

Abstract
The gastrointestinal tract harbors large and diverse populations of bacteria that vary among individuals and within individuals over time. Numerous internal and external factors can influence the contents of these microbial communities, including diet, geography, physiology, and the extent of contact among hosts. To investigate the contributions of such factors to the variation and changes in gut microbial communities, we analyzed the distal gut microbiota of individual chimpanzees from two communities in Gombe National Park, Tanzania. These samples, which were derived from 35 chimpanzees, many of whom have been monitored for multiple years, provide an unusually comprehensive longitudinal depth for individuals of known genetic relationships. Although the composition of the great-ape microbiota has been shown to codiversify with host species, indicating that host genetics and phylogeny have played a major role in its differentiation over evolutionary timescales, the geneaological relationships of individual chimpanzees did not coincide with the similarity in their gut microbial communities. However, the inhabitants from adjacent chimpanzee communities could be distinguished based on the contents of their gut microbiota. Despite the broad similarity of community members, as would be expected from shared diet or interactions, long-term immigrants to a community often harbored the most distinctive gut microbiota, suggesting that individuals retain hallmarks of their previous gut microbial communities for extended periods. This pattern was reinforced in several chimpanzees sampled over long temporal scales, in which the major constituents of the gut microbiota were maintained for nearly a decade.
Complex consortia of microbes colonize the mammalian digestive tract at birth and appear to be critical to the health, growth, and development of the hosts (1–3). Although these microbial communities are continually seeded from external sources and can change drastically over the lifetime of an individual (4, 5), the gut microbial communities of conspecifics tend to be more similar to one another than to those of other species (6–9). Moreover, the relationships of the gut microbial communities within great apes are concordant with the phylogeny of their host species, suggesting that features characteristic of a host species promote the specificity and codiversification of bacterial communities with hosts (10).
Despite the distinctiveness of gut microbial communities at the level of host species, there is considerable variation in the gut microbiota both among the members of a species (7, 11–13) and within individuals over time (14–17). Because of the constant influx of new microbes into hosts and the high microbial diversity maintained in the gastrointestinal tract, it is not surprising that variation in microbial community contents stems from several sources. For example, the gut microbiota of closely related individuals are more similar to one another than are those of unrelated or cohabiting individuals, suggesting an influence of host genetic factors (6, 15, 18,). However, comparisons of age-matched children from different continents gave indication that broad dietary and geographic differences are also associated with the composition of gut microbial communities (13).
Although numerous internal and external factors undoubtedly shape the contents and composition of the gut microbiota, the contribution of each has been difficult to untangle because of the highly dynamic nature of these complex communities (5, 12, 16). Most previous analyses of gut microbial communities have focused on hosts who differed with respect to one specific variable or whose gut microbiotae were sampled only once (e.g., refs. 11, 13, 18, and 19). The most comprehensive longitudinal surveys have focused on individual hosts sampled at daily-to-weekly intervals for timespans of up to 2.5 y (5, 17). These studies have divulged the short-term temporal dynamics of microbial communities but have only begun to disentangle the many factors that can contribute to the variation in the microbiota within and among hosts. Such questions might best be resolved through the long-term analysis of populations of hosts of known genealogical relationships and demographic histories. In this regard, the samples collected from chimpanzees in Gombe National Park provide an extraordinary resource for understanding the interplay of multiple factors on the variation of gut microbial communities within a species. These chimpanzee hosts are represented by fecal specimens sampled for more than a decade from individuals of known identity, genealogy, provenance, health, and community status (20–22). These samples combine the host diversity and temporal depth to allow us address questions about the dynamics and persistence of gut microbes, many of which have never been addressed in any species. And because chimpanzees are our closest relatives, the dynamics of their gut microbial communities can serve as models for understanding the evolution our own microbiota.
Results
We examined the gut microbial communities of chimpanzees (Pan troglodytes schweinfurthii) of known identity and kinship from Gombe National Park in Tanzania (Fig. 1 and SI Appendix, Table S1) (20–22). The ability to assign samples to specific hosts allowed us both to distinguish among factors that contribute variation among individuals and to investigate the long-term stability of gut microbiota within individuals.
