

Abstract
The role of inflammation in neurodegenerative diseases has been widely demonstrated. Intraneuronal protein accumulation may regulate microglial activity via the fractalkine (CX3CL1) signaling pathway that provides a mechanism through which neurons communicate with microglia. CX3CL1 levels fluctuate in different stages of neurodegenerative diseases and in various animal models, warranting further investigation of the mechanisms underlying microglial response to pathogenic proteins, including Tau, β-amyloid (Aβ), and α-synuclein. The temporal relationship between microglial activity and localization of pathogenic proteins (intra- versus extracellular) likely determines whether neuroinflammation mitigates or exacerbates disease progression. Evidence in transgenic models suggests a beneficial effect of microglial activity on clearance of proteins like Aβ and a detrimental effect on Tau modification, but the role of CX3CL1 signaling in α-synucleinopathies is less clear. Here we review the nature of fractalkine-mediated neuronmicroglia interaction, which has significant implications for the efficacy of anti-inflammatory treatments during different stages of neurodegenerative pathology. Specifically, it is likely that anti-inflammatory treatment in early stages of disease during intraneuronal accumulation of proteins could be beneficial, while anti-inflammatory treatment in later stages when proteins are secreted to the extracellular space could exacerbate disease progression.
1. Introduction
Increased microglial activity facilitates beneficial responses to central nervous system (CNS) injuries, including phagocytosis of debris and clearance of apoptotic cells; however, unregulated microglial activity can lead to production of neurotoxic factors that worsen CNS pathology and cause neuronal degeneration [1–7]. Microglia constitute the main immune cells in the CNS and provide innate immunity under physiological conditions and adaptive immunity under stress, promoting inflammation in response to various signals from apoptotic cells [1–4]. The phenotype of CNS resident macrophages is considered activated and designated M1 or “classical activation,” which describes the proinflammatory phenotypic response. M2 or “alternative activation” describes phenotypic responses to cytokines, such as Interleukin-(IL-) 4 and IL-13 [8]. In many neurodegenerative diseases, persistent injury (such as intraneuronal protein accumulation) promotes the production of proinflammatory molecules (Figure 1), like tumor necrosis factor (TNF)-α, Interleukin (IL)-1β, IL-6, reactive oxygen species (ROS), and nitric oxide (NO) [9]. Proinflammatory factors activate microglia [10, 11], which may remove not only apoptotic or damaged neurons, but also healthy neurons, aggravating the pathogenic process [5].
Figure 1.
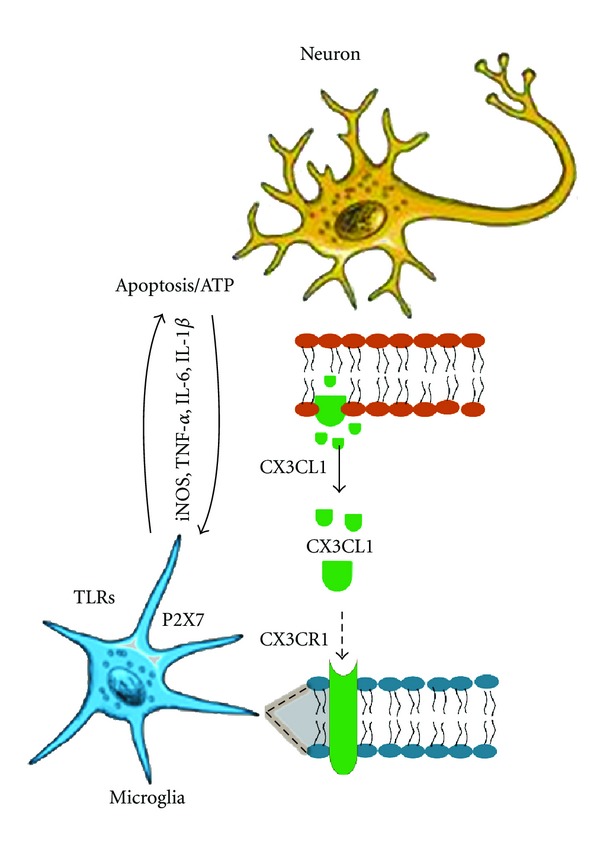
Initiation of inflammatory response following intraneuronal protein accumulation. Intraneuronal accumulation of pathogenic proteins causes ATP release by apoptotic neurons to activate purinergic microglia P2X7 receptors or TLRs. Activated microglia release proinflammatory cytokines (TNF-α, IL-16, IL-1) and iNOS to activate astrocytes (via MCP-1 chemotaxis) and increase apoptosis in stressed neurons. To initiate a neuroprotective immune response, injured neurons may communicate via fractalkine (CX3CR1) and suppress inflammation.
