

Abstract
Inelastic neutron scattering, far-infrared spectroscopy, and cryogenic nuclear magnetic resonance are used to investigate the quantized rotation and ortho–para conversion of single water molecules trapped inside closed fullerene cages. The existence of metastable ortho-water molecules is demonstrated, and the interconversion of ortho-and para-water spin isomers is tracked in real time. Our investigation reveals that the ground state of encapsulated ortho water has a lifted degeneracy, associated with symmetry-breaking of the water environment.
Keywords: cryogenic NMR, endofullerene, magic-angle-spinning NMR, rotational spectroscopy
The synthetic procedure known as “molecular surgery” (1–3) involves opening an orifice in fullerene (C60) cages by a series of chemical reactions, insertion of small molecules into the cavity, and resealing of the cage by further reactions. This procedure has made available, in macroscopic quantities, a class of pure, stable, substances in which closed fullerene cages encapsulate small molecules. These systems provide “nano-laboratories” in which the small molecules are isolated in a geometrically well-defined, highly symmetrical environment. Confined small molecules such as H2 behave as quantum rotors, in which the quantized translational and rotational levels are mixed by the confinement, generating a rich energy level structure (4–12).
The recently synthesized water-endofullerene H2O @ C60 (3) is particularly intriguing (see Fig. 1A). Like H2, water exhibits spin isomerism, with ortho- and para-forms, but unlike H2, water has an electric dipole moment. Do the electric dipoles of encapsulated water molecules rotate freely, reflecting the absence of hydrogen bonding? Do the ortho- and para-spin isomers interconvert? Can energetic ortho-water molecules be trapped in a metastable state? What is the potential describing the interaction of water with the curved carbon surface? Do the electric dipole moments of water molecules in neighboring cages line up cooperatively, exhibiting ferroelectricity (13)?
Fig. 1.

(A) Molecular structure of the water-endofullerene H2O @ C60, using a ball-and-stick representation for the C60 cage and space-filling spheres for water; (B) Energy level diagram, showing the principal transitions detected by INS (grey arrows) and IR spectroscopy (dashed arrows). The three-fold rotational degeneracy of the ortho-H2O ground state is lifted. The upper level of the split 101 state is doubly degenerate. Unresolved fine structure is indicated by shading.
We study the energy levels, spin isomerism and quantum dynamics of single water molecules enclosed in fullerene cages, using three complementary physical methods: inelastic neutron scattering (INS), far-infrared spectroscopy (FIR), and nuclear magnetic resonance (NMR). In all cases, the homogeneous and symmetric environment provided by the fullerene cages isolates the ortho and para spin isomers, and gives rise to relatively narrow and unambiguous spectral features. We observe the existence of metastable ortho-water molecules, follow the slow conversion of ortho-water into para-water, and detect an energy splitting in the ortho-water rotational ground state, which is a signature of broken symmetry.
A simplified energy level diagram of endohedral water, based on free water in the gas phase, is shown in Fig. 1B. The endohedral water molecule behaves as an asymmetric top, with rotational energy levels indexed by the three numbers JKaKc where J is the quantum number for the total angular momentum. The indices Ka and Kc refer to the angular momentum quantum numbers in the corresponding prolate and oblate symmetric tops (14). In an environment with tetrahedral symmetry or higher, each JKaKc level is (2J + 1)-fold degenerate, with an additional quantum number MJ∈{-J,-J + 1…+J} defining the angular momentum projection on an external axis. The rotational states are correlated to the proton spin states through the Pauli principle: ortho-water states have total nuclear spin I = 1, and odd parity for Ka + Kc in the ground vibrational state. The para-water states have total nuclear spin I = 0, and even parity for Ka + Kc in the ground vibrational state.
Results
Inelastic Neutron Scattering.
Fig. 2 shows INS spectra of H2O @ C60 as a function of neutron energy transfer ΔE, at two different sample temperatures. The intense peak centred at ΔE = 0 represents elastically scattered neutrons. The inelastic neutron scattering peaks appear as smaller features on either side, with the sign convention that ΔE represents the energy loss or gain relative to the molecule. Therefore in the scattering event between the neutron and the nuclei of the molecule, negative ΔE corresponds to neutron energy (NE) gain transitions while positive ΔE represents NE loss.