Fig. 1.

Pedigree of sampled chimpanzees from Gombe National Park. Genealogies of chimpanzees (circles, females; squares, males) from the Kasekela (KK, blue) and Mitumba (MT, red) communities whose gut microbial communities were analyzed. Individual chimpanzees are categorized by their current community affiliation. Individuals were sampled in 2008 (green), 2001 (orange), and/or 2000 (yellow). Those left uncolored are individuals whose gut microbiotae were not surveyed. For all sampled individuals, ages in 2008 are provided next to each name. Question marks indicate fathers of unknown identity, and asterisks denote individuals shown more than once.
Placement of Gombe Chimpanzee Microbiotae Among Great Apes.
Our previous analysis of distal gut microbiota within great apes established that the branching order of the phylogeny based on the composition of gut microbial communities mirrored the established relationships of the great ape hosts (10). To ascertain the distribution of samples from Gombe chimpanzees within this great ape phylogeny, we integrated the phylotype information for the Gombe chimpanzees along with that recovered previously for 24 individuals of five species of great apes (SI Appendix, Table S2). Using both phylogenetic and sample ordination approaches, we find that the topology and relatedness among samples generally recapitulates what is known about the species-level relationships among hosts (Fig. 2). (i) The samples from the 34 Gombe chimpanzees form a single clade that includes the Gombe sample previously examined but excludes all other great apes. (ii) The gut microbial communities within Gombe chimpanzees are most similar to those of other chimpanzees. (iii) Although Gombe chimpanzees are classified as subspecies P.t. schweinfurthii, their microbial communities do not cluster with those of P.t. schweinfurthii from the Democratic Republic of the Congo (DRC). This incongruity has been noted (10) and could be caused by the relatively low sampling of the microbial communities available for the DRC apes. (The mean number of pyrotags for DRC P.t. schweinfurthii samples was 4,217, whereas for every clade of Pan spp., there were at least 12,000.) This analysis reinforces the presence of a strong signal of host phylogeny on the composition of the great ape gut microbiota. Therefore, our analyses of factors influencing the microbial communities in Gombe chimpanzees examine the variation that occurs at the very tips of the tree: As such, they must be interpreted in the context of a monophyletic group of samples of recent origin and of high similarity relative to that observed in the species at large.
Fig. 2.

Relationship of gut microbial communities in Gombe chimpanzees to those of other great apes. (A) Neighbor-joining phylogenetic reconstruction of log abundances of microbial phylotypes (99% OTUs) present in Gombe chimpanzees and other great apes. Phylogenies and the bootstrap support for branching orders in these phylogenies were generated by using default parsimony and neighbor-joining parameters in PAUP 4b10 (30) as in ref. 10. Colored triangles delineate species or subspecies boundaries, and bootstrap values ≥75 are marked with asterisks. (B) Nonmetric multidimensional scaling ordination of Sørensen distances of the microbial phylotypes detected in great ape guts. Colors match the species and subspecies designations in A and in ref. 10 with points representing previously analyzed samples labeled. The cluster of points corresponding to the gut microbial communities of Gombe chimpanzees are outlined with a dashed line.
Microbial Community Profiles.
We generated a total of 1,206,387 pyrosequencing reads for 48 samples: For each host (Fig. 1 and SI Appendix, Table S1), we examined the microbial diversity within a sample collected in October to December 2008; and for seven of the hosts, we examined additional samples collected during the same period in 2000 and/or in 2001. More than one-half of the sequencing reads (687,499) passed the initial quality and length filters and, of these, 31,837 were either chimeric, or of chloroplast or eukaryotic origins, and removed. This filtering yielded an average of 13,113 reads per sample (8,323–22,563), which were clustered into phylotypes applying 0.01% divergence cutoff [99% operational taxonomic units (OTUs)]. After removal of OTUs present in only one sample, the 5,788 phylogenetically informative phylotypes were assigned to taxonomic groups, and a total of 14 phyla were represented (SI Appendix, Fig. S1). The majority of phylotypes were classified as either Firmicutes (59.8%) or Bacteriodetes (17.9%), as observed in other mammals (7, 9, 10), and representatives of six other phyla were also present in all samples, but at substantially lower frequencies. Approximately 13% could not be assigned to a particular phylum; however, the majority of these unassigned phyla (476/784) have ≥95% BLAST similarity to 16S rRNA sequences retrieved from human gut samples.