The inflammatory response is generally localized to areas of CNS injury via communication between immune cells and stressed neurons. Innate inflammation is reported in Alzheimer's disease (AD), Parkinson's disease (PD), and the Tauopathies (reviewed in [12]). In the healthy brain, microglia have a resting “deactivated” phenotype (ramified) [10]. Activated microglia are present in human postmortem brain tissues of patients with tauopathies, including AD, frontotemporal dementia with parkinsonism linked to chromosome-17 (FTDP), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) [13–15]. Also associated with neurodegeneration in these diseases are hyperphosphorylated Tau (p-Tau) deposits [16–23]. It has been demonstrated in animal models of AD that the endotoxin lipopolysaccharide (LPS) promotes both inflammation and the accumulation of p-Tau [24] and that suppression of microglial activity prolongs survival in FTDP-associated P301L transgenic mice [25]. Our laboratory has previously shown a differential increase in microglial activity in response to accumulation of p-Tau in lentiviral wild type Tau versus mutant P301L mice at 1 month after-injection [26]. Cell culture models also demonstrate that proinflammatory cytokines can induce p-Tau [27–29]. These data suggest that microglial activity aggravates p-Tau through a common underlying mechanism moderating communication between microglia and neurons. Determining how this mechanism is temporally altered in response to p-Tau is critical to understanding the beneficial or detrimental role of microglial activity in different stages of disease pathology [30–32].
2. Fractalkine in Human Disease and Animal Models of Neurodegeneration
A central question in current research pertains to how communication between microglia and neurons, in which pathogenic proteins accumulate, affects the progression of inflammation. One inducer through which neurons and microglia can communicate to regulate inflammation is fractalkine (CX3CL1) (Figure 1). CX3CL1 is a 373-amino acid protein that has a chemokine domain located on top of a mucin-like stalk [33, 34]. Neurons secrete CX3CL1 [34], which exists in both membrane-bound and soluble forms [35]. The membrane-bound CX3CL1 can serve as an adhesion molecule for leukocytes expressing the fractalkine receptor (CX3CR1) [36] and soluble CX3CL1 can function as both a proinflammatory chemoattractant that activates receptive inflammatory cells [33, 37] and an anti-inflammatory [38], neuroprotective agent that reduces neuronal apoptosis [39]. The relationship between soluble CX3CL1 in peripheral blood and inflammatory diseases of the CNS is unclear. Several findings suggest that deletion of CX3CR1 increases microglial activity in various models of acute and chronic neuronal injury [40–43]. Fluctuations in CX3CL1 levels are also observed in many neurodegenerative diseases. Increased levels of serum CX3CL1 are reported in patients with multiple sclerosis [39, 44], traumatic brain injury [45], and human immunodeficiency virus (HIV) with CNS complications [46], but increased levels of serum CX3CL1 are not observed in patients with Guillain-Barré Syndrome and viral and bacterial meningitis [44]. Genetic variants with reduced levels of CX3CR1 are linked to age-related macular degeneration in humans [47].
CX3CL1 and its cognate receptor CX3CR1 may play an important role in immunoregulation in animal models of neurodegeneration. CX3CL1 expression is decreased in the cerebral cortex and hippocampus in the aged brains of amyloid precursor protein (APP) transgenic mice [48]. Decreased CX3CL1 levels are also observed in aged AD transgenic mouse models (Tg2576) in association with increased Aβ levels [48]. Microglial activity was increased while the levels of Aβ load and CX3CR1 were decreased in MyD88−/− mice, suggesting CX3CL1 involvement in Aβ clearance [49]. CX3CR1 deficiency leads to decreased levels of Aβ deposition and protects against Aβ toxicity in transgenic mouse models of AD [50, 51]. LPS induces p-Tau of both endogenous and transgene-derived Tau in nontransgenic mice and in a humanized mouse model of Tauopathy, depending on LPS dose and CX3CR1 deficiency [40]. Additionally, impairment of CX3CL1 signaling pathway leads to deterioration in cognitive function and synaptic plasticity via alteration of IL-1β function [52]. Although CX3CR1 deficiency exacerbates AD-related neuronal and behavioral pathologies in mice overexpressing human Aβ, these effects are likely to be associated with the level of cytokine production and not Aβ plaque load, suggesting that alteration of proinflammatory factors, including TNF-α and IL-6 may modulate CX3CL1 signaling [43]. Conversely, production of NO, IL-6, and TNF-α may be inhibited by CX3CL1 [53, 54].
Exogenous CX3CL1 is neuroprotective in some other models of neuroinflammation [55, 56], and disruption of CX3CL1-CX3CR1 communication by deletion of the CX3CR1 gene causes neurotoxicity in mouse models of systemic inflammation, PD, and amyotrophic lateral sclerosis [57] but protects against neuronal loss in a mouse model of focal cerebral ischemia [58]. CX3CR1 knockout mice show more toxicity and substantia nigra (SN) degeneration in response to LPS treatment following administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxic precursor of 1-methyl-4-phenylpyridinium (MPP+) [57]. Together, these studies suggest altered microglial activity through CX3CL1 signaling, which may play a direct role in immunoregulation depending upon the CNS insult. CX3CL1-CX3CR1 signaling is therefore a possible mediator of communication between injured neurons and microglia and may play a significant role in the regulation of microglial activity in response to pathogenic protein accumulation in early, or protein secretion, in later stages of disease.