Fig. 2.

INS spectra recorded on the time-of-flight spectrometers IN4 and IN5. The 10 K spectrum is an average over 5, 10, and 15 K data. The dashed lines show the best fit components where the transitions are labeled according to Fig. 1B.
INS transitions are mediated by a spin dependent interaction and are dominated by scattering from 1H nuclei. This has significant consequences. Firstly, unlike photon spectroscopy, INS can drive transitions between different nuclear spin-isomers of H2O. Therefore, INS peaks that connect ortho-H2O and para-H2O appear with significant intensity. Secondly, since para-water has spin 0, transitions between different states of para-H2O have negligible intensity. However, since the ortho spin isomer has spin 1, ortho–ortho transitions appear with similar intensity to ortho–para transitions.
Even at the lowest temperature T = 1.5 K, an INS peak (labeled “1”) appears with NE gain at ΔE = -2.4 ± 0.1 meV; this is generated by scattering events in which the sample has undergone a transition from a high energy state to one of lower energy, raising the energy and speed of the scattered neutron. Since the energy of the transition ΔE/kB = 28 K is much larger than the available thermal energy at 1.5 K, this peak must arise from a meta-stable state that is not at thermal equilibrium. Peak “1” is therefore assigned to the transition from the metastable ground state of ortho-H2O to the ground state of para-H2O, 101 → 000 (see Fig. 1B). The observation of this peak provides compelling evidence that the ortho-H2O spin isomer is long-lived inside the fullerene cage and, upon cooling, does not convert completely to para-H2O, over the timescale of the experiment (6 h).
Increasing the sample temperature to T = 10 K gives rise to a second peak in NE gain at ΔE = -2.9 ± 0.1 meV (labeled “2”). This peak becomes populated at temperatures as low as 5 K and the temperature dependence of the peak amplitude indicates that it corresponds to an excited state of ortho-water that is approximately 0.6 meV above the ortho-H2O ground state. We postulate that the three-fold degenerate 101 ortho ground state is split, and assign peak “2” to transitions between the upper sublevel of 101 and the para ground state 000, while peak “1” is assigned to transitions between the lower sublevel of 101 and 000 (Fig. 1B). The existence of a splitting in the ortho ground state is supported by the NMR data (see below).
We can be confident that the NE gain spectrum arises only from transitions originating in metastable ortho-H2O molecules. Librational and vibrational modes of C60 and phonon modes above approximately 0.2 meV are not populated in the NE gain spectrum at a temperature of 1.5 K.
More transitions are observed in the NE loss spectrum, including the transitions 1 and 2 in the opposite direction to those observed in NE gain. Transition 3 in Fig. 1B appears in neutron energy loss, but it overlaps with transitions 1 and 2 (see infrared data, below). As indicated above, the para–para transition 4 is absent in INS.
The small feature in the 10 K spectrum at a neutron energy gain of ΔE ≅ -0.6 meV (indicated by the arrow) is provisionally attributed to transitions from the upper to the lower sublevel of 101.
Far-Infrared Spectroscopy.
Far-infrared (FIR) spectroscopy detects transitions between the rotational levels of H2O. The absorption coefficient of radiation is proportional to the difference in the populations of the initial and final states. Unlike INS, transitions between ortho and para states are highly forbidden. Since each spin isomer attains internal thermal equilibrium very rapidly, the slow time evolution of the peak intensities is due to ortho–para conversion. This allows us to identify the ortho and para lines unambiguously, and to determine the kinetics of ortho–para conversion.
The FIR spectrum of 10 mg of H2O @ C60 is shown in Fig. 3. The three peaks in the displayed spectral region are assigned to the rotational transitions shown in Fig. 1B. Peak “4” is a rotational transition of para-H2O (000 → 111), while peaks “3” and “5” are rotational transitions of ortho-H2O (101 → 110 and 101 → 212, respectively). The assignments are supported by the effect of ortho–para conversion (see below). Peak “4” provides strong absorption of the infrared radiation, which saturates the absorption curve under the experimental conditions. On thinner samples, the absorption maximum is observed at 4.55 ± 0.2 meV. The energy of transition “3” lies between that of “1” and “2”, which explains why it is unresolved in neutron energy gain (see above).
