

Abstract
We surveyed endophytic fungal communities in leaves of a single tree species (Metrosideros polymorpha) across wide environmental gradients (500–5,500 mm of rain/y; 10–22 °C mean annual temperature) spanning short geographic distances on Mauna Loa Volcano, Hawai’i. Using barcoded amplicon pyrosequencing at 13 sites (10 trees/site; 10 leaves/tree), we found very high levels of diversity within sites (a mean of 551 ± 134 taxonomic units per site). However, among-site diversity contributed even more than did within-site diversity to the overall richness of more than 4,200 taxonomic units observed in M. polymorpha, and this among-site variation in endophyte community composition correlated strongly with temperature and rainfall. These results are consistent with suggestions that foliar endophytic fungi are hyperdiverse. They further suggest that microbial diversity may be even greater than has been assumed and that broad-scale environmental controls such as temperature and rainfall can structure eukaryotic microbial diversity. Appropriately constrained study systems across strong environmental gradients present a useful means to understand the environmental factors that structure the diversity of microbial communities.
Keywords: Mauna Loa matrix, microbial biogeography, beta diversity
The rapid advance of sequencing technologies has facilitated research describing the extent of microbial richness in hundreds of environments around the world. Work using culture-independent, high-throughput sequencing methods has found very high levels of diversity, much of it resulting from unknown species, many of which are rare (1, 2). Despite these new tools and a growing appreciation for the diversity of the microbial world, it has proved challenging to understand spatial patterns in microbial diversity and their controls (3–5). It is clear that microbial communities can vary at different scales than do plant and animal communities (5) and that substantial changes in soil communities can occur over very short distances (6). Questions that remain include: How does the number of microbial species within a community differ from site to site, and to what extent does environmental variation contribute to these differences? What proportion of the diversity of a region is associated with differences in community composition among sites (as opposed to species richness within sites), and how does environmental variation contribute to regional diversity?
The phyllosphere (the leaf surface and particularly its interior) represents a useful and tractable context for examining relationships between microbes and their environment (7, 8), and several factors enhance its utility for understanding microbial biogeography in particular. First, the phyllosphere carries out the majority of terrestrial carbon fixation and, thus, represents one of the strongest biotic links between the biosphere and the atmosphere (9). Second, the phyllosphere provides habitat for complex assemblages of microorganisms (10–12). Third, leaves represent largely discrete, relatively uniform, bounded habitats to the microbes that inhabit them. Finally, leaves allow for a high level of structure to sampling: within and between leaves of the same tree, as well as within and between individual trees of the same species across a landscape. This nested replication is particularly helpful when seeking to understand complex microbial communities.
Despite these experimental incentives, and the fact that its surface area is approximately twice as great as the land surface (6, 13), the phyllosphere remains one of the least-studied environments of the Earth. Within this environment, some of the most diverse and potentially influential members of the biotic community are the fungal endophytes (FEs) (7, 8, 14–16). FEs inhabit the asymptomatic aboveground tissues of their hosts (9, 17–19) and are found in all species and in all divisions of land plants (20). These organisms have been the subjects of significant research for several decades (e.g., refs. 21–23). In comparison with FEs in agricultural grasses, research on endophytes in woody plants has been relatively sparse (14), attributable, in part, to much higher fungal species heterogeneity within and among hosts (24–26). Foliar endophytes of woody plants appear to be highly diverse (13, 27), particularly in the tropics (8, 19), and this diversity has inspired recent studies by mycologists, ecologists, and natural products researchers (e.g., refs. 28–30).
In this study, we sought to acquire basic information on the diversity of FEs within individual trees, within sites, between adjacent sites that differ in nutrient availability, and across a landscape encompassing wide environmental gradients over small geographic distances in a tropical system. In addition, we asked:
i) How does variation in endophyte diversity among sites reflect environmental characteristics? The diversity of most plant and animal communities is much greater in tropical than temperate environments and greater in lowland rainforests than other tropical environments. When sampling is constrained to a single host species, do individuals in lowland wet rainforests support the highest diversity of FEs?
ii) Does the composition of the FE community vary among sites? Less diverse communities could be subsets of more diverse communities (as is often observed when comparing the communities of different-sized islands) or each community could support a different group of species. If the latter is observed, how much does variation among sites (or β-diversity) contribute to the overall species richness in the landscape?
iii) Most importantly, to what extent is spatial variation in the composition of FE communities associated with environmental variation? A substantial degree of environmental control over these microbial communities (all resident in leaves of a single tree species) would indicate that coarse-scale environmental variation could play a substantial role in structuring microbial communities regionally and globally.
To answer these questions, we used culture-independent, high-throughput barcoded amplicon pyrosequencing to quantify patterns of variation in FE communities within a single host tree species (Metrosideros polymorpha) across an extraordinarily broad range of environments (ranging in elevation from 100 to 2,400 m and in rainfall from <500 to >5,500 mm/y) on the Island of Hawai’i. At seven locations, we collected samples from paired early-successional (ca. 150 y-old) and later-successional (ca. 3,500 y-old) sites, which enabled us to examine independently the effects of rainfall, elevation, and substrate age on endophyte communities.
Results
Diversity of Fungal Endophytes.