Factors Influencing Microbial Communities.
Estimates of diversity within (alpha) and between (beta) gut microbial communities were assessed for 34 chimpanzees that were each sampled in October to December 2008 (SI Appendix, Table S3). Individual samples contained 518–1,389 phylotypes, with high overall alpha diversity (Shannon’s H′, 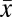 = 5.09 ± 0.30) and sampling evenness
= 5.09 ± 0.30) and sampling evenness 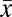 (E,
(E, 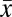 = 0.76 ± 0.031). There are no significant differences in Shannon diversity indices, sample evenness, or Chao1 species richness estimators with respect to sex, geographic community affiliation, or matrilines. However, in contrast to what has been reported in humans (5), infants (0–5 y) exhibited significantly greater within sample diversity (H′) than did adults (16–33 y) [nonparametric Wilcoxon test; P = 0.044 (infant-adult)]. In addition, adolescents have significantly greater within sample diversity (P = 0.032) and evenness (P = 0.011); infants harbor significantly greater phylotype diversity than adults (P = 0.031) (SI Appendix, Fig. S2). These statistical differences persist after applying a resampling procedure that accounts for variation in sequence-read numbers among samples.
= 0.76 ± 0.031). There are no significant differences in Shannon diversity indices, sample evenness, or Chao1 species richness estimators with respect to sex, geographic community affiliation, or matrilines. However, in contrast to what has been reported in humans (5), infants (0–5 y) exhibited significantly greater within sample diversity (H′) than did adults (16–33 y) [nonparametric Wilcoxon test; P = 0.044 (infant-adult)]. In addition, adolescents have significantly greater within sample diversity (P = 0.032) and evenness (P = 0.011); infants harbor significantly greater phylotype diversity than adults (P = 0.031) (SI Appendix, Fig. S2). These statistical differences persist after applying a resampling procedure that accounts for variation in sequence-read numbers among samples.
We performed an ordination analysis by using nonmetric multidimensional scaling (NMS) with a Sørensen distance matrix generated for the 34 chimpanzees to assess beta diversity among samples. The two major axes, which together represent the majority of the observed variation (cumulative R2 = 0.765), separate samples into two broad groups that roughly correspond to geographic community affiliation (Fig. 3). Results of multiresponse permutation procedures (MRPP) corroborated the NMS analyses: There is a significant chance-corrected within-group agreement (A) values when the sample is classified by community (A = 0.0182, P = 0.0003), but not by any of the other parameters tested (e.g., sex, P = 0.38; age class, P = 0.10). A total of 108 phylotypes were significantly associated with separating hosts according to community affiliation (P < 0.05, indicator value ≥50%), and it is noteworthy that several of the borderline cases represent samples from hosts that switched community affiliation (Fig. 3).
Fig. 3.

Chimpanzee microbiota assort by community affiliation of host. Nonmetric multidimensional scaling ordination analysis of 34 chimpanzees sampled in 2008 distinguishes two groups that correspond to the two (KK and MT) chimpanzee communities. Individuals are labeled according to the key, with year and direction of immigration included when applicable.
The distributions of several beta diversity measures were compared within and between (i) geographic communities, (ii) hosts of different sexes, and (iii) individuals with different degrees of relatedness. Sørensen distances corroborated the observed distinction between the gut microbiota of hosts from the Kasekela (KK) and Mitumba (MT) communities but also indicate a significant difference between the sexes (Fig. 4). Alternative estimators of beta diversity (e.g., Morisita–Horn) yielded qualitatively similar results.