3. Intraneuronal Aβ and Inflammation in Early Stages of AD
A primary feature of AD is the presence of extracellular aggregates of Aβ peptide (plaques) and intracellular inclusions (tangles) containing p-Tau [59–61]. Variants of Aβ peptide, including Aβ42 and Aβ40, are produced by the cleavage of APP and subsequent cleavage of an intermediate fragment, APP C-terminal fragments (CTFs) [62]. Cleavage of APP at an alternative site within the Aβ region by the cleaving enzyme α-secretase precludes Aβ formation [63, 64]. The causal association between mutations in APP and the onset of familial AD supports the role of Aβ in AD pathogenesis [65]. It is likely that in the early stages of disease, Aβ accumulates intraneuronally prior to the formation of extracellular plaques [66]. It is also likely that the intracellular pool of Aβ is externalized as neurons die, contributing to the formation of senile plaques [67–70]. The presence of intraneuronal Aβ is significant in that such a presence constitutes a preplaque stage of AD pathology. Our laboratory has previously shown that intraneuronal Aβ induces microglial and astrocyte activation and increases inflammatory markers in gene transfer models [71, 72]. Furthermore, it has been shown that intraneuronal Aβ can cause apoptosis and cell death, which stimulate microglial and astrocyte activation independently of extracellular plaques [73]. These results implicate communication between microglia and Aβ expressing neurons in the onset of inflammation in AD. Inflammation has been associated with neurodegenerative disease etiology in AD, in which Aβ and Tau can act as inflammatory stimuli to promote microglial activity [1, 74–76]. Therefore, inflammation in AD may arise not only from extracellular plaque formation, but also as a consequence of communication between microglia and Aβ-expressing neurons. Accumulation of intraneuronal Aβ can induce damage to lysosomes and multivesicular bodies, leading to leakage of Aβ from vesicles into the cytosol and activation of inflammatory mechanisms without extracellular accumulation of amyloid plaques. Several studies have suggested that manipulation of chemokines and/or their receptors may be a therapeutic target in neurodegenerative diseases, including AD [77–79]. Microglia treated with recombinant CX3CL1 or IL-34 partially protect against Aβ toxicity via enhancement of Aβ clearance and antioxidant production [80]. Significant differences in CX3CL1 levels were detected in a cohort of 51 patients with mild cognitive impairment (MCI), 51 AD patients and 57 controls [81]. However, the increase in plasma CX3CL1 levels is not congruent with tissue levels, which are decreased in the hippocampus and frontal cortex of advanced AD cases [43], suggesting variable roles of CX3CL1 in different stages of AD pathogenesis. The level of plasma soluble fractalkine was significantly higher in MCI and moderate AD patients compared to severe AD, suggesting that higher levels of soluble plasma fractalkine is associated with greater cognitive impairment [81]. Therefore, the fractalkine signaling pathway that mediates communication between microglia and neurons is deficient in AD brains and downregulated by Aβ.
4. Fractalkine in PD-Related Inflammation
The characteristics of PD include death of dopaminergic neurons in the SN [50, 58, 82] and formation of Lewy bodies (LBs) [83–92], or inclusions comprised mainly of α-synuclein [83–99]. The simultaneous occurrence of α-synuclein and Tau pathology is observed in multiple system atrophy (MSA), though the mechanisms underlying a possible connection between the two proteins are unknown [100, 101]. Early onset familial PD arises from mutations in the autosomal recessive genes PARKIN, PTEN-induced kinase-1 (PINK1), and DJ-1 [94] while late onset PD is associated with dominantly-inherited mutations in leucine-rich repeat kinase 2 (LRRK2) and α-synuclein.
Aggregation of α-synuclein is implicated in the activation of microglia and subsequent inflammation associated with PD. It was previously thought that α-synuclein-related pathology was confined to within neurons, but recent research suggests that microglia are activated following the release of α-synuclein aggregates into the extracellular space by apoptotic cells [102]. However, extracellular α-Synuclein has not been found in PD brains. Aggregated forms of α-synuclein induce microglial activation [99, 103]. Several microglia-derived inflammatory factors (ROS, NO, TNF-α, and IL-1β), as well as LPS, promote death of dopaminergic neurons [104–106]. The phagocytosis of α-synuclein by microglia induces NADPH oxidase activity and the production of ROS [103]. These neurotoxic effects signify a contributory role of microglia and inflammation in PD pathology. Inflammation has also been detected in PD brains lacking LBs, such as parkin-linked autosomal recessive early onset PD [9]. These cases, as well as the role of Tau as a risk factor for PD, suggest that additional mechanisms regulate inflammation. For example, CX3CL1 suppresses microglial activation and protects against neuronal loss and striatal lesion in 6-hydroxydopamine (6-OHDA) rat model of PD [107]. MPP+ increases neuronal CX3CL1 levels in rat SN, but administration of CX3CR1 antagonists blocks PD-like pathology, including loss of dopaminergic neurons and motor behavior [108], suggesting that fractalkine can modulate microglial activation in PD models. Deletion of CX3CR1 aggravates microglial neurotoxicity in response to LPS in the MPTP model of PD and in the superoxide dismutase 1 (SOD1) G93A model of ALS [57], suggesting that CX3CL1 signaling may limit microglial toxicity [57]. The level of plasma soluble CX3CL1 also correlates positively with disease severity and progression in human PD patients, suggesting that CX3CL1 can be used as a biomarker to differentiate between neurodegenerative diseases [109].