During the fall of 2009, we sampled the endophytic fungal communities of trees spanning 13 sites on Mauna Loa Volcano, Hawai’i (Fig. 1). Ten leaves were pooled from each of these trees (n = 130 pooled samples; 10 trees/site). Genomic (g)DNA was extracted from surface-sterilized leaves and amplified with fungal-specific primers for the nuclear ribosomal (nr)DNA internal transcribed spacer region 1 (ITS1) region and sequenced with barcoded titanium pyrosequencing (see Methods). After quality control and trimming, we had 665,155 high-quality sequences that were used for all primary analyses (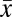 tree = 5,117 sequences;
tree = 5,117 sequences; 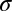 tree= 1,574; mean trimmed ITS1 length: 157 bp). The putative identities of the most common operational taxonomic units (OTUs), as determined via a BLAST search of GenBank are summarized in Table S1.
tree= 1,574; mean trimmed ITS1 length: 157 bp). The putative identities of the most common operational taxonomic units (OTUs), as determined via a BLAST search of GenBank are summarized in Table S1.
Fig. 1.

Study site locations on the Island of Hawai’i.
Grouping sequences into OTUs at 95% similarity [more or less corresponding to species-level similarity (31)], per-tree (n = 130) richness across the landscape ranged from 40 to 257 (16–190 nonsingletons). Sequence-based rarefaction curves based on observed OTUs (Sobs) and the Chao1 richness estimator were asymptotic or nearly asymptotic for most trees (Fig. 2 and Fig. S1), implying that per-tree sequencing depth was adequate to capture the diversity of FEs present in samples pooled from individual trees. However, these communities are heterogeneous, and it is likely that sampling additional leaf tissue would lead to increased sequence diversity. Per-site (n = 13) richness ranged from 401 to 829 (255–550 nonsingletons), per location (colocated sites that differed in substrate age at the same elevation, n = 6) from 708 to 1,229 (417-817 nonsingletons), and overall fungal OTU richness across the landscape was 4,253 (2,552 nonsingletons). At the site level and above (Fig. 2), rarefaction curves did not approach asymptotes. The failure to reach asymptotes was likely attributable to insufficient sampling depth (too few trees sampled), rather than to insufficient sequencing depth. Richness estimators and nonsingleton counts by level of analysis are summarized in Tables S2–S5.
Fig. 2.
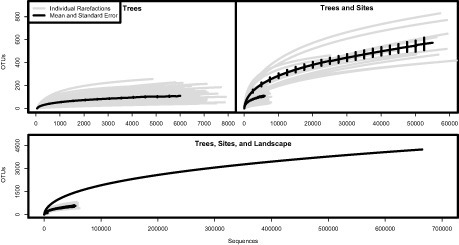
Rarefaction curves of observed OTU richness at the tree, site, and landscape level. OTUs were designated based on 95% similarity of the ITS1 rDNA region.
Species Richness Within Sites.
Mean OTU richness was similar at most sites and locations, with a mean of 105 ± 42 OTUs per tree (68 ± 30 nonsingletons), except for the wet low elevation (100 m) sites, where we observed significantly greater richness of ca. 160 OTUs per tree [Tables S2–S5; Tukey honestly significant difference (HSD): P < 0.05].
Variation in Fungal Endophyte Communities Among Sites.
We found substantial among-site and among-location variation in FE communities. Fungal communities of trees in a given location were significantly different from those in other locations [analysis of similarities (ANOSIM): R = 0.66; P < 0.001]. The inverse of Whittaker’s multiplicative measure (32, 33) of among-site variation or β-diversity, in which mean within-site diversity (α-diversity) is divided by overall regional diversity (γ-diversity), provides an index of among-site diversity that ranges from 0 to 1. Results close to 1 indicate most diversity is within sites; results close to 0 indicate the importance of among-site diversity. With the most conservative assumptions (that within-location diversity is the Chao estimated value at the most speciose location, and that total richness across the landscape is the observed value), among-site diversity contributed more to total diversity than did within-site diversity in this study (the inverse of Whittaker’s β = 0.42). Additional diversity calculations are summarized in Tables S2–S5.
Environment and Community Structure.
We evaluated the direct contributions of three specific factors to FE community differences: elevation, rainfall, and lava-flow age/nutrient availability. Our study system was chosen to enable us to decouple the effects of these from one another. Based on the Chao estimated Jaccard distance (34), a permutational multivariate analysis of variance (PERMANOVA) using distance matrices found each of these three factors to be significant in structuring endophyte communities (PERMANOVA: P < 0.01; combined coefficient of determination R2 = 0.56). Elevation and rainfall explained most of the variation (combined R2 = 0.53). Although the amount of community variation explained by each factor depended on the distance metric used, these three factors always remained significant (Tables S6–S9). Analyses that controlled for geographic distance also supported the significance of elevation and rainfall (see Methods for details). Whereas Mantel correlograms showed no significant spatial autocorrelation within transects, distance covaried with rainfall and elevation across portions of the Mauna Loa matrix.
We find further evidence for strong structuring of these endophyte communities by macroscale environmental variables, based on the placement and dispersion of locations in ordination [nonmetric multidimensional scaling (NMDS)] space (Fig. 3). Patterns of community similarity were strongly associated with rainfall and elevation; the rainfall and elevation parameters of the different locations were fit to linear vectors in ordination space and these vectors were nearly orthogonal and highly correlated with the placement of location centroids (rainfall: r2 = 0.95, P < 0.01; elevation: r2 = 0.93, P < 0.01).
Fig. 3.