Fig. 4.

Gut microbiota differentiate by chimpanzee community affiliation and sex but not degree of genetic relatedness. Estimates of microbial community similarity (Sørensen distances) of Gombe chimpanzees within and between communities (A), sexes (B), and coefficients of relatedness (C) were compared by using nonparametric comparisons of each pair using the Wilcoxon method. Levels of significance are only reported for comparisons reaching significant P values (as observed in samples sorted by community affiliation and sex).
Given the comprehensive pedigree available for the chimpanzees (Fig. 1), we evaluated the effect of kinship and genetic relatedness on beta diversity. Whereas in humans, increased levels of relatedness are associated with greater similarity in gut microbial communities (6, 18), this pattern is not the case in Gombe chimpanzees (Fig. 4C). We analyzed a total of 17 parent–offspring pairs (r = 0.5), one verified set of full siblings (r = 0.5), 12 pairs of half-siblings (r = 0.25), and several other pairs of r = 0.25 (grandparent–grandchild, uncle/aunt–nephew/niece), and there were no significant differences in Sørensen distances between the microbial communities of chimpanzees of differing levels of relatedness.
Temporal Changes in Microbial Communities as Revealed by Pyrotags.
To examine the long-term stability of gut microbial communities, we first used the pyrotag approach to interrogate the microbial communities in samples from 2000, 2001, and 2008 for multiple chimpanzee hosts. Among the chimpanzees sampled longitudinally, between 170 and 350 of the phylotypes detected in the initial year (2000) were present in both of the later samples (2001 and 2008) from an individual host. The conserved phylotypes were usually the most abundant microbes in a host’s gut microbial communities (constituting 40–70% of the total sequence reads). Within each of the longitudinally sampled hosts, there were a few (5–30) unique phylotypes maintained at low frequencies over the entire 8-y sampling period, suggesting that certain host-specific strains persist rather than originate from repeated reinfection (Fig. 5 and SI Appendix, Fig. S3). The taxonomic distribution of phylotypes was virtually identical in the samples from each year, but there is long-term turnover in the microbial community constituents at the level of bacterial phylotype. Samples collected in 2000 and 2001 were, for most chimpanzees, two to four times more similar to one another than either was to the 2008 sample. The greater similarity of the samples from 2000 and 2001 is due to the maintenance of particular phylotypes, which are subsequently replaced by new phylotypes from the same taxonomic division.
Fig. 5.

Temporal changes in chimpanzee gut microbial communities. NMS ordination of seven chimpanzees sampled through time can be distinguished according to the sampling year (2000, yellow; 2001, orange; 2008, green). Sample identifiers (individual and year) are presented in the key, and dashed lines connect samples derived from a single chimpanzee host. Ellipse encompasses all samples from 2008. Note that several of these chimpanzees are related: 1Sparrow is the mother of Sheldon and Sandi; 2Darbee and Tubi are presumed to be half-siblings.
The nature of the temporal changes in phylotype diversity becomes apparent when the gut microbial communities were subjected to ordination methods. Considering the two major axes (cumulative R2 = 0.775), collection year is the main factor driving the stratification of samples (A = 0.0203, P = 0.0038), with the 2008 samples from all seven hosts converging into a single cluster (Fig. 5). The similarity among samples from 2008 is due primarily to the increased abundance of a group of phylotypes classified as Prevotellaceae, Erysipelotrichaceae, and Coriobacteriaceae, which are taxonomically distinct from the indicator phylotypes from previous years.
Microbial Community Continuity as Revealed by iTags.
To examine long-term changes in the gut microbiota at a finer level of resolution, we implemented an Illumina-based 16S-rDNA profiling on multiple samples collected from individual hosts during the interval spanning 2000–2009 (SI Appendix, Table S4). After quality filtering and the removal of reads unique to a single sample, the 39 samples each averaged nearly 50,000 iTag reads, which were clustered into 99% OTUs.