Fig. 3.

Far-infrared spectra of H2O @ C60 at 3.5 K. The times indicate the elapsed intervals after cooling from 77 K. The circled numbers refer to the transition assignments in Fig. 1B. The time-dependence of the peaks indicates slow ortho-to-para conversion. Peaks 4 and 5 are initially saturated due to the strong absorption and thick sample.
The spectra shown in Fig. 3 were all obtained at a sample temperature of 3.5 K, and were taken after slow cooling from a temperature of 77 K. The spectrum shown in black was obtained immediately after cooling, while the spectra shown by the red and blue curves were obtained after the indicated waiting periods. Peak “5” is initially saturated due to its strong absorption, but the saturation disappears at later times, as the peak becomes less intense. Although the amplitude of peak “4” cannot be measured directly, the movement of the peak edges indicate that peak “4” increases in amplitude as peaks “3” and “5” become weaker.
The changing intensity of the FIR peaks is due to ortho-to-para conversion, which strongly depletes the ortho state in favor of para at the sample temperature of 3.5 K. The time dependence, shown in Fig. S1, is almost mono-exponential with a time constant of 12 ± 1.3 h at the temperature of 3.5 K. This is similar to the ortho–para conversion rate for water in argon matrices at 4.2 K (15).
The splitting in the ortho ground state, which is resolved by INS, is not visible in the FIR, presumably due to unresolved structure in the higher energy levels. The ortho–ortho transition 101 → 110 (transition “3” in Fig. 1B) has similar energy to the transition from the para ground state 000 to the lower sublevel of the ortho ground state 101(transition “1”). This coincidence explains why transition “3” does not separately show up in NE loss in the INS spectrum (Fig. 2); the NE loss spectrum evidently contains multiple transitions, not all of which are thermally populated in NE gain at the temperatures studied.
Magic-Angle-Spinning NMR.
Magic-angle-spinning (MAS) NMR is used to provide high-resolution spectra of solid samples; rapid spinning of the sample around an axis subtending the angle 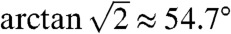 averages out many anisotropic nuclear spin interactions, providing narrow spectral lines in the solid state (16). Recent technological advances have made it possible to perform MAS-NMR experiments reliably in the cryogenic temperature regime (4, 17–20).
averages out many anisotropic nuclear spin interactions, providing narrow spectral lines in the solid state (16). Recent technological advances have made it possible to perform MAS-NMR experiments reliably in the cryogenic temperature regime (4, 17–20).
MAS proton NMR spectra of 9 mg of H2O @ C60 are shown in Fig. 4. These NMR signals come exclusively from the ortho spin isomer: the para spin isomer has zero nuclear spin and does not provide an NMR signal. In the light of the INS and FIR results, the 9.6 K NMR spectra must originate from metastable ortho-H2O molecules.
Fig. 4.

Magic-angle-spinning proton NMR spectra of H2O @ C60, taken in a magnetic field of 14.1T (proton precession frequency ≈-600.0 MHz). All spectra were obtained by taking the Fourier transform of a single transient after a 90° excitation pulse and subjecting it to Lorentzian linebroadening with a linewidth of 200 Hz followed by baseline correction. The sample temperatures and spinning frequencies are (A) 22.8 K, 8.86 kHz; (B) 18.5 K, 8.38 kHz; (C) 13.0 K, 7.12 kHz; (D) 9.6 K, 5.04 kHz. The peaks indicated by asterisks are spinning sidebands. The grey line is a simulation for randomly oriented pairs of protons with a magnetic dipole–dipole interaction of -5.5 kHz.