Two-dimensional ordination using NMDS (stress, 18.1). Each point (n = 130) represents the fungal endophyte community of an individual tree and colors signify different locations across the Mauna Loa matrix. Labels represent the rainfall (W, wet; D, dry) and elevation (in meters) of the locations. Open shapes signify trees on young substrate; filled shapes signify those on older substrate. Environmental vectors were fit to the centroids (n = 7) of each location in ordination space (rainfall: r2 = 0.95, P < 0.01; elevation: r2 = 0.93, P < 0.01).
Discussion
To understand FE richness across the wide environmental gradients of Mauna Loa, we relied on a sequencing depth several times higher than used by any fungal pyrosequencing study thus far. In doing so, we documented the presence of a remarkable number of fungal species: a richness exceeding 4,200 OTUs across thirteen sites, all within the same host, on the same island, and within 80 km of each other. When rarefied to a comparable number of sequences, this richness represents roughly half of the number of OTUs found in a global study of indoor fungi (35) and a greater richness than found in the entire phyllosphere of the temperate Quercus macrocarpa (27); for more details, see Table S10. Earlier work by Arnold and colleagues using culture and cloning-based methods documented the high species richness of tropical endophytes (19, 28, 36) relative to those in temperate and boreal locations. As with other microbial communities, the advent of next-generation pyrosequencing greatly increases our ability to recognize diversity in comparison with earlier studies.
Although recent studies have pointed out that PCR, sequencing, and OTU clustering error can all contribute to inflated richness in pyrosequencing data (37–39), two lines of evidence indicate that this study does not exaggerate endophyte richness. First, the patterns of OTU accumulation and richness estimation for individual trees were in most cases nearly asymptotic and did not display accumulation patterns characteristic (38) of PCR and/or sequencing error (Fig. 2 and Fig. S1). Second, the high β-diversity we observed is predictably structured: elevation and rainfall are highly correlated with community composition at the landscape scale. Finally, although it is possible that a small number of leaf epiphytic fungi were incorporated into these analyses, we have evidence that many of these organisms were removed before DNA extraction (40) (Table S11).
Arnold and colleagues sought to understand global patterns of FE distribution (19, 28) and found much higher richness in the tropics, even though phylogenetic diversity was greater given the same sampling effort in temperate and boreal sites. Within the constrained set of sites we examined in Hawai’i, we find no strong patterns in within-site species richness beyond high richness observed at the low elevation wet site. However, we did find substantial variation among sites; indeed, most of richness we detected reflects among-location differences. Moreover, the true levels of among-site differences are larger still; despite high sequencing depth, OTU richness curves for fungal endophytes at the site and landscape level were not asymptotic (Fig. 2). The failure to reach asymptotes was likely attributable to insufficient sampling depth (i.e., number of trees sampled); we believe this is a result of the high among-site diversity we observed, as detailed above, rather than from insufficient sequencing depth. Furthermore, the 95% ITS1 cutoff we used in this study is likely a conservative estimate for true species numbers (41). These results indicate that assessments of the hyperdiversity of fungi globally by Hawksworth (42) and, more recently, Blackwell (43) may prove to be conservative. Whereas those analyses were based on fungal communities across different hosts, a factor not addressed here, the among-site differences presented in this study suggest even greater endophytic diversity on single hosts than previously suspected.
Additionally, the variation we found among sites is strongly associated with environmental factors. Our result is in general agreement with the description of variation with environment in the endophytes associated with conifers across sites in the Pacific Northwest by Carroll and Carroll (21), as well as a recent culture-based study by U’Ren et al. (44) of endophytes at the continental scale, which suggested that climate could shape endophyte community similarity. However, our much deeper sampling and perhaps differences between temperate and tropical environments accentuated the pattern of among-site variation in our study. Beyond environmental factors, host genotype can also have an effect on endophyte communities (45, 46), but this effect may be relatively less important in our results: population genetic studies of M. polymorpha have shown a high level of gene flow between populations and no significant correlation between M. polymorpha genetic distance and geographic distance on the Island of Hawai’i (47, 48).
Our results for among-site variation contrast with analyses of soil microbes, in which strongest associations are not tied to climate but to soil pH and C:N ratios (49, 50). There are several possible reasons for this difference. First, and most importantly, our study is focused exclusively on fungi and is several times deeper than previous studies with similar focus. Second, endophytes may be subject to a greater extent of environmental variation than soil microorganisms; temperature extremes experienced by leaves are somewhat buffered in soils (51). Finally, it is possible that the broad extent of environmental variation on Mauna Loa reveals the potential for environmental control of microbial diversity that is masked in other, less-variable study systems. If the last is the case, microbial communities elsewhere may also be controlled by factors similar to plant/animal communities, and strong gradients like those presented by the Mauna Loa environmental matrix may offer a useful system for understanding these controls.
Methods
Host and Sites.
We focused on the endophytic fungal communities of M. polymorpha (Myrtaceae), a tree endemic to Hawai’i. This tree species has a remarkably wide native environmental range (400–11,000 mm rainfall/y; 10–24 °C mean annual temperature), and previous research has characterized its successional patterns (52–54), physiology (55–59), nutrient utilization patterns (60, 61), and population-level genetic structure (47, 48). At each of seven locations, paired early-successional (ca. 150-y-old) and later-successional (ca. 3,500-y-old) sites were identified using flow designations developed by the US Geological Survey (USGS) (62) and Geographical Information Systems (GIS) (Fig. 1). At the 1,700-m dry site, the older flow lacked Metrosideros and was not sampled, leaving a total of 13 sites. In addition to some of the background research on M. polymorpha referenced above, previous work in these same sites has evaluated patterns of productivity and litter decomposition (63–65). Site characteristics are summarized in Table S12.