To determine whether the 99% OTUs based on iTags, which assayed a different region of the 16S rDNA gene, recapitulate the patterns observed for pyrotags, we applied analogous ordination methods to the iTag dataset. Again, we observed that the samples collected in 2008 tended to cluster together and apart from those from the same chimpanzees sampled in 2000 and 2001, even when we include samples from intervening and later years (SI Appendix, Fig. S4).
It is possible that long-term sample storage could either increase sequence diversity (because of DNA damage) or alternatively reduce diversity (through cellular degradation and decomposition), but there were no clear trends among samples. To determine how variation in bacterial OTUs within an individual changed through time, and whether there were systematic increases or decreases in bacterial diversity that might be attributable to sample storage, we examine the continuity of these 99% OTUs across multiple time-points for each chimpanzee host. On average, 40% of the OTUs identified from an individual are shared across every sample collected from the 9-y period, with the proportion of the shared OTUs representing as much as 94% to as few as 37% of the reads from a single sample (SI Appendix, Fig. S5). Furthermore, the amount of diversity was not associated with the age of a sample, and large proportions of very low frequency OTUs were maintained within hosts for close to a decade (SI Appendix, Fig. S6). For example, more than 25% of the OTUs that were present at frequencies of <0.1% (corresponding to an average of only 22 reads per samples) were detected within hosts over the entire sampling period.
Although the number of OTUs (SI Appendix, Table S4) and the phylogenetic distribution of OTUs remained remarkably consistent over the sampling period, we examined the patterns of change in the frequencies of each OTU detected in all longitudinal samples from a given host. As expected, if the long-term storage of samples has no affect on the recovery and recognition of OTUs, there was no consistent change in the frequency of persistent OTUs among hosts: In four hosts, a majority of the persistent OTUs decreased through time, in two hosts, a majority increased, and in one host, there was no appreciable difference between the number of OTUs that increased or decreased in frequency.
Discussion
We examined a large and diverse set of chimpanzees from Gombe National Park in Tanzania and found that their gut microbial communities represent a discrete, phylogenetic clade distinct from those present in other chimpanzees and great apes (Fig. 2). Previous analysis of the gut microbiota of several species of great apes (including humans) revealed that the branching order of phylogenies based on microbial community compositions paralleled the evolutionary relationships of great ape species (10). The phylogenetic concordance between gut communities and host species, which resembles a pattern of vertical transmission, is surprising given that gut microbes are acquired from external sources each generation. This observation suggests that genetically determined factors (such as the host immune system), which accumulate differences over evolutionary timescales, have played a major role in the diversification of the great ape gut microbial communities.
Despite the concordance of the species-level phylogenies, which indicate that each great ape species has signature gut microbiota that is most similar to that in its most closely related species, the majority of gut community variation occurs among individuals within each species. Such within-species variation has been observed in comparisons of distal gut microbial communities of human hosts (7, 12, 13, 18) and is evident from the long terminal branches in the microbial community phylogenies (Fig. 2). Because numerous factors can potentially produce the large within-species diversity, we assayed samples collected over the past decade from Gombe chimpanzees to determine the contribution of genetic, social, geographic, and temporal variables on gut microbial community composition and continuity.
Given the strong phylogenetic signal at the core of the relationships among the gut microbiotae of great apes, one might expect that the degree of genetic relatedness between individuals would dictate the level of similarity in the compositions of their gut microbial communities. However, we detected no association between coefficients of relatedness between individuals and the overall similarity in their gut microbial communities (Fig. 4). This situation in Gombe chimpanzees differs from previous findings on humans in which the gut microbial communities of twins and of parent-offspring pairs were more similar than were those of unrelated individuals (6, 18). Within the Gombe chimpanzees, there are significant sex-specific differences in the beta diversity of their gut microbial communities. Such dissimilarities have been observed (8) but are not common, which suggests that they stem not from physiological differences but from differences in the feeding behaviors of male and female chimpanzees. Male chimpanzees tend to seek food over the entire range of their community, whereas females typically feed in small, confined areas (23, 24), a pattern that might contribute to disparity between the sexes. However, the difference between males and females is observed in infants/juveniles (who feed with their mothers) as well as in adults, suggesting that other sex-specific traits contribute to differences in the gut microbiota.