The NMR spectra do not exhibit strong spinning sidebands at temperatures above ∼20 K. This is a signature of rapid isotropic molecular motion, which is consistent with molecular dynamics simulations (21). However, at low temperature, spinning sidebands develop in the MAS NMR spectrum, and an unresolved shoulder appears on the centerband. Apart from the asymmetric centerband, the 9.6 K spectrum is a good match to a simulation for randomly oriented pairs of spins-1/2 experiencing a through-space dipole–dipole coupling of -5.5 kHz (grey curve in Fig. 4). For comparison, no spinning sidebands are observed in the MAS proton spectrum of H2 @ C60 under similar conditions (5). The slight asymmetry of the centreband at 9.6 K is currently unexplained.
Ortho-to-para transitions are expected to lead to a slow decay in the NMR signal intensity at low temperatures, since para-H2O provides no NMR signal. We did not observe this decay, presumably because our NMR experiments were performed at the lowest temperature for approximately 90 min, as compared to the approximately 12-h ortho–para conversion time observed in infrared spectroscopy (see above).
Internuclear dipole–dipole couplings must be interpreted by a quantum treatment which takes into account the spatial delocalization of the rotational wavefunctions in rotational quantum states (4, 22). For J = 1 states, the nuclear dipole–dipole coupling is scaled by a factor of 2/5 compared to a classical model, with the same internuclear distance (4, 22). In addition, the nuclear dipole–dipole coupling tensor is averaged over the populated quantum states, with populations governed by the Boltzmann distribution. At high temperature, all 2J + 1 components of a level with rotational quantum number J are equally populated, leading to a vanishing dipole–dipole coupling. This corresponds to the case of isotropic classical rotation. An energy splitting between the sublevels causes unequal populations in thermal equilibrium, and generates a finite dipole–dipole coupling tensor. Conversely, the existence of a finite dipole–dipole coupling is a signature of an energy splitting between the sublevels.
The -5.5 kHz internuclear dipolar coupling at 9.6 K leads to an estimate of the ortho ground state splitting of approximately 0.9 meV. The analysis (see SI Text) makes the following approximations and assumptions: rigid water molecules, with a proton-proton distance of 1.515 Å (see Fig. S2); splitting of the three 101 sublevels into a nondegenerate lower level with MJ = 0 and a doubly degenerate upper level with MJ = ± 1; and neglect of higher rotational and translational states of ortho-water. The estimated splittings from cryogenic NMR (approximately 0.9 meV) and INS (approximately 0.6 meV) are reasonably consistent, given the many approximations that are involved. The discrepancy is attributed to contributions from intermolecular couplings, chemical-shift anisotropy, spin-rotation effects, and the influence of higher rotational and translational levels.
The approximately 1 kHz linewidth of the experimental 1H MAS peaks is greater than that anticipated for isolated water molecules. The origin of the linewidths is currently unknown, but may be associated with the presence of a protonated impurity in the sample, see Fig. S3.
Discussion
The energy level spacings of the lowest rotational sublevels of water in H2O @ C60, as determined by INS and FIR spectroscopy, are compared with those of gas-phase water vapor (23, 24) in Table 1. The most striking difference between encapsulated water and free water is the 0.6 meV splitting in the ortho-water ground state. Apart from this splitting, the encapsulation in C60 does not change the lower rotational energies of water by more than 0.2 meV (taking the average of the split 101 levels, and accounting for the double degeneracy of the upper level). Unlike the case of H2 @ C60 (6, 7), there is no obvious signature of strong translation-rotation coupling.
Table 1.
Rotational transitions of water in H2O @ C60 and in the gas phase
| H2O @ C60 [meV (cm-1)] (this work) | ||||||
| Transition | Quantum states | Type | IR | INS | Gas phase H2O [meV (cm-1)] (ref. (24)) | |
| 1 | 000 | 101 (lower) | p↔o | — | 2.3 ± 0.1 (18.6 ± 0.8) | 2.95* (23.8)* |
| 2 | 000 | 101 (upper) | p↔o | — | 2.9 ± 0.1 (23.4 ± 0.8) | |
| 3 | 101 | 110 | o↔o | 2.5 ± 0.6 (20 ± 5) | — | 2.30 (18.6) |
| 4 | 000 | 011 | p↔p | 4.6 ± 0.6 (37 ± 5) | — | 4.60 (37.1) |
| 5 | 101 | 212 | o↔o | 7.0 ± 0.6 (56 ± 5) | — | 6.91 (55.7) |
The transition energies are given in meV and in cm-1 (in parentheses). The gas phase data is from ref. (24), except for the ortho–para transition 000 → 101 (marked with an asterisk), which is inferred by modeling the water molecule as a rigid asymmetric rotor.