Field Sample Collection.
We targeted our methods on endophytes resident in leaves and not on incidental phyllosphere species. We collected 10 mature, asymptomatic sun leaves from 10 randomly selected trees in each of the 13 sites described above between July and November 2009. We randomly located 100-m transects via GIS to be within substrate boundaries and to avoid edge effects. A canopy Metrosideros was selected every 10 m along each transect and leaves were taken at random from multiple aspects of each tree. At the 2,400 m elevation sites, transect sampling was not possible because tree density was low, so all available individuals were sampled. The height at which leaves were sampled ranged from less than 0.5 m above the ground at the highest elevation sites to ∼5 m at the lower sites; this height also varied to a smaller extent within sites.
Laboratory Methods.
Sample preparation.
All leaves were surface sterilized within 72 h after collection to reduce the presence of surface microorganisms. The leaves were rinsed in deionized (di)H2O, immersed in 95% (vol/vol) ethanol (5 s), 0.5% NaOCl (2 min), and 70% vol/vol ethanol (2 min), and finally received three sequential 1 min rinses in diH2O (66). We believe that this procedure removed most surface microorganisms, so we refer throughout this report to the organisms remaining after surface sterilization as fungal endophytes (see ref. 40 and Table S11). The leaves were dried at 40 °C for 48 h and then pooled and ground using a Spex CertiPrep 8000 ball-mill with AISI 316 SS bearings (Glen Mills) before analysis. Bearings were sterilized after each use following a modification of Qiagen standard protocol: sequential rinses of diH2O, 1 min in 0.5 N HCl, then 5 min in 0.5% NaOCl (67), and then dried for a minimum of 24 h at 60 °C.
DNA extraction, amplification, and sequencing.
Total gDNA was extracted from subsamples of plant tissue pooled from 10 leaves from each tree using Qiagen Plant DNeasy kits with a modified protocol on a Qiagen QiaCube. We pooled leaves at the tree level; our primary interest was in variation of communities across a landscape instead of variation leaf-to-leaf. The standard Qiagen extraction protocol was modified as follows: dry tissue weight used was 50 mg, the recommended amounts of buffers AP1 and AP2 were doubled, 8 μL of 1 M sodium metabisulfite was added, fifteen 2.3 mm Zirconia/Silica beads were used to improve vortexing, and the incubation step was extended to 60 min. Extracts were then PCR amplified on a PTC-0225 tetrad thermal cycler (MJ Research) in triplicate to alleviate the effects of differential amplification from environmental samples. The 18.75-μL PCR reactions were prepared on ice in individually capped PCR tubes to reduce formation of primer-dimers and to prevent sample cross-contamination. Reaction components included 9.3 μL of Sigma REDtaq ReadyMix, 6 μL of DNA template, 0.6 μL each of 20 μM forward and reverse primers, 1.5 μL of 16 mg/mL BSA, and 0.75 μL of 10 mM MgCl2. The primers used were HPLC-purified 454 Fusion Primers that incorporated the fungal-specific primer ITS1-F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) and reverse primer ITS2 (5′-GCT GCG TTC TTC ATC GAT GC-3′) and one of twelve 10-bp multiplex identifier sequences (MIDs), as provided by Roche. To allow postsequencing differentiation of samples, each reaction included a distinct combination of forward and reverse MIDs. The hot-start PCR reactions were cycled for 3 min at 94 °C; 30 cycles of 1 min at 94 °C, 30 s at 54 °C, and 1 min at 72 °C, and, finally, 7 min at 72 °C. Negative controls were run for both extraction and PCR; these remained free of amplicons. Each triplicate reaction was pooled and PCR cleanup was performed on each with AMPure beads (Beckman Coulter Genomics). After testing cleaned PCR products for concentration via PicoGreen quantitation (Invitrogen) on a Gemini XPS fluorometer (Molecular Devices) and size via gel electrophoresis, amplicon samples were standardized by molarity, pooled into a single sample, and sent for bidirectional 454 FLX Titanium pyrosequencing on half of a pico-titer plate at the Duke Institute for Genomic Sciences and Policy (Durham, NC) using Lib-A reagents. Standard Flowgram Format amplicon sequence data have been deposited in National Center for Biotechnology Information (NCBI) Sequence Read Archive under accession number SRX153137.
Bioinformatics.
Although bioinformatics pipelines for processing data from amplicon pyrosequencing have progressed rapidly (68, 69), they are often targeted toward amplicons of the 16S region from bacteria or archaea. We found none that incorporated all of the analyses in which we were interested: individuation based on forward and reverse Fusion Primer tags, extraction of the ITS1 region from its flanking bases, denoising and clustering that was rigorous yet computationally feasible, and graphical analysis of BLAST results at multiple taxonomic levels. We used programs and code available from a variety of sources for our bioinformatics processing (see below); where code did not exist, we wrote our own using Perl.
Sequence quality control.
Amplicons were checked for length and quality: sequences with errors or missing sections in the primer region or barcodes, any ambiguous bases, with low average read score (<30) or with a total length of <100 bp were discarded. The remaining sequences were individuated via the unique combination of forward and reverse MIDs present in each sequence using Perl. Primers and barcodes were trimmed with a Perl script. Any remaining conserved SSU or 5.8S bases were removed using hidden Markov models (HMMs) for the ITS region (70). Those sequences that diverged substantially from the HMM predictions in the SSU or 5.8S regions were aligned via MUSCLE (71) and manually trimmed in Geneious v5 (72). Although the ITS region is highly variable and thus notoriously difficult to align across broad taxa, the conserved regions on each end aligned well and enabled manual sequence trimming.