Several aspects of the variation in gut microbial communities of the Gombe chimpanzees support the view that the differentiation (or convergence) in their gut microbial communities is shaped by inputs from environmental sources (such as diet, provenance, and/or social contact). For example, gut microbial communities differentiate according to the geographic origin and community affiliation of the host (Fig. 3), with most of the borderline cases being females who switched communities or immigrated from outside territories. The differentiation of individuals from the two communities is caused by approximately 100 phylotypes, which were either unique to members of a particular community or exhibited up to a 100-fold difference in mean abundance between communities. Despite the similarities in the gut microbiota in the members of a chimpanzee community, which could be due to a common diet and/or increased social interactions, higher-order factors are also contributing to the contents and composition of the gut microbiota. By examining the fecal microbiota of individual chimpanzees sampled over 8 y, we detected both host-specific and large-scale components to the differentiation of gut microbial communities.
The longitudinally sampled chimpanzees harbored broadly overlapping sets of core phylotypes that were maintained at relatively high frequencies throughout the entire sampling period. However, samples taken 1 y apart were, on average, twice as similar as those separated by 7 or 8 y, reflecting the perpetuation of resident phylotypes and gradual turnover of transient members. This investigation represents the longest temporal sampling of an individual’s gut microbiota, although such long-term studies of the microbial communities on and within humans are beginning to accumulate. Another level of temporal structuring to these microbial communities was observed: The microbiota from all seven longitudinally sampled chimps converge in 2008, even those derived from chimpanzees from different communities. Moreover, even after implementing a method that returned a higher resolution of 16S rDNA phylotypes, we found no evidence indicating either that these temporal patterns of variation among hosts or that the changes in microbial communities over time stem from the use of samples that have been stored frozen for more than a decade. The most likely explanation for the convergence in the gut microbiotae of chimpanzees sampled 2008 comes from field observations that members from the Kasekela community were frequently ranging into the Mitumba community territory in 2007 and 2008, which would increase similarity in the dietary sources of microbes among members of the two communities.
The identification of core groups of resident taxa in the Gombe chimpanzee is reminiscent of the recent description of human enterotypes, which correspond to groups of individuals clustered in characteristic gut microbiota profiles (25). We find that multiple factors—including geographic, temporal, sex-, and age-specific—are associated with the long-term composition and diversity of the gut microbial communities harbored by Gombe chimpanzees, but that despite the variation over space and time, their gut communities remain distinct from those of other great apes, including other subspecies of chimpanzees.
Materials and Methods
The microbial community diversity present in 47 fecal specimens from 35 SIVcpz negative chimpanzees (P.t. schweinfurthii) at the Gombe National Park (Tanzania) was assayed by 16S rDNA pyrotag and iTag sequencing procedures (10, 26, 27). The individual source of all samples was determined by genetic markers and could be unequivocally assigned to hosts, such that the sex, age at time of sampling, community affiliation, and genealogical relationships are known. For each chimpanzee, we analyzed a sample collected between October and December 2008 with few exceptions (Tubi_2008, Darbee_2008). For seven chimpanzee hosts, we selected additional samples from several additional years collected during the same 3-mo interval. Total DNA was extracted and used as a template to amplify fragments corresponding to the V6–V9 region of 16S ribosomal RNA by using the universal primers 926F and 1492R, which were barcoded for multiplexing samples into a single pyrosequencing run. Raw pyrosequencing reads were quality-trimmed as in ref. 10 by using an error rate of 0.5% (equivalent to Phred quality score of 23), and reads <260 nucleotides in length, or that contained undefined bases or lacked perfect barcode and primer sequences, were removed. Taxonomic assignments of the quality-trimmed reads (also known as “pyrotags”) were performed with the RDP classifier (28). Trimmed, taxonomically assigned sequences were dereplicated, chimera-checked, aligned, and clustered at a range of similarity values (90–99%). Identical procedures were performed on 454 flowgrams that were denoised in QIIME (29). Pyrotag analyses of microbial communities were based on occurrences and abundances of 99% OTUs (i.e., clusters of pyrotag sequences that differ by 1% or less) that were phylogentically informative (i.e., present in two or more samples). To examine changes in gut microbial communities at higher levels of resolution, we implemented an Illumina-based 16S-tag (iTag) approach that was analogous to the pyrotag procedure but differs by interrogating a 100-nt region from the 5′-end of amplicons spanning the V4 region of 16S rDNA (27). iTags were generated for a total of 39 samples, which represent annual samples spanning 10 consecutive years for the seven chimpanzees that were surveyed at multiple time-points by the pyrotag procedure.
