

Abstract
The concept of a folding funnel with kinetic traps describes folding of individual proteins. Using in situ Atomic Force Microscopy to investigate S-layer assembly on mica, we show this concept is equally valid during self-assembly of proteins into extended matrices. We find the S-layer-on-mica system possesses a kinetic trap associated with conformational differences between a long-lived transient state and the final stable state. Both ordered tetrameric states emerge from clusters of the monomer phase, however, they then track along two different pathways. One leads directly to the final low-energy state and the other to the kinetic trap. Over time, the trapped state transforms into the stable state. By analyzing the time and temperature dependencies of formation and transformation we find that the energy barriers to formation of the two states differ by only 0.7 kT, but once the high-energy state forms, the barrier to transformation to the low-energy state is 25 kT. Thus the transient state exhibits the characteristics of a kinetic trap in a folding funnel.
Keywords: biological assembly, protein crystal growth, two-step crystallization, protein folding funnel
Proteins that naturally self-assemble into extended ordered structures often adopt conformations that are distinct from those of the individual monomeric proteins (1–4). For example, collagen matrices, which constitute the organic scaffolds of bones and teeth in all higher organisms, are constructed from triple helices of the individual collagen monomers (4). These helices further assemble into highly organized twisted fibrils exhibiting a pseudohexagonal symmetry. In some cases, assembly is inexorably linked to folding transformations, as in the case of prion or amyloid fibrils, where misfolding of the monomers triggers assembly, which in turn drives misfolding of new monomers (1).
When individual proteins transform from an unstructured state to the final equilibrium state, the concept of a folding funnel characterized by a large number of potential initial states higher in energy than the final state is often invoked (5, 6). The walls of the funnel are presumed not to be smooth and the resulting bumps and valleys define kinetic traps where the protein exhibits nonequilibrium structures for extended periods of time. These transient structures can be disordered “molten globules” or partially ordered intermediates (7). This physical picture of folding in which one fraction of a protein population promptly reaches the folded state while the remaining fraction gets trapped in metastable states has been referred as the kinetic partitioning mechanism (8). The energy landscape that defines the funnel as well as the pathways through it have been explored in some detail at the single molecule level for a number of small proteins and protein subdomains through both simulations (8–10) and experiments (11–13). However, despite the fact that conformational transformations are part and parcel of protein self-assembly into extended architectures, this concept of the folding funnel has not been considered in that context and little is known about either the existence of partially ordered intermediate states or the energetics that define the pathways. Here, we investigate the connection between these two phenomena and explore the dynamics and energetics of conformational transformations during protein assembly by utilizing the inherent single molecule resolution of in situ Atomic Force Microscopy (AFM) to directly observe kinetic trapping during 2D self-assembly of S-layer protein structures on mica surfaces.
S-layers are a class of proteins that assemble into 2D crystalline sheets that form the outermost membrane of many bacteria and Archaea (14, 15). The crystalline lattice is typically built from multimeric growth units of the individual S-layer monomers, which in some cases can adopt more than one structural conformation within a single membrane (16). In our previous work (17), we discovered that the S-layer SbpA from Lysinibacillus sphaericus (ATCC 4525, 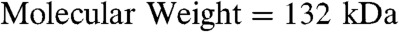 ) assembles on supported lipid bilayers (SLBs) via a multistage process that includes: (1) adsorption of monomers with an extended conformation to form a mobile adsorbed phase, (2) condensation into amorphous 3D clusters, (3) transformation through S-layer folding into 2D crystalline clusters composed of compact tetramers, and (4) cluster growth by formation of new tetramers exclusively at lattice sites along the cluster edges. Analysis of the growth kinetics showed that the growth stage was limited by the energy barrier to creation of new tetramers and that the barrier was significantly reduced by the presence of the prefolded tetramers at the edges of the crystalline clusters.
) assembles on supported lipid bilayers (SLBs) via a multistage process that includes: (1) adsorption of monomers with an extended conformation to form a mobile adsorbed phase, (2) condensation into amorphous 3D clusters, (3) transformation through S-layer folding into 2D crystalline clusters composed of compact tetramers, and (4) cluster growth by formation of new tetramers exclusively at lattice sites along the cluster edges. Analysis of the growth kinetics showed that the growth stage was limited by the energy barrier to creation of new tetramers and that the barrier was significantly reduced by the presence of the prefolded tetramers at the edges of the crystalline clusters.
In this study, we show that assembly on mica—which lacks the fluidic nature of the SLB—also begins with condensation of the monomers into clusters. However, from that initial condensate the systems tracks along two different pathways. One path leads directly to the low-energy final, ordered state. The other leads to a kinetic trap occupied by a long-lived transient state that is more disordered. This higher-energy state transforms into the stable state with an exponential dependence on time. Moreover, we find that the energy barriers to formation of the two states differ by approximately 1.6 kJ/mol, whereas the barrier to transformation from the high-to low-energy state is approximately 61 kJ/mol. Consequently, although either state is easily accessible during nucleation, if the system falls into the high-energy state, escape to the final, low-energy state is strongly impeded at room temperature. The results demonstrate that the concept of folding funnels is equally valid for self-assembly of extended protein structures.
Results
Fig. 1A shows an in situ AFM micrograph and its height profile demonstrating that the S-layer forms two types of compact crystalline domains on mica. These domains have an average size of approximately 150 nm and are randomly distributed and oriented (see Fig. 1A, Inset). The two types of domains, labeled short (S) and tall (T), exhibit almost identical lattice spacing (approximately 15.4 nm) and symmetry (P4), but have different heights (SI Appendix, Fig. S3). As shown by the height profile, the average height of the tall domains is 8–9 nm, which corresponds to a single layer of S-layer tetramers (17), and the average height difference between the short and tall domains is consistently approximately 2–3 nm. A native protein gel analysis confirms that the purified protein has a homogeneous native state in solution (SI Appendix, Fig. S1), and control experiments with SLBs- and poly-lysine-coated mica surfaces produced crystalline S-layer domains of uniform height (SI Appendix, Fig. S2). Therefore, the observed phenomenon was neither a result of protein inhomogeneity nor an artifact of imaging, but rather a result of the interactions between the S-layers and the mica surface.
Fig. 1.