All three forms of spectroscopy reveal the presence of metastable ortho-water molecules at low temperature which undergo slow transitions to the low-energy para-spin isomer. FIR spectroscopy provides the most detailed picture of ortho–para conversion, allowing the spin isomerization process to be followed in real time on the timescale of hours.
The INS and NMR data are both consistent with a splitting in the triply degenerate ground state of ortho-water by around 0.6 meV. This lifting of degeneracy may be due to crystal packing effects which break the local symmetry, or to a spontaneous distortion of the fullerene cage induced by the rotationally degenerate water molecule, analogous to the Jahn-Teller effect. The observation of this phenomenon in the magic-angle-spinning NMR spectra indicates that the symmetry-breaking distortion must have a lifetime of at least several tens of milliseconds. An intriguing possibility, which requires deeper investigation, is that the breaking of symmetry reflects cooperative electric dipole alignment; i.e., ferroelectricity (13).
Materials and Methods
Synthesis.
H2O @ C60 was synthesized by the published procedure (3). Solution-state proton NMR spectra of the last open-cage derivative before the final closing reaction indicated that about 60% of the C60 cages are occupied by water molecules. The compound, which was purified by column chromatography eluted with CS2/pentane = 2/1, was dissolved in CS2. The solution was dropped into pentane to give a precipitate. The precipitate was collected by centrifuge and then heated at 180 °C under vacuum for 3 d. The resulting powder was used for the measurements. Solid-state NMR measurements indicated the presence of a protonated impurity with a proton concentration of about 3 times the endohedral water molecules (see Fig. S3).
Inelastic Neutron Scattering.
The experiments were conducted using two different time-of-flight INS spectrometers, IN4 and IN5, at the Institut Laue-Langevin. In both instruments, the sample is irradiated by a pulsed beam of monochromatic neutrons. The energy of the scattered neutrons is then determined from the time taken between the arrival of the neutron pulse at the sample and the arrival of a neutron in a detector that is situated a known distance away. The momentum transfer may be inferred from the geometry of the scattering event, and the angular position of the neutron detector. The INS spectra in Fig. 2 are obtained from a sum of detectors covering a momentum transfer range approximately 1.5 Å ≤ Q ≤ 8 Å. On IN4, Bragg diffraction from a curved monochromator is used to select the energy/wavelength of the incident neutrons, with a Fermi chopper being used to provide the pulsed structure of the beam. The flight path between sample and detectors is 2 m. On IN5 the pulsed monochromatic structure is obtained using six pairs of counter-rotating choppers, with their phases and frequencies being set to determine the energy/wavelength of the beam. IN5 has a 4-m flight path between the sample and an array of pixilated position sensitive detectors.
The sample used for the INS experiments was 95 mg of H2O @ C60 powder packed in an aluminum foil container. The sample was mounted in the cryostat under a reduced pressure of He gas.
Far Infrared Spectroscopy.
The sample was pressed under vacuum from a powder of 60% of H2O @ C60 and 40% of empty C60 into a d = 0.83 mm thick and 3-mm diameter pellet of mass 10 mg.
The pellet was pumped at room temperature for several hours inside the sample compartment of a liquid He cryostat before cooling and adding the He heat exchange gas to the sample compartment. Light was guided from the Sciencetech SPS Martin-Puplett type spectrometer to the sample cryostat using a light pipe and detected with a 0.3 K bolometer located inside a vacuum chamber in the same liquid He cryostat. A motorized sample changer inside the cryostat was used to change between the sample and a reference hole of 3 mm diameter. The absorption coefficient was calculated by 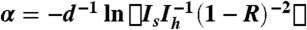 , where Is and Ih are the light intensities transmitted by the sample and by the reference hole. This formula accounts for one back reflection from the pellet front and back face, where the reflection coefficient R = (n - 1)2(n + 1)-2 and constant index of refraction n = 2 is assumed. The plotted spectra in Fig. 3 are not corrected for the 65% content of H2O @ C60 in the sample.