Alignment and clustering.
Two methods that are gaining widespread use in reducing artifactual OTUs generated by pyrosequencing and/or clustering error (noise) in short-read amplicon pyrosequencing data are AmpliconNoise by Quince et al. (39, 73) and the single-linkage preclustering (SLP) method of Huse et al. (37). Although marked improvements have been made in the speed of the AmpliconNoise (previously PyroNoise) algorithm (39), it is still intensive computationally, particularly with large datasets, so we used SLP to process our pyrosequences. To do so, we followed the SLP protocol as described (37, 74), with modifications for the ITS1 region. Unique sequences across all sites were found using mothur (75, 76). Pairwise 6-mer distances were then calculated using the computer cluster version of ESPRIT (77, 78) using default parameters, with the exception of a 6-mer distance cutoff of 0.6. This cutoff was chosen after analysis of the relationship between 6-mer distance and Needleman–Wunsch (NW) pairwise distance for ITS1 amplicons. We used the needledist algorithm, again from the ESPRIT package, to calculate the NW pairwise distance for each of the sequence pairs that had a 6-mer distance of less than 0.6. These distances were preclustered with the SLP algorithm using a cutoff distance of 0.02. The resulting clusters from the SLP algorithm were then clustered at 95% similarity using the average distance clustering method of mothur. A similarity cutoff of 95% was chosen following the rationale in (31, 79). This cutoff allowed comparison with other fungal pyrosequencing studies (27, 80, 81).
Taxonomic identification.
Representative sequences from each OTU were selected based on abundance with a python script from QIIME v. 1.2 (34, 68) and were checked for their closest match in GenBank via BLASTn with default parameters (82, 83). The BLAST results were analyzed in MEGAN (74, 84), the output from which was also used in conjunction with a Perl script that queried the NCBI taxonomy database to generate full lineages of each of the representative sequences for each OTU that had a match. Although BLAST searches of ITS sequences against the NCBI database can be inaccurate (75, 85–87), we were primarily interested in gaining a broad qualitative sense of the identities of the taxa present and believe that these methods provide a reasonable preliminary grouping at higher taxonomic levels (Table S1).
Statistics
Richness and community analyses.
OTU richness and rarefaction statistics were calculated using the vegan (77) package in R (79). To assess the significance of community similarity among vs. within sites, we used ANOSIM as described (80). We used NMDS ordinations to visualize community similarities across the landscape; ordination plots were created using the “metaMDS” function in vegan, which incorporated a square-root transform and Wisconsin double-standardization of OTU abundances. To take into account the effects of undersampling and rare species on community similarity, we used estimated abundance-based Jaccard similarity as described by Chao et al. (34), which is robust for communities with long-tail species abundance distributions or uneven sampling depth. We also compared our ordination results to those generated from OTU tables rarefied to 700 sequences per tree (the lowest number of sequences in any tree); with all singletons removed, all singletons and doubletons removed, and all singletons, doubletons, and tripletons removed; and to results calculated with presence-absence distance metrics (standard Jaccard), abundance-based metrics (Bray–Curtis and Morisita–Horn), and the probabilistic Raup–Crick metric. All distance measures and other comparisons provided qualitatively similar patterns across the landscape and produced very similar NMDS ordination plots, ANOSIM, PERMANOVA using distance matrices, and distance-based redundancy analyses (dbRDA) results (see Tables S6–S9).
Spatial autocorrelation and distance.
To understand patterns of spatial autocorrelation, we used the ecodist (82) package in R to do mantel and partial mantel tests and to visualize their associated correlograms (84). To quantify the contribution of environmental variables to the makeup of the endophyte communities, we used PERMANOVA, as implemented in the “adonis” function in the vegan R library and described by (85–87). Although environmental factors partially covary across the Mauna Loa matrix, we sought to test the significance of environmental control on communities while statistically constraining the variation attributable to distance alone. We used partial mantel tests and dbRDA constrained using principal components of neighbor matrices (PCNM), implemented in R as the “capscale” function and as described (86, 88, 89). All tests provided similar and significant results regarding the importance of environmental variables for the structuring of endophytic communities (see Tables S6–S9).
Supplementary Material
Acknowledgments
We thank Mali'o Kodis, Kolea Zimmerman, Eric Slessarev, Kye Epps, Jennifer Johnson, Elizabeth Kodis, Robert Zimmerman, and Heraldo Farrington for help in the field; Don Hemmes, Ward Watt, and Dmitri Petrov for use of laboratory facilities; and Josefa González for technical help with molecular methods. We thank Nikhil Narahari and the US Army Pōhakuloa Training Area for access to sites. We thank Jennifer Johnson, Gabriel Maltais-Landry, Tadashi Fukami, Dmitri Petrov, and two formal reviewers for helpful comments on earlier versions of this manuscript. This work was funded, in part, by a National Science Foundation (NSF) graduate research fellowship (to N.B.Z.) and NSF Doctoral Dissertation Improvement Grant (DDIG) DEB-1010504 (to P.V. and N.B.Z.). Supercomputer access was made possible by a computing resources grant to Stanford University (NSF Grant CNS-0619926).