5. The Effects of Microglial Activation Depend on Disease Stage
Whether inflammation rescues or exacerbates cell death in neurodegenerative disease likely depends on the stage of disease progression. Microglial activation facilitates the removal of apoptotic cells and toxins from the CNS, releasing neurotrophic factors that aid in repair following injury [5]. However, microglia also release inflammatory markers that can induce apoptosis. The apparent ambivalence of increased microglial activity is associated with unsuccessful attempts to provide anti-inflammatory treatment in human clinical trials. Preliminary clinical trials in which nonsteroidal anti-inflammatory drugs (NSAIDs) were administered before the development of neurodegeneration suggested that disease risk was reduced by inhibition of the immune response [110, 111]. However, later trials found that anti-inflammatory drugs were harmful in AD patients [110]. These conflicting data reflect the current lack of understanding of the role of the immune response in CNS diseases and point to the importance of the temporal relationship between the disease stage and the anti-inflammatory intervention.
The timing of the immune response in relation to disease progression complicates the use of anti-inflammatory treatment in various CNS diseases. For example, the permanence of brain damage following stroke or ischemia depends on the activity of proinflammatory cytokines, the activation of microglia, and the recruitment of leukocytes [112, 113]. It has been found that inhibiting TNF-α and IL-1, which mediate postischemic activity by attracting leukocytes to the injury or by damaging cells directly, confers neuroprotection in animal models of stroke [112, 113]. In AD models, the involvement of innate immunity via microglial activation and phagocytosis of Aβ renders anti-inflammatory therapy particularly relevant to the study of AD [114–116]. In AD patients, however, deficits in Toll-like receptors (TLRs) expression inhibit the removal of Aβ from the brain and result in lack of Aβ clearance by macrophages [117], and TLR2 deficiency in AD mouse models is associated with severe cognitive impairment [118]. In addition to anti-inflammatory treatment, intervention in the hematopoietic system has been suggested as a possible model of treatment for AD. The administration of macrophage colony-stimulating factor, a hematopoietic cytokine, to mouse microglia promotes degradation of internalized Aβ in vitro [119] and protects against cognitive decline in vivo when administered prior to the development of learning and memory deficits [120], supporting the importance of timing of anti-inflammatory treatment relative to disease progression. Taken together, these findings support the targeting of innate immune cells as a therapeutic approach for AD and other neurodegenerative diseases. However, conflicting data from clinical trials necessitate further investigation of the role of the immune response in disease development and progression.
6. Putting It into Perspective
Research on the suppression of microglial activity has been actively pursued with limited success [121] and strategies to manipulate the protective role of microglia—the detection and removal of apoptotic cells—have not been fully investigated [122–124]. These strategies warrant further research, as apoptotic cells that enter secondary necrosis [125] and trigger inflammation [126, 127] increase tissue damage. In this context, the role of CX3CL1 in mediating communication between preapoptotic neurons and microglia becomes greatly important. Such intervention would be relevant in early stages of disease progression, during which intracellular accumulation of pathogenic proteins anticipates apoptosis and the formation of extracellular protein aggregates. In later stages of disease pathology, decreased CX3CL1 signaling may activate microglia and induce p-Tau (Figure 2), which exacerbates disease progression by promoting apoptosis. Additionally, the use of NSAIDs to restrain microglial activity may exacerbate pathology due to lack of phagocytic clearance of secreted extracellular amyloids, including α-Synuclein, Aβ and p-Tau. In this context, targeting microglial activity in later disease stages may be detrimental and contributory to disease progression. However, targeting the CX3CL1 pathway in early disease stages could be beneficial, at least in delaying disease progression via restraint of microglial activity. Along this line of thought, NSAIDs administration could regulate key proinflammatory cytokines (Figure 2) that would modulate CX3CL1 signaling and microglial activity. It remains to be fully elucidated when and how alteration of proinflammatory markers may increase or decrease CX3CL1 signaling, which may either activate or suppress microglia. One possibility is increased CX3CL1 levels to restrain microglial activity and prevent the exacerbation of p-Tau damage. However, this intervention should be timed to avoid interference with microglial activity when patients progress into more advanced stages of disease, during which removal of extracellular deposits becomes necessary. Therefore, understanding the critical interplay between proinflammatory (TNF-α, IL-6, IL-1β, and IL-1α), anti-inflammatory cytokines (IL-10, TGF-β, IL-34, etc.), and fractalkine levels to modulate microglial activity is highly significant. Furthermore, whether the activation of microglia in the context of neurodegenerative disease is beneficial or detrimental may also depend upon the type of disease. Successful anti-inflammatory treatments of CNS diseases will likely be specific not only to the stage of disease pathology, but also to the type of disease. It has been found that many of the same cytokines are implicated in the pathology of AD, PD, and ALS despite distinct patterns of neuronal loss in each disease [9]. Previous literature presents contradictory evidence regarding the effects of targeting microglia in various CNS diseases. Glass et al. [128], for example, suggest that targeting microglia in PD and ALS is detrimental while other studies suggest that targeting microglia aids in Aβ clearance in AD. Further investigation of the role of the inflammatory response in each disease will determine the potential for anti-inflammatory treatments. Here we suggest a temporally-defined strategy of intervention in which early targeting of CX3CL1 signaling slows disease progression and prevents p-Tau formation.
