagastrin and histamine-stimulated acid secretion were not elevated compared with controls and no effect was observed 13 days after discontinuing treatment with JNJ-26070109. Co-administration of JNJ-26070109 with omeprazole prevented the acid rebound that was observed when omeprazole was administered alone. These effects were observed for both acid secretion and also markers of ECL cell proliferation. JNJ-26070109 was also demonstrated to ‘treat’ acid rebound when administered to rats with established omeprazole-induced acid rebound. This demonstrates that short-term administration of JNJ-26070109 could have significant effects on the acid secretory capacity of the stomach even after the trophic effects of omeprazole were established. It is known that serum gastrin concentration remains elevated for 3–5 days after discontinuing omeprazole treatment (Tielemans et al., 1992). Thus short-term treatment with a CCK2 receptor antagonist could be a useful therapy to wean patients who find cessation of PPI treatment difficult due to the occurrence of acid rebound (see the acid rebound argument presented by McColl and co-workers: Waldum et al., 1996; Gillen et al., 1999; Gillen and McColl, 2001). The safety of PPIs has been questioned by some (Opar, 2009). Discontinuing acid suppression therapy may be beneficial as a recent analysis has shown that there is an increase in the incidence of hip fracture (Yang et al., 2006) and osteoporosis-related fracture in patients treated for >7 years with PPIs (Targownik et al., 2008).
Inhibition of acid secretion in human volunteers has been evaluated using the CCK2 receptor antagonist YF476 (published in abstract form, Boyce et al., 2000). Interestingly, in that study, the effect of YF476 appeared to diminish over time, so that by day 7 of treatment no significant effect on acid secretion was observed. As far as we are aware, the mechanism of this ‘tachyphylaxis’ has not been established, however, there are a number of differences between JNJ-26070109 and YF476. These include the observation that while YF476 has very high affinity for the human CCK2 receptor, it is only ∼60-fold selective over the related human CCK1 receptor, whereas JNJ-26070109 is >1200-fold selective for the human CCK2 over the CCK1 receptor (Morton et al., 2005a,b; 2011b). This may be of functional significance as gastrin/cholecystokinin dual knockout mice have a phenotype where acid secretion is similar to controls. This is in contrast to the CCK2 receptor and gastrin knockout mice both of which have a phenotype consistent with the chronic effect of JNJ-26070109 observed in the current study. In the gastrin/cholecystokinin dual knockout mice it was argued that gastric acid secretion is driven by vagal input in compensation for the lack of CCK1 and CCK2 receptor activation (Chen et al., 2004). The pharmacokinetics of these two compounds are also different as demonstrated by the finding that a single subcutaneous dose of 300 µmol kg−1 YF476 suppressed HDC activity for 8 weeks (Kitano et al., 2000). Thus, in these studies the actions of YF476 were only assessed at a high and sustained exposure level in rats (Kitano et al., 2000). The current study demonstrates that doses of a reversible CCK2 receptor antagonist that are modestly effective in terms of inhibition of gastric acid secretion have relevant anti-trophic actions. Furthermore, the complex and long-lived pharmacokinetic profile of YF476 in rats makes it difficult to assess the effects of stopping treatment and acid rebound with this CCK2 receptor antagonist. Although the mechanism for the loss of efficacy of YF476 in humans remains unknown, the unique pharmacological properties of JNJ-26070109 make it worthy of clinical investigation.
In summary, the CCK2 receptor antagonist JNJ-26070109 was found to provide effective inhibition of acid secretion following sustained administration in rats without the potential for acid rebound. JNJ-26070109 may have an advantage over current therapies due to its rapid onset of action and down-regulation of the acid secretory capacity of the stomach.
Acknowledgments
The authors would like to thank the La Jolla bioanalytical group of Janssen Pharmaceutical Companies of Johnson & Johnson for their support.