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the Sequence Read Archive database, www.ncbi.nlm.nih.gov/sra (accession no. SRX153137).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1209872109/-/DCSupplemental.
References
- 1.Huse SM, et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sogin ML, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor JW, Turner E, Townsend JP, Dettman JR, Jacobson D. Eukaryotic microbes, species recognition and the geographic limits of species: Examples from the kingdom Fungi. Philos Trans R Soc Lond B Biol Sci. 2006;361:1947–1963. doi: 10.1098/rstb.2006.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorsten Lumbsch H, Buchanan PK, May TW, Mueller GM. Phylogeography and biogeography of fungi. Mycol Res. 2008;112:423–424. doi: 10.1016/j.mycres.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Martiny JBH, et al. Microbial biogeography: Putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 6.Franklin RB, Mills AL. Multi-scale variation in spatial heterogeneity for microbial community structure in an eastern Virginia agricultural field. FEMS Microbiol Ecol. 2003;44:335–346. doi: 10.1016/S0168-6496(03)00074-6. [DOI] [PubMed] [Google Scholar]
- 7.Meyer KM, Leveau JHJ. Microbiology of the phyllosphere: A playground for testing ecological concepts. Oecologia. 2012;168:621–629. doi: 10.1007/s00442-011-2138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold AE. In: Tropical Forest Community Ecology. Schnitzer S, Carson W, editors. Oxford: Blackwell Scientific; 2008. pp. 254–271. [Google Scholar]
- 9.Waring RH, Schlesinger WH. Forest Ecosystems: Concepts and Management. Orlando, FL: Academic; 1985. [Google Scholar]
- 10.Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine MC. Drivers of bacterial beta-diversity depend on spatial scale. Proc Natl Acad Sci USA. 2011;108:7850–7854. doi: 10.1073/pnas.1016308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leveau JHJ. Microbiology: Life on leaves. Nature. 2009;461:741–742. doi: 10.1038/461741a. [DOI] [PubMed] [Google Scholar]
- 12.Andrews JH, Harris RF. The ecology and biogeography of microorganisms of plant surfaces. Annu Rev Phytopathol. 2000;38:145–180. doi: 10.1146/annurev.phyto.38.1.145. [DOI] [PubMed] [Google Scholar]
- 13.Bailey MJ, Lilley AK, Timms-Wilson TM, Spencer-Phillips PTN. Microbial Ecology of Aerial Plant Surfaces. Cambridge, MA: Centre for Agricultural Bioscience International Publishing; 2006. [Google Scholar]
- 14.Rodriguez RJ, White JF, Jr, Arnold AE, Redman RS. Fungal endophytes: Diversity and functional roles. New Phytol. 2009;182:314–330. doi: 10.1111/j.1469-8137.2009.02773.x. [DOI] [PubMed] [Google Scholar]
- 15.Saikkonen K, Faeth SH, Helander M, Sullivan TJ. Fungal endophytes: A continuum of interactions with host plants. Annu Rev Ecol Syst. 1998;29:319–343. [Google Scholar]
- 16.Porras-Alfaro A, Bayman P. Hidden fungi, emergent properties: Endophytes and microbiomes. Annu Rev Phytopathol. 2011;49:291–315. doi: 10.1146/annurev-phyto-080508-081831. [DOI] [PubMed] [Google Scholar]
- 17.Petrini O. In: Microbial Ecology of Leaves. Andrews JH, Hirano SS, editors. New York: Springer; 1991. pp. 179–197. [Google Scholar]
- 18.Wilson D. In: Microbial Endophytes. Bacon CW, White JF, editors. New York: Marcel Dekker; 2000. pp. 389–420. [Google Scholar]
- 19.Arnold AE, Lutzoni F. Diversity and host range of foliar fungal endophytes: Are tropical leaves biodiversity hotspots? Ecology. 2007;88:541–549. doi: 10.1890/05-1459. [DOI] [PubMed] [Google Scholar]
- 20.Bacon CW. In: Microbial Endophytes. White JF, editor. New York: Marcel Dekker; 2000. [Google Scholar]
- 21.Carroll G, Carroll F. Studies on the incidence of coniferous needle endophytes in the Pacific Northwest. Can J Bot. 1978;56:3034–3043. [Google Scholar]
- 22.Carroll G, Petrini O. Patterns of substrate utilization by some fungal endophytes from coniferous foliage. Mycologia. 1983;75:53–63. [Google Scholar]
- 23.Baker G, Dunn P, Sakai W. Fungus communities associated with leaf surfaces of endemic vascular plants in Hawai'i. Mycologia. 1979;71:272–292. [Google Scholar]
- 24.Saikkonen K. Forest structure and fungal endophytes. Fungal Biol Rev. 2007;21:67–74. [Google Scholar]
- 25.Arnold AE, et al. A phylogenetic estimation of trophic transition networks for ascomycetous fungi: Are lichens cradles of symbiotrophic fungal diversification? Syst Biol. 2009;58:283–297. doi: 10.1093/sysbio/syp001. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman MT, Arnold AE. Geographic locality and host identity shape fungal endophyte communities in cupressaceous trees. Mycol Res. 2008;112:331–344. doi: 10.1016/j.mycres.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Jumpponen A, Jones KL. Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere. New Phytol. 2009;184:438–448. doi: 10.1111/j.1469-8137.2009.02990.x. [DOI] [PubMed] [Google Scholar]
- 28.Arnold AE, Maynard Z, Gilbert GS, Coley P, Kursar T. Are tropical fungal endophytes hyperdiverse? Ecol Lett. 2000;3:267–274. [Google Scholar]