Figure 2.
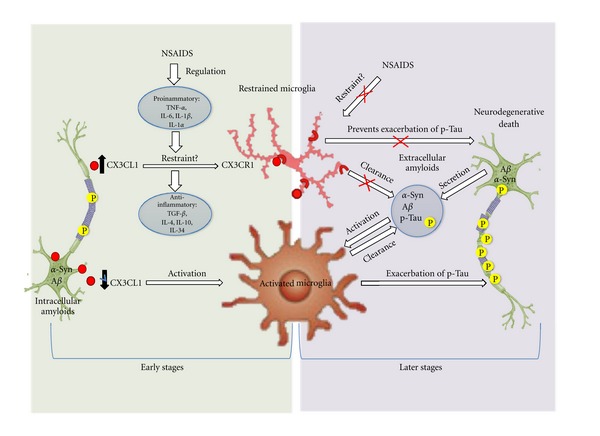
Modulation of CX3CL1 in early versus late disease stages. The success of anti-inflammatory treatment in neurodegenerative diseases likely depends on the stage of disease progression. Treatment with NSAIDS early in disease pathology may alter the levels of various proinflammatory markers, including TNF-α, IL-6, IL-1β, and IL-1α, and anti-inflammatory markers, including TGF-β, IL-4, IL-10, and IL-34. The changes in the levels of these cytokines may lead to altered CX3CL1 signaling, which would either increase microglial activity (if CX3CL1 were reduced) or restrain microglia (if CX3CL1 levels were increased). In later stages of disease, secretion of pathogenic proteins like Aβ, α-synuclein, and p-Tau to the extracellular space increases microglial activation. Microglial activity promotes p-Tau, which destabilizes microtubules and leads to cell death. Treatment with NSAIDS in later stages of disease would likely be detrimental, as restraining microglia would weaken the immune response to remove extracellular protein aggregates.
Acknowledgments
This work was supported by NIH Grant AG 30378 and Georgetown University support to C. E.-H. Moussa.
References
- 1.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 2.Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. GLIA. 2002;40(2):133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Di Virgilio F, Ceruti S, Bramanti P, Abbracchio MP. Purinergic signalling in inflammation of the central nervous system. Trends in Neurosciences. 2009;32(2):79–87. doi: 10.1016/j.tins.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Molecular Neurobiology. 2010;41(2-3):242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- 6.Balistreri CR, Colonna-Romano G, Lio D, Candore G, Caruso C. TLR4 polymorphisms and ageing: implications for the pathophysiology of age-related diseases. Journal of Clinical Immunology. 2009;29(4):406–415. doi: 10.1007/s10875-009-9297-5. [DOI] [PubMed] [Google Scholar]
- 7.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. GLIA. 2002;40(2):195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 8.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Khandelwal PJ, Herman AM, Moussa CEH. Inflammation in the early stages of neurodegenerative pathology. Journal of Neuroimmunology. 2011;238(1):1–11. doi: 10.1016/j.jneuroim.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 12.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiology of Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microscopy Research and Technique. 2001;54(1):47–58. doi: 10.1002/jemt.1120. [DOI] [PubMed] [Google Scholar]
- 14.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology of Disease. 2006;21(2):404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. Journal of Neuropathology and Experimental Neurology. 2001;60(6):647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- 16.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302(5646):819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 17.Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Annals of Neurology. 2000;47(3):374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Dickson DW. Tau and synuclein and their role in neuropathology. Brain Pathology. 1999;9(4):657–661. doi: 10.1111/j.1750-3639.1999.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lippa SM, Lippa CF, Mori H. α-synuclein aggregation in pathological aging and Alzheimer’s disease: the impact of β-amyloid plaque level. American Journal of Alzheimer’s Disease and other Dementias. 2005;20(5):315–318. doi: 10.1177/153331750502000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pletnikova O, West N, Lee MK, et al. Aβ deposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases. Neurobiology of Aging. 2005;26(8):1183–1192. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Popescu A, Lippa CF, Lee VMY, Trojanowski JQ. Lewy bodies in the amygdala: increase of α-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Archives of Neurology. 2004;61(12):1915–1919. doi: 10.1001/archneur.61.12.1915. [DOI] [PubMed] [Google Scholar]
- 22.Yancopoulou D, Xuereb JH, Crowther RA, Hodges JR, Spillantini MG. Tau and α-synuclein inclusions in a case of familial frontotemporal dementia and progressive aphasia. Journal of Neuropathology and Experimental Neurology. 2005;64(3):245–253. doi: 10.1093/jnen/64.3.245. [DOI] [PubMed] [Google Scholar]
- 23.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Research Reviews. 2000;33(1):95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 24.Kitazawa M, Yamasaki TR, LaFerla FM. Microglia as a potential bridge between the amyloid β-peptide and tau. Annals of the New York Academy of Sciences. 2004;1035:85–103. doi: 10.1196/annals.1332.006. [DOI] [PubMed] [Google Scholar]
- 25.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE-H. Wild type and P301L mutant Tau promote neuro-inflammation and α-Synuclein accumulation in lentiviral gene delivery models. Molecular and Cellular Neuroscience. 2012;49(1):44–53. doi: 10.1016/j.mcn.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Liu L, Barger SW, Griffin WST. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. Journal of Neuroscience. 2003;23(5):1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Experimental Cell Research. 2004;295(1):245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Saez ET, Pehar M, Vargas M, Barbeito L, Maccioni RB. Astrocytic nitric oxide triggers tau hyperphosphorylation in hippocampal neurons. In Vivo. 2004;18(3):275–280. [PubMed] [Google Scholar]