, where Is and Ih are the light intensities transmitted by the sample and by the reference hole. This formula accounts for one back reflection from the pellet front and back face, where the reflection coefficient R = (n - 1)2(n + 1)-2 and constant index of refraction n = 2 is assumed. The plotted spectra in Fig. 3 are not corrected for the 65% content of H2O @ C60 in the sample.
Nuclear Magnetic Resonance.
The equipment for cryogenic magic-angle spinning NMR uses three separate cryogenic streams of supercritical He for the bearing system, the turbine system, and for sample cooling. The separate streams of supercritical helium were generated from liquid in a large custom-built cryogenic vessel, and kept below 7 K at a stable controlled pressure of 2.5–4.5 bar. The flow rates and temperatures of all three streams are independently controlled by using cryogenic needle valves and electrical heaters. The temperatures of the three gas streams are measured on entry to the probe cryostat.
The cryogenic spinning system uses a zirconia rotor with an outer diameter of 2 mm and length of 16.2 mm (including the turbine wheel inserts), provided by Revolution NMR. The rotor was packed with 9 mg of H2O @ C60 powder. The temperature of the gas exiting the spinning assembly was monitored by using a Cernox temperature sensor mounted in the exhaust pipe about 2 cm from the rotor with an accuracy of 0.15 K. The real sample temperature was cross-checked against the exhaust gas temperature in separate experiments using the 127I nuclear spin-lattice relaxation in CsI as an internal thermometer (20).
The 9.6 K spectrum shown in Fig. 4 was obtained at the end of a long experimental series involving slow cooling of the magic-angle-spinning sample from room temperature over approximately 10 h. The sample spent approximately 90 min below 20 K during the acquisition of this spectrum. This interval is reasonably short compared to the ortho–para conversion rates measured by infrared spectroscopy.
The chemical shift scale in the proton spectra was calibrated by using the 1.8 ppm proton NMR peak of adamantane as a reference. The endohedral water peak is at -4.6 ppm, which is close to the position observed in room temperature liquid state NMR (3).
Supplementary Material
ACKNOWLEDGMENTS.
The following funding agencies are acknowledged: EPSRC (UK); National Science Foundation (USA); MEXT, JSPS, and JURC at ICR, Kyoto University (Japan). M.C. acknowledges a University Research Fellowship from the Royal Society (UK); U.N. and T.R. acknowledge the Estonian Ministry of Education and Research Grant No. SF0690029s09 and the EstonianScience Foundation Grant Nos. ETF8170, ETF8703. We also thank M. al-Mosawi and M. Webb (Southampton, UK) for experimental help.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1210790109/-/DCSupplemental.
References
- 1.Rubin Y. Ring opening reactions of fullerenes: Designed approaches to endohedral metal complexes. Top Curr Chem. 1999;199:67–91. [Google Scholar]
- 2.Komatsu K, Murata M, Murata Y. Encapsulation of molecular hydrogen in fullerene C60 by organic synthesis. Science. 2005;307:238–240. doi: 10.1126/science.1106185. [DOI] [PubMed] [Google Scholar]
- 3.Kurotobi K, Murata Y. A single molecule of water encapsulated in fullerene C60. Science. 2011;333:613–616. doi: 10.1126/science.1206376. [DOI] [PubMed] [Google Scholar]
- 4.Carravetta M, et al. Cryogenic NMR spectroscopy of endohedral hydrogen-fullerene complexes. J Chem Phys. 2006;124:104507. doi: 10.1063/1.2174012. [DOI] [PubMed] [Google Scholar]
- 5.Carravetta M, et al. Solid-state NMR of endohedral hydrogen-fullerene complexes. Phys Chem Chem Phys. 2007;9:4879–4894. doi: 10.1039/b707075f. [DOI] [PubMed] [Google Scholar]