- 29.Murali TS, Suryanarayanan TS, Venkatesan G. Fungal endophyte communities in two tropical forests of southern India: Diversity and host affiliation. Mycol Prog. 2007;6:191–199. [Google Scholar]
- 30.Suryanarayanan T, Venkatesan G, Murali TS. Endophytic fungal communities in leaves of tropical forest trees: Diversity and distribution patterns. Curr Sci India. 2003;85:489–493. [Google Scholar]
- 31.U’ren JM, et al. Diversity and evolutionary origins of fungi associated with seeds of a neotropical pioneer tree: A case study for analysing fungal environmental samples. Mycol Res. 2009;113:432–449. doi: 10.1016/j.mycres.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Whittaker R. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol Monogr. 1960;30:280–338. [Google Scholar]
- 33.Anderson MJ, et al. Navigating the multiple meanings of β diversity: A roadmap for the practicing ecologist. Ecol Lett. 2011;14:19–28. doi: 10.1111/j.1461-0248.2010.01552.x. [DOI] [PubMed] [Google Scholar]
- 34.Chao A, Chazdon RL, Colwell RK, Shen T-J. A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol Lett. 2005;8:148–159. [Google Scholar]
- 35.Amend AS, Seifert KA, Samson R, Bruns TD. Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc Natl Acad Sci USA. 2010;107:13748–13753. doi: 10.1073/pnas.1000454107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold AE, Herre EA. Canopy cover and leaf age affect colonization by tropical fungal endophytes: Ecological pattern and process in Theobroma cacao (Malvaceae) Mycologia. 2003;95:388–398. [PubMed] [Google Scholar]
- 37.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dickie IA. Insidious effects of sequencing errors on perceived diversity in molecular surveys. New Phytol. 2010;188:916–918. doi: 10.1111/j.1469-8137.2010.03473.x. [DOI] [PubMed] [Google Scholar]
- 39.Quince C, Lanzén A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallery RE, Dalling JW, Arnold AE. Diversity, host affinity, and distribution of seed-infecting fungi: A case study with Cecropia. Ecology. 2007;88:582–588. doi: 10.1890/05-1207. [DOI] [PubMed] [Google Scholar]
- 41.Gazis RO, Rehner S, Chaverri P. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol Ecol. 2011;20:3001–3013. doi: 10.1111/j.1365-294X.2011.05110.x. [DOI] [PubMed] [Google Scholar]
- 42.Hawksworth DL. The magnitude of fungal diversity: The 1.5 million species estimate revisited. Mycol Res. 2001;105:1422–1432. [Google Scholar]
- 43.Blackwell M. The fungi: 1, 2, 3 … 5.1 million species? Am J Bot. 2011;98:426–438. doi: 10.3732/ajb.1000298. [DOI] [PubMed] [Google Scholar]
- 44.U’ren JM, Lutzoni F, Miadlikowska J, Laetsch AD, Arnold AE. Host and geographic structure of endophytic and endolichenic fungi at a continental scale. Am J Bot. 2012;99:898–914. doi: 10.3732/ajb.1100459. [DOI] [PubMed] [Google Scholar]
- 45.Elamo P, Helander M, Saloniemi I, Neuvonen S. Birch family and environmental conditions affect endophytic fungi in leaves. Oecologia. 1999;118:151–156. doi: 10.1007/s004420050713. [DOI] [PubMed] [Google Scholar]
- 46.Bailey JK, et al. Host plant genetics affect hidden ecological players: Links among Populus, condensed tannins, and fungal endophyte infection. Can J Bot. 2005;83:356–361. [Google Scholar]
- 47.Percy DM, et al. Progressive island colonization and ancient origin of Hawaiian Metrosideros (Myrtaceae) Proc Biol Sci. 2008;275:1479–1490. doi: 10.1098/rspb.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harbaugh DT, Wagner WL, Percy DM, James HF, Fleischer RC. Genetic structure of the polymorphic metrosideros (Myrtaceae) complex in the Hawaiian islands using nuclear microsatellite data. PLoS ONE. 2009;4:e4698. doi: 10.1371/journal.pone.0004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC. Global patterns in belowground communities. Ecol Lett. 2009;12:1238–1249. doi: 10.1111/j.1461-0248.2009.01360.x. [DOI] [PubMed] [Google Scholar]
- 51.Monteith JL, Unsworth MH. Principles of Environmental Physics. Burlington, MA: Academic; 2008. [Google Scholar]
- 52.Drake D, Mueller-Dombois D. Population development of rain-forest trees on a chronosequence of Hawaiian lava flows. Ecology. 1993;74:1012–1019. [Google Scholar]
- 53.Aplet G, Hughes RF, Vitousek PM. Ecosystem development on Hawaiian lava flows: Biomass and species composition. J Veg Sci. 1998;9:17–26. [Google Scholar]
- 54.Zimmerman N, et al. Patterns of primary succession of native and introduced plants in lowland wet forests in eastern Hawai'i. Biotropica. 2008;40:277–284. [Google Scholar]
- 55.Cordell S, Goldstein G, Mueller-Dombois D, Webb D, Vitousek PM. Physiological and morphological variation in Metrosideros polymorpha, a dominant Hawaiian tree species, along an altitudinal gradient: The role of phenotypic plasticity. Oecologia. 1998;113:188–196. doi: 10.1007/s004420050367. [DOI] [PubMed] [Google Scholar]
- 56.Fisher JB, Goldstein G, Jones TJ, Cordell S. Wood vessel diameter is related to elevation and genotype in the Hawaiian tree Metrosideros polymorpha (Myrtaceae) Am J Bot. 2007;94:709–715. doi: 10.3732/ajb.94.5.709. [DOI] [PubMed] [Google Scholar]