- 30.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Molecular Neurodegeneration. 2009;4(1, article 47) doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 32.Cameron B, Landreth GE. Inflammation, microglia, and alzheimer’s disease. Neurobiology of Disease. 2010;37(3):503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640–642. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 34.Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and Cx3cr1-expressing microglia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. Journal of Neuroscience Research. 2002;69(3):418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 36.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor Cx3cr1, which mediates both leukocyte migration and adhesion. Cell. 1997;91(4):521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 37.Pan Y, Lloyd C, Zhou H, et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387(6633):611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- 38.Yoneda O, Imai T, Goda S, et al. Fractalkine-mediated endothelial cell injury by NK cells. Journal of Immunology. 2000;164(8):4055–4062. doi: 10.4049/jimmunol.164.8.4055. [DOI] [PubMed] [Google Scholar]
- 39.Tong N, Perry SW, Zhang Q, et al. Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. Journal of Immunology. 2000;164(3):1333–1339. doi: 10.4049/jimmunol.164.3.1333. [DOI] [PubMed] [Google Scholar]
- 40.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cardona AE, Sasse ME, Liu L, et al. Scavenging roles of chemokine receptors: chemokine receptor deficiency is associated with increased levels of ligand In circulation and tissues. Blood. 2008;112(2):256–263. doi: 10.1182/blood-2007-10-118497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Condello C, Schain A, Harb R, Grutzendler J. Cx3cr1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. Journal of Neuroscience. 2010;30(50):17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho S-H, Sun B, Zhou Y, et al. Cx3cr1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. Journal of Biological Chemistry. 2011;286(37):32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW. CSF and serum levels of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system. Journal of Neuroimmunology. 2003;137(1-2):210–217. doi: 10.1016/s0165-5728(03)00085-7. [DOI] [PubMed] [Google Scholar]
- 45.Rancan M, Bye N, Otto VI, et al. The chemokine Fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. Journal of Cerebral Blood Flow and Metabolism. 2004;24(10):1110–1118. doi: 10.1097/01.WCB.0000133470.91843.72. [DOI] [PubMed] [Google Scholar]
- 46.Sporer B, Kastenbauer S, Koedel U, Arendt G, Pfister HW. Increased intrathecal release of soluble fractalkine in HIV-infected patients. AIDS Research and Human Retroviruses. 2003;19(2):111–116. doi: 10.1089/088922203762688612. [DOI] [PubMed] [Google Scholar]
- 47.Combadière C, Feumi C, Raoul W, et al. Cx3cr1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. Journal of Clinical Investigation. 2007;117(10):2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duan RS, Yang X, Chen ZG, et al. Decreased fractalkine and increased IP-10 expression in aged brain of APPswe transgenic mice. Neurochemical Research. 2008;33(6):1085–1089. doi: 10.1007/s11064-007-9554-z. [DOI] [PubMed] [Google Scholar]
- 49.Lim J-E, Kou J, Song M, et al. MyD88 deficiency ameliorates β-amyloidosis in an animal model of Alzheimer's disease. American Journal of Pathology. 2011;179(3):1095–1103. doi: 10.1016/j.ajpath.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee S, Varvel NH, Konerth ME, et al. Cx3cr1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. American Journal of Pathology. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuhrmann M, Bittner T, Jung CKE, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nature Neuroscience. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tromba C, Cohen IS. A novel action of isoproterenol to inactivate a cardiac K+ current is not blocked by beta and alpha adrenergic blockers. Biophysical Journal. 1990;58(3):791–795. doi: 10.1016/S0006-3495(90)82422-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. GLIA. 2000;29(4):305–315. [PubMed] [Google Scholar]
- 54.Limatola C, Lauro C, Catalano M, et al. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. Journal of Neuroimmunology. 2005;166(1-2):19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 55.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(24):14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Research. 2003;979(1-2):65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- 57.Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 58.Denes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of Cx3cr1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. Journal of Cerebral Blood Flow and Metabolism. 2008;28(10):1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 59.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 60.Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nature Cell Biology. 2004;6(11):1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 61.Hardy J. Alzheimer’s disease: genetic evidence points to a single pathogenesis. Annals of Neurology. 2003;54(2):143–144. doi: 10.1002/ana.10624. [DOI] [PubMed] [Google Scholar]
- 62.Jarrett JT, Berger EP, Lansbury PT. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 63.Allinson TMJ, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein α-secretases. Journal of Neuroscience Research. 2003;74(3):342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- 64.Li QX, Maynard C, Cappai R, et al. Intracellular accumulation of detergent-soluble amyloidogenic Aβ fragment of Alzheimer’s disease precursor protein in the hippocampus of aged transgenic mice. Journal of Neurochemistry. 1999;72(6):2479–2487. doi: 10.1046/j.1471-4159.1999.0722479.x. [DOI] [PubMed] [Google Scholar]