- 6.Mamone S, et al. Rotor in a cage: Infrared spectroscopy of an endohedral hydrogen-fullerene complex. J Chem Phys. 2009;130:081103–081104. doi: 10.1063/1.3080163. [DOI] [PubMed] [Google Scholar]
- 7.Horsewill AJ, et al. Inelastic neutron scattering of a quantum translator-rotator encapsulated in a closed fullerene cage: Isotope effects and translation-rotation coupling in H_{2}@C_{60} and HD@C_{60} Phys Rev B. 2010;82:081410. [Google Scholar]
- 8.Mamone S, et al. Theory and spectroscopy of an incarcerated quantum rotor: The infrared spectroscopy, inelastic neutron scattering and nuclear magnetic resonance of H2@C60 at cryogenic temperature. Coord Chem Rev. 2011;255:938–948. [Google Scholar]
- 9.Ge M, et al. Interaction potential and infrared absorption of endohedral H_{2} in C_{60} J Chem Phys. 2011;134:054507–054513. doi: 10.1063/1.3535598. [DOI] [PubMed] [Google Scholar]
- 10.Ge M, et al. Infrared spectroscopy of endohedral HD and D2 in C60. J Chem Phys. 2011;135:114511. doi: 10.1063/1.3637948. [DOI] [PubMed] [Google Scholar]
- 11.Xu M, Sebastianelli F, Bacic Z, Lawler R, Turro NJ. H-2, HD, and D-2 inside C-60: Coupled translation-rotation eigenstates of the endohedral molecules from quantum five-dimensional calculations. J Chem Phys. 2008;129:064313. doi: 10.1063/1.2967858. [DOI] [PubMed] [Google Scholar]
- 12.Xu M, et al. Coupled translation-rotation eigenstates of H-2 in C-60 and C-70 on the spectroscopically optimized interaction potential: Effects of cage anisotropy on the energy level structure and assignments. J Chem Phys. 2009;130:224306. doi: 10.1063/1.3152574. [DOI] [PubMed] [Google Scholar]
- 13.Cioslowski J, Nanayakkara A. Endohedral fullerites: A new class of ferroelectric materials. Phys Rev Lett. 1992;69:2871–2873. doi: 10.1103/PhysRevLett.69.2871. [DOI] [PubMed] [Google Scholar]
- 14.Bunker PR, Jensen P. Molecular Symmetry and Spectroscopy. Ottawa: NRC Research Press; 2006. [Google Scholar]
- 15.Abouaf-Marguin L, Vasserot AM, Pardanaud C, Michaut X. Nuclear spin conversion of water diluted in solid argon at 4.2K: Environment and atmospheric impurities effects. Chem Phys Lett. 2007;447:232–235. [Google Scholar]
- 16.Mehring M. High Resolution NMR in Solids. 2nd Ed. Berlin: Springer; 1982. [Google Scholar]
- 17.Samoson A, et al. New horizons for magic-angle spinning NMR. Top Curr Chem. 2005;246:15–31. doi: 10.1007/b98647. [DOI] [PubMed] [Google Scholar]
- 18.Thurber KR, Tycko R. Biomolecular solid state NMR with magic-angle spinning at 25 K. J Magn Reson. 2008;195:179–186. doi: 10.1016/j.jmr.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thurber KR, Tycko R. Measurement of sample temperatures under magic-angle spinning from the chemical shift and spin-lattice relaxation rate of Br-79 in KBr powder. J Magn Reson. 2009;196:84–87. doi: 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarkar R, et al. An NMR thermometer for cryogenic magic-angle spinning NMR: The spin-lattice relaxation of 127I in cesium iodide. J Magn Reson. 2011;212:460–463. doi: 10.1016/j.jmr.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Bucher D. Orientational relaxation of water trapped inside C60 fullerenes. Chem Phys Lett. 2012;534:38–42. [Google Scholar]
- 22.Tomaselli M. Dynamics of diatomic molecules in a chemical trap I. NMR experiments on hydrogen in solid C60. Mol Phys. 2003;101:3029–3051. [Google Scholar]
- 23.Hall RT, Dowling JM. Pure rotational spectrum of water vapor. J Chem Phys. 1967;47:2454–2461. [Google Scholar]
- 24.Tennyson J, Zobov NF, Williamson R, Polyansky OL, Bernath PF. Experimental energy levels of the water molecule. J Phys Chem Ref Data. 2001;30:735–831. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.