Glossary
- CGA
chromogranin A
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GORD
gastro-oesophageal reflux disease
- HDC
histidine decarboxylase
- JNJ-26070109
(R)4-bromo-N-[1-(2,4-difluoro-phenyl)-ethyl]-2-(quinoxaline-5-sulfonylamino)-benzamide, PPI, proton pump inhibitor
Conflicts of interest
These studies were solely funded by Janssen Pharmaceutical Companies of Johnson & Johnson, 3210 Merryfield Row, San Diego, CA92101, USA. All authors were employees of Janssen Pharmaceutical Companies of Johnson & Johnson with the exception of Luc Andries, HistogeneX N.V., who was contracted by Janssen Pharmaceutical Companies of Johnson & Johnson to perform the histological examination of the gastric mucosa samples.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson E, Abrahamsson H, Simrén M, Mattsson N, Jensen C, Agerforz P, et al. Discontinuation of proton pump inhibitors in patients on long-term therapy: a double-blind, placebo-controlled trial. Aliment Pharmacol Ther. 2006;24:945–954. doi: 10.1111/j.1365-2036.2006.03084.x. [DOI] [PubMed] [Google Scholar]
- Black JW, Shankley NP. How does gastrin act to stimulate oxyntic cell secretion? Trends Pharmacol Sci. 1987;8:486–490. [Google Scholar]
- Boyce M, Warrington S, Johnston A, Harris A. Effect on gastric pH of repeated doses of YF476, a new gastrin antagonist, compared with omeprazole and placebo. Br J Clin Pharmacol. 2000;49:383–384. [Google Scholar]
- Cawston EE, Miller LJ. Therapeutic potential for novel drugs targeting the type 1 cholecystokinin receptor. Br J Pharmacol. 2010;159:1009–1021. doi: 10.1111/j.1476-5381.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhao C, Håkanson R, Samuelson L, Rehfeld J, Friis-Hansen L. Altered control of gastric acid secretion in gastrin-cholecystokinin double mutant mice. Gastroenterology. 2004;126:476–487. doi: 10.1053/j.gastro.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Dockray GJ, Varro A, Dimaline R, Wang T. The gastrins: their production and biological activities. Annu Rev Physiol. 2001;63:119–139. doi: 10.1146/annurev.physiol.63.1.119. [DOI] [PubMed] [Google Scholar]
- Driman DK, Wright C, Tougas G, Riddell R. Omeprazole produces parietal cell hypertrophy and hyperplasia in humans. Dig Dis Sci. 1996;41:2039–2047. doi: 10.1007/BF02093608. [DOI] [PubMed] [Google Scholar]
- Gillen D, McColl K. Problems associated with clinical use of proton pump inhibitors. Pharmacol Toxicol. 2001;89:281–286. doi: 10.1034/j.1600-0773.2001.d01-161.x. [DOI] [PubMed] [Google Scholar]
- Gillen D, Wirz A, Ardill J, McColl K. Rebound hypersecretion after omeprazole and its relation to on treatment acid suppression and helicobacter pylori status. Gastroenterology. 1999;116:239–247. doi: 10.1016/s0016-5085(99)70118-6. [DOI] [PubMed] [Google Scholar]
- Kanai S, Hosoya H, Akimoto S, Ohta M, Matsui T, Takiguchi S, et al. Gastric acid secretion in cholecystokinin-1 receptor, -2 receptor, and -1, -2 receptor gene knockout mice. J Physiol Sci. 2009;59:23–29. doi: 10.1007/s12576-008-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano M, Norlen P, Ding X-Q, Nakamura S, Hakanson R. Long-lasting cholescystokinin 2 receptor blockade after a single subcutaneous injection of YF476 or YM022. Br J Pharmacol. 2000;130:699–705. doi: 10.1038/sj.bjp.0703342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberts R, Creutzfeldt W, Stockmann F, Jacubaschke U, Maas S, Brunner G. Long-term omeprazole treatment in man: effects on gastric endocrine cell populations. Digestion. 1988;39:126–135. doi: 10.1159/000199615. [DOI] [PubMed] [Google Scholar]
- Larsson H, Carlson E, Hakanson R, Mattsson H, Nilsson G, Seensalu R, et al. Time-course of development and reversal of gastric endocrine cell hyperplasia after inhibition of acid secretion. Studies with omeprazole and ranitidine in intact and antrectomized rats. Gastroentrology. 1988;95:1477–1486. doi: 10.1016/s0016-5085(88)80066-0. [DOI] [PubMed] [Google Scholar]