Additional details about the source of samples, sample preparation, pyrotag sequencing and processing, statistical analyses, iTag sequencing and processing, as well as associated references, are provided in SI Appendix.
Supplementary Material
Acknowledgments
PCR and sequencing primers for iTag analysis were generously provided by Rob Knight (University of Colorado) and Integrated DNA Technologies. We thank the Jane Goodall Institute and the National Science Foundation through Grant BSC-0648481 (to M.L.W.) for supporting collection of behavioral data and fecal samples from chimpanzees at the Gombe Stream Research Centre; and the Tanzania Commission for Science and Technology, the Tanzania Wildlife Research Institute, and the Tanzania National Parks for permission to conduct research in Gombe. This work was supported in part by National Institutes of Health Grants R01 AI50529, R01 AI58715 (to B.H.H.), and R01 GM74738 and R01 GM101209 (to H.O.); University of Alabama at Birmingham Center for AIDS Research Grant P30 AI 27767; and the National Science Foundation (A.E.P.). R.S.R. was funded by a Howard Hughes Medical Institute Med-into-Grad Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110994109/-/DCSupplemental.
References
- 1.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 2.Mackie RI, Sghir A, Gaskins HR. Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr. 1999;69:1035S–1045S. doi: 10.1093/ajcn/69.5.1035s. [DOI] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariat D, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoetendal EG, Akkermans ADL, Akkermans van-Vliet WM, de Visser JAGM, de Vos WM. The host genotype affects the bacterial community in the human gastrointestinal tract. Microb Ecol Health Dis. 2001;13:129–134. [Google Scholar]
- 7.Ley RE, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. and erratum (2008) 322:1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna P, et al. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 2008;4:e20. doi: 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yildirim S, et al. Characterization of the fecal microbiome from non-human wild primates reveals species specific microbial communities. PLoS ONE. 2010;5:e13963. doi: 10.1371/journal.pone.0013963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ochman H, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claesson MJ, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caporaso JG, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benson AK, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Constable JL, Ashley MV, Goodall J, Pusey AE. Noninvasive paternity assignment in Gombe chimpanzees. Mol Ecol. 2001;10:1279–1300. doi: 10.1046/j.1365-294x.2001.01262.x. [DOI] [PubMed] [Google Scholar]
- 21.Keele BF, et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature. 2009;460:515–519. doi: 10.1038/nature08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wroblewski EE, et al. Male dominance rank and reproductive success in chimpanzees, Pan troglodytes schweinfurthii. Anim Behav. 2009;77:873–885. doi: 10.1016/j.anbehav.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrangham RW, Smuts BB. Sex differences in the behavioral ecology of chimpanzees in the Gombe National Park, Tanzania. J Reprod Fertil. 1980;28(Suppl):13–31. [PubMed] [Google Scholar]
- 24.Williams JM, Pusey AE, Carlis JV, Farm B, Goodall J. Female competition and male territorial behaviour influence female chimpanzees’ ranging patterns. Anim Behav. 2002;63:347–360. [Google Scholar]
- 25.Arumugam M, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity. ISME J. 2012;6:183–194. doi: 10.1038/ismej.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporaso JG, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole JR, et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods) Sunderland, MA: Sinauer Associates, Inc.; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.