- 57.Martin RE, Asner GP, Sack L. Genetic variation in leaf pigment, optical and photosynthetic function among diverse phenotypes of Metrosideros polymorpha grown in a common garden. Oecologia. 2007;151:387–400. doi: 10.1007/s00442-006-0604-z. [DOI] [PubMed] [Google Scholar]
- 58.Vitousek PM, Field CB, Matson PA. Variation in foliar δ13C in Hawaiian metrosideros polymorpha: A case of internal resistance. Oecologia. 1990;84:362–370. doi: 10.1007/BF00329760. [DOI] [PubMed] [Google Scholar]
- 59.Baruch Z, Goldstein G. Leaf construction cost, nutrient concentration, and net CO2 assimilation of native and invasive species in Hawai'i. Oecologia. 1999;121:183–192. doi: 10.1007/s004420050920. [DOI] [PubMed] [Google Scholar]
- 60.Vitousek PM, Aplet G, Turner D, Lockwood J. The Mauna Loa environmental matrix: Foliar and soil nutrients. Oecologia. 1992;89:372. doi: 10.1007/BF00317415. [DOI] [PubMed] [Google Scholar]
- 61.Treseder KK, Vitousek PM. Potential ecosystem-level effects of genetic variation among populations of Metrosideros polymorpha from a soil fertility gradient in Hawaii. Oecologia. 2001;126:266–275. doi: 10.1007/s004420000523. [DOI] [PubMed] [Google Scholar]
- 62.Trusdell F, Wolfe E, Morris JH. 2005. Data series 144. Digital Database of the Geologic Map of the Island of Hawai'i. Available at: http://pubs.usgs.gov/ds/2005/144. Accessed June, 2009.
- 63.Vitousek PM, Turner D, Parton W, Sanford R. Litter decomposition on the Mauna Loa environmental matrix, Hawai'i: Patterns, mechanisms, and models. Ecology. 1994;75:418. [Google Scholar]
- 64.Raich J, Russell A, Vitousek PM. Primary productivity and ecosystem development along an elevational gradient on Mauna Loa, Hawai'i. Ecology. 1997;78:707. [Google Scholar]
- 65.Scowcroft P, Turner D, Vitousek PM. Decomposition of Metrosideros polymorpha leaf litter along elevational gradients in Hawai'i. Glob Change Biol. 2000;6:73–85. [Google Scholar]
- 66.Arnold AE, Henk DA, Eells RL, Lutzoni F, Vilgalys R. Diversity and phylogenetic affinities of foliar fungal endophytes in loblolly pine inferred by culturing and environmental PCR. Mycologia. 2007;99:185–206. doi: 10.3852/mycologia.99.2.185. [DOI] [PubMed] [Google Scholar]
- 67.Prince AM, Andrus L. PCR: How to kill unwanted DNA. Biotechniques. 1992;12:358–360. [PubMed] [Google Scholar]
- 68.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giongo A, et al. PANGEA: Pipeline for analysis of next generation amplicons. ISME J. 2010;4:852–861. doi: 10.1038/ismej.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nilsson RH, et al. An open source software package for automated extraction of ITS1 and ITS2 from fungal ITS sequences for use in high-throughput community assays and molecular ecology. Fungal Ecol. 2010;3:284–287. [Google Scholar]
- 71.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drummond AJ, et al. 2011. Geneious v5.4. Available at: http://www.geneious.com.
- 73.Quince C, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 74.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nilsson RH, et al. Taxonomic reliability of DNA sequences in public sequence databases: A fungal perspective. PLoS ONE. 2006;1:e59. doi: 10.1371/journal.pone.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schloss PD, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oksanen J, et al. 2010. vegan: Community ecology package. R package version 1.17-4. Available at http://CRAN.R-project.org/package=vegan.
- 78.Sun Y, et al. ESPRIT: Estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009;37:e76. doi: 10.1093/nar/gkp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. R Development Core Team (2010) R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria)
- 80.Clarke K. Nonparametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–143. [Google Scholar]
- 81.Jumpponen A, Jones KL, David Mattox J, Yaege C. Massively parallel 454-sequencing of fungal communities in Quercus spp. ectomycorrhizas indicates seasonal dynamics in urban and rural sites. Mol Ecol. 2010;19(Suppl 1):41–53. doi: 10.1111/j.1365-294X.2009.04483.x. [DOI] [PubMed] [Google Scholar]
- 82.Goslee S, Urban D. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22:1–19. [Google Scholar]
- 83.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 84.Smouse P, Long J, Sokal R. Multiple-regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool. 1986;35:627–632. [Google Scholar]
- 85.McArdle B, Anderson MJ. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology. 2001;82:290–297. [Google Scholar]
- 86.Legendre P, Anderson MJ. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol Monogr. 1999;69:1–24. [Google Scholar]
- 87.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 88.Borcard D, Legendre P. All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol Modell. 2002;153:51–68. [Google Scholar]
- 89.Dray S, Legendre P, Peres-Neto PR. Spatial modelling: A comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM) Ecol Modell. 2006;196:483–493. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.