- Morton M, Liu P, Reik A, de la Rosa R, Mendel M, Li X, et al. Pharmacological analysis of CCK2 receptors up-regulated using engineered transcription factors. Regul Pept. 2005a;129:227–232. doi: 10.1016/j.regpep.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Morton M, Pyati J, Dai H, Li L, Moreno V, Shankley N. Molecular cloning, expression and pharmacological characterization of the canine cholecystokinin 1 receptor. Br J Pharmacol. 2005b;145:374–384. doi: 10.1038/sj.bjp.0706196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton M, Prendergast C, Barrett TD. Targeting gastrin for the treatment of gastric acid related disorders and pancreatic cancer. Trends Pharmacol Sci. 2011a;32:201–205. doi: 10.1016/j.tips.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Morton MF, Barrett TD, Freedman JM, Li L, Rizzolio MC, Prendergast CE, et al. JNJ-26070109 [(R) 4-Bromo-N-[1-(2, 4-difluoro-phenyl)-ethyl]-2-(quinoxaline-5-sulfonylamino)-benzamide]: a novel, potent and selective cholecystokinin 2 receptor antagonist with good bioavailability in rats and dogs. J Pharmacol Exp Ther. 2011b;338:328–336. doi: 10.1124/jpet.110.178483. [DOI] [PubMed] [Google Scholar]
- Nishida A, Kobayashi-Uchida A, Akuzawa S, Takinami Y, Shishido T, Kamato T, et al. Gastrin receptor antagonist YM022 prevents hypersecretion after long-term acid suppression. Am J Physiol. 1995;269:G699–G705. doi: 10.1152/ajpgi.1995.269.5.G699. [DOI] [PubMed] [Google Scholar]
- Noble F, Wank S, Crawley J, Bradwejn J, Seroogy K, Hamon M, et al. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev. 1999;51:745–781. [PubMed] [Google Scholar]
- Opar A. A close look at acid reflux drugs points to possible risks. Nat Med. 2009;15:710. doi: 10.1038/nm0709-710. [DOI] [PubMed] [Google Scholar]
- Reimer C, Søndergaard B, Hilsted L, Bytzer P. Proton pump inhibitor therapy induces acid-related symptoms in healthy volunteers after withdrawal of therapy. Gastroenterology. 2009;137:80–87. doi: 10.1053/j.gastro.2009.03.058. [DOI] [PubMed] [Google Scholar]
- Targownik L, Lix L, Metge C, Prior H, Leung S, Leslie W. Use of proton pump inhibitors and risk of osteoporosis-related fractures. Can Med Assoc J. 2008;179:319–326. doi: 10.1503/cmaj.071330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielemans Y, Chen D, Sundler F, Hakanson R, Willens G. Reversability of the cell kinetic changes induced by omeprazole in the rat oxyntic mucosal. Scand J Gastroenterol. 1992;27:155–160. doi: 10.3109/00365529209165437. [DOI] [PubMed] [Google Scholar]
- Waldum H, Arnestad J, Brenna E, Eide I, Syvernen U, Sandvik A. Marked increase in gastric acid secretory capacity after omeprazole treatment. Gut. 1996;39:649–653. doi: 10.1136/gut.39.5.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldum HL, Lehy T, Brenna E, Sandvik AK, Petersen H, Sognen BS, et al. Effect of the histamine-1 antagonist astemizole alone or with omeprazole on rat gastric mucosa. Scand J Gastroenterol. 1991;26:23–35. doi: 10.3109/00365529108996480. [DOI] [PubMed] [Google Scholar]
- Weinstein W, Ippoliti A, Lee S. Acid hypersecretion, parietal cell hyperplasia and endoscopic changes after withdrawal of long-term high dose omeprazole therapy: a prospective study. Gastroenterology. 1996;110:A294. [Google Scholar]
- Wettstein JG, Bueno L, Junien JL. CCK antagonists: pharmacology and therapeutic interest. Pharmacol Ther. 1994;62:267–282. doi: 10.1016/0163-7258(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lewis J, Epstein S, Metz D. Long-term proton pump inhibitor therapy and risk of hip fracture. J Am Med Assoc. 2006;296:2947–2953. doi: 10.1001/jama.296.24.2947. [DOI] [PubMed] [Google Scholar]
- Zhao C, Chen D, Kimura K, Hakanson R. Reversibility of omeprazole-evoked changes in the structure of ECL cells in the rat stomach. Cell Tissue Res. 1998;291:91–95. doi: 10.1007/s004410050982. [DOI] [PubMed] [Google Scholar]