- 65.Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nature Reviews Neuroscience. 2008;9(10):768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 66.Von Kienlin M, Künnecke B, Metzger F, et al. Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiology of Disease. 2005;18(1):32–39. doi: 10.1016/j.nbd.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 67.Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Aβ . American Journal of Pathology. 2006;168(1):184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal Aβ-amyloid precedes development of amyloid plaques in Down syndrome. Archives of Pathology and Laboratory Medicine. 2001;125(4):489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- 69.Mori C, Spooner ET, Wisniewski KE, et al. Intraneuronal Aβ42 accumulation in Down syndrome brain. Amyloid. 2002;9(2):88–102. [PubMed] [Google Scholar]
- 70.Ohyagi Y, Tsuruta Y, Motomura K, et al. Intraneuronal amyloid β42 enhanced by heating but counteracted by formic acid. Journal of Neuroscience Methods. 2007;159(1):134–138. doi: 10.1016/j.jneumeth.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 71.Burns MP, Zhang L, Rebeck GW, Querfurth HW, Moussa CE-H. Parkin promotes intracellular Aβ 1-42 clearance. Human Molecular Genetics. 2009;18(17):3206–3216. doi: 10.1093/hmg/ddp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rebeck GW, Hoe HS, Moussa CEH. β-Amyloid1-42 gene transfer model exhibits intraneuronal amyloid, gliosis, tau phosphorylation, and neuronal loss. Journal of Biological Chemistry. 2010;285(10):7440–7446. doi: 10.1074/jbc.M109.083915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pereira C, Agostinho P, Moreira PI, Cardoso SM, Oliveira CR. Alzheimer’s disease-associated neurotoxic mechanisms and neuroprotective strategies. Current Drug Targets. 2005;4(4):383–403. doi: 10.2174/1568007054546117. [DOI] [PubMed] [Google Scholar]
- 74.Brown AM, Finch SJ, Gordon D. Genome-wide association study of genetic loci and Alzheimer disease. Journal of the American Medical Association. 2010;304(8):p. 858. doi: 10.1001/jama.2010.1172. [DOI] [PubMed] [Google Scholar]
- 75.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics. 2011;43(5):429–436. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rogers J, Webster S, Lue L-F, et al. Inflammation and Alzheimer's disease pathogenesis. Neurobiology of Aging. 1996;17(5):681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- 77.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4(4):590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoarau JJ, Krejbich-Trotot P, Jaffar-Bandjee MC, et al. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg) CNS & Neurological Disorders Drug Targets. 2011;10(1):25–43. doi: 10.2174/187152711794488601. [DOI] [PubMed] [Google Scholar]
- 79.Rezai-Zadeh K, Gate D, Gowing G, Town T. How to get from here to there: macrophage recruitment in Alzheimer’s disease. Current Alzheimer Research. 2011;8(2):156–163. doi: 10.2174/156720511795256017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mizuno T. The biphasic role of microglia in Alzheimer's disease. International Journal of Alzheimer's Disease. 2012;2012 doi: 10.1155/2012/737846.737846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim TS, Lim HK, Lee JY, et al. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neuroscience Letters. 2008;436(2):196–200. doi: 10.1016/j.neulet.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 82.Younkin SG. The role of Aβ42 in Alzheimer’s disease. Journal of Physiology Paris. 1998;92(3-4):289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- 83.Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and α-synucleinopathies. Philosophical Transactions of the Royal Society B. 1999;354(1386):1101–1118. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nature Reviews Neuroscience. 2001;2(7):492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 85.Lundvig D, Lindersson E, Jensen PH. Pathogenic effects of α-synuclein aggregation. Molecular Brain Research. 2005;134(1):3–17. doi: 10.1016/j.molbrainres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 86.Grazia Spillantini M, Anthony Crowther R, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neuroscience Letters. 1998;251(3):205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 87.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spillantini MG, Goedert M. The α-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Annals of the New York Academy of Sciences. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 89.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 90.Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Masliah E. C-terminal α-synuclein immunoreactivity in structures other than Lewy bodies in neurodegenerative disorders. Acta Neuropathologica. 2000;99(3):296–304. doi: 10.1007/pl00007441. [DOI] [PubMed] [Google Scholar]
- 91.Trojanowski JQ, Lee VMY. Parkinson’s disease and related α-synucleinopathies are brain amyloidoses. Annals of the New York Academy of Sciences. 2003;991:107–110. doi: 10.1111/j.1749-6632.2003.tb07468.x. [DOI] [PubMed] [Google Scholar]
- 92.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in lewy bodies in Parkinson’s disease. Neuroscience Letters. 1997;239(1):45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- 93.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Reviews in Molecular Medicine. 2009;11:p. e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 94.Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Human Molecular Genetics. 2010;19(1):R21–R27. doi: 10.1093/hmg/ddq167.ddq167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Healy DG, Abou-Sleiman PM, Lees AJ, et al. Tau gene and Parkinson’s disease: a case-control study and meta-analysis. Journal of Neurology, Neurosurgery and Psychiatry. 2004;75(7):962–965. doi: 10.1136/jnnp.2003.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martin-Villalba A, Hahne M, Kleber S, et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death and Differentiation. 2001;8(7):679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- 97.Benner EJ, Banerjee R, Reynolds AD, et al. Nitrated α-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001376.e1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Annals of the New York Academy of Sciences. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- 99.Reynolds AD, Kadiu I, Garg SK, et al. Nitrated alpha-synuclein and microglial neuroregulatory activities. Journal of NeuroImmune Pharmacology. 2008;3(2):59–74. doi: 10.1007/s11481-008-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chin SSM, Goldman JE. Glial inclusions in CNS degenerative diseases. Journal of Neuropathology and Experimental Neurology. 1996;55(5):499–508. doi: 10.1097/00005072-199605000-00001. [DOI] [PubMed] [Google Scholar]
- 101.Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VMY. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. Journal of Cell Biology. 1995;129(6):1629–1640. doi: 10.1083/jcb.129.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roodveldt C, Christodoulou J, Dobson CM. Immunological features of α-synuclein in Parkinson’s disease. Journal of Cellular and Molecular Medicine. 2008;12(5B):1820–1829. doi: 10.1111/j.1582-4934.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang W, Wang T, Pei Z, et al. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. The FASEB Journal. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 104.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 105.Castaño A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. Journal of Neurochemistry. 1998;70(4):1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 106.Hunter RL, Dragicevic N, Seifert K, et al. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. Journal of Neurochemistry. 2007;100(5):1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- 107.Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. Journal of Neuroinflammation. 2011;8, article 9 doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shan S, Hong-Min T, Yi F, et al. NEW evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiology of Aging. 2011;32(3):443–458. doi: 10.1016/j.neurobiolaging.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 109.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Annals of Neurology. 2011;69(3):570–580. doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer’s disease anti-inflammatory prevention trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Archives of Neurology. 2008;65(7):896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson’s disease. Trends in Pharmacological Sciences. 2003;24(8):395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- 112.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nature Reviews Neuroscience. 2001;2(10):734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 113.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nature Reviews Immunology. 2005;5(8):629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 114.Liu Y, Walter S, Stagi M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128(8):1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 115.Combarros O, Infante J, Rodríguez E, et al. CD14 receptor polymorphism and Alzheimer’s disease risk. Neuroscience Letters. 2005;380(1-2):193–196. doi: 10.1016/j.neulet.2005.01.082. [DOI] [PubMed] [Google Scholar]
- 116.Chen K, Iribarren P, Hu J, et al. Activation of toll-like receptor 2 on microglia promotes cell uptake of alzheimer disease-associated amyloid β peptide. Journal of Biological Chemistry. 2006;281(6):3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 117.Fiala M, Liu PT, Espinosa-Jeffrey A, et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(31):12849–12854. doi: 10.1073/pnas.0701267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. Journal of Neuroscience. 2008;28(22):5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Majumdar A, Cruz D, Asamoah N, et al. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Molecular Biology of the Cell. 2007;18(4):1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on β-amyloid deposition and cognitive impairment in Alzheimers disease. Brain. 2009;132(4):1078–1092. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- 121.Glezer I, Bittencourt JC, Rivest S. Neuronal expression of Cd36, Cd44, and Cd83 antigen transcripts maps to distinct and specific murine brain circuits. Journal of Comparative Neurology. 2009;517(6):906–924. doi: 10.1002/cne.22185. [DOI] [PubMed] [Google Scholar]
- 122.Garden GA, Möller T. Microglia biology in health and disease. Journal of Neuroimmune Pharmacology. 2006;1(2):127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 123.Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. The FASEB Journal. 2004;18(6):743–745. doi: 10.1096/fj.03-0374fje. [DOI] [PubMed] [Google Scholar]
- 124.Witting A, Müller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by microglia/brain macrophages in vitro: Involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. Journal of Neurochemistry. 2000;75(3):1060–1070. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- 125.Silva MT, do Vale A, dos Santos NMN. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis. 2008;13(4):463–482. doi: 10.1007/s10495-008-0187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Molecular Cell. 2004;14(3):277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 127.Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death and Differentiation. 1998;5(7):563–568. doi: 10.1038/sj.cdd.4400407. [DOI] [PubMed] [Google Scholar]
- 128.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]