(A) AFM height profile and height image of 2D S-layers assembled on mica showing the two types of domains [incubation temperature (Ti) = 25 °C, protein concentration (CP) = 41 μg/mL in a buffer solution of 10 mM tris(hydroxymethyl)aminomethan (Tris), pH 7.2, 50 mM CaCl2, 100 mM NaCl]. The height profile (Top) measured along the black dotted line in the height image shows the average difference in domain height (Δh) as measured between the blue (tall phase: T) and red (short phase: S) dotted lines is approximately 3 nm. The average height of the tall domains measured from the mica surface (highlighted by white dotted circles) corresponds to ca. 8–9 nm, which is consistent with measurements of S-layers on SLBs. (B–D) Time sequence of in situ AFM images and height profiles showing the early stage of S-layer assembly (CP = 70 μg/mL, Ti = 25 °C). White circles highlight initial nuclei formed from the adsorbed proteins. Height profiles were measured along the horizontal dotted black lines in each image. Blue and red circles in each image highlight a pair of clusters, one tall (blue) and the other short (red). They are denoted in the height profiles by the blue and red stars, respectively, and maintain a consistent height difference of approximately 3 nm during the observed growth period. The nonzero baselines in each height profiles denote the level of the adsorbed monomers, which have an average height of approximately 2 nm. The images were captured at (B) 37, (C) 48, and (D) 59 min after start of the in situ growth experiment.
To understand the origin of the two crystalline S-layer domains, we investigated the assembly process via in situ AFM imaging. Fig. 1B–D presents a series of in situ AFM images and height profiles showing an early stage of S-layer growth on mica. It reveals a simultaneous emergence of clusters of the two types of crystalline domains, two of which are highlighted by blue and red circles. A few unique aspects of the early growth process are worth addressing. The average number and size of the short clusters (white circles in Fig. 1B–D) are initially smaller than those of the tall clusters by approximately 38% (by area). Once nucleated, both types of clusters continuously grow laterally while maintaining their height difference. They cease their growth phase when expansion becomes physically hindered by neighboring domains. Analysis of cluster sizes shows the majority of short clusters grow slightly faster than the tall clusters so that, by the end of the growth phase, the size distribution of short and tall domains is quite similar. However, as shown in Fig. 2A, the ratio of tall-to-short domains at the end of the growth phase increases with increasing temperature. These observations show: (1) The pathway of individual cluster evolution is set at the time of nucleation of the crystalline phase; (2) There is a preference for nucleating tall domains and it increases with temperature; and (3) The probability of forming new tetramers at a domain edge is greater for short domains.
Fig. 2.

Temperature dependence of ratio between number of short and tall domains of S-layer crystals on mica. (A) Relative populations by area fraction of tall (T), short (S), and empty (NC) domains for three different incubation temperatures (Ti). Relative populations depend on Ti, with higher temperatures producing larger populations of the more stable tall domains. S-layers were grown at CP = 40 μg/mL and Ti = 4, 25 °C, and 37 °C for approximately 4 h and were imaged ex-situ. From these data, the difference between the barriers to formation of the tall and short domains is approximately 1.6 kJ/mol. (See SI Appendix for the detailed analysis.) (B) Kinetics of short-to-tall domain transformation. The plot shows the percent area covered by tall domains as a function of time. Labeling of data points (i–iv) corresponds to the image in Fig. 2A on which the analysis was performed. Solid line gives a guide to the eye. Times at which images were captured are (i) 4.5, (ii) 6.8, (iii) 10.0, and (iv) 29.2 h. (C) Plot of relative number ratio of short phase domains (fS) vs. time showing a simple exponential dependence. The best linear fit is shown as a solid line. From these data, the energy barrier to transformation of a domain from short to tall was found to be about approximately 61 kJ/mol (see SI Appendix for details of the analysis).
As Fig. 3 shows, after the end of the growth phase, the short domains gradually transformed into tall domains (see SI Appendix, Fig. S5 and Movie S1). This occurred irreversibly and without any detachment of tetramers. By Δt ∼ 2.3 h (Fig. 3A, ii), more than approximately 25% of the short domains had transformed into tall domains. Close examination of the two areas highlighted by blue and red boxes (Fig. 3A, i and ii) shows that, in some instances, domain transformation was accompanied by coalescence of smaller domains into one larger domain. By Δt ∼ 19.2 h, approximately 95% of the short domains had transformed into tall domains [Note that the transformation between the two types of clusters was not observed in the early stages of growth (Fig. 1B–D), because the timescale for the transformation is long compared to that for nucleation and growth]. The time dependence of the transformation at fixed temperature (25 °C) is given in Fig. 2B.
Fig. 3.

Sequences of in situ AFM images showing transformation from short to tall domains. Δt indicates time elapsed since collection of images i and i*. S-layer crystals were initially grown in 5 mL solution at 25 °C and further development of the domains was monitored in the same solution. Series in A shows evolution in distribution of domains in protein solution with CP = 41 μg/mL (10 mM Tris pH 7.2, 50 mM CaCl2, 100 mM NaCl) following an incubation time of 4.5 h. (i) Initial ratio of tall to short domains by area was 1.3. (ii) By Δt ∼ 2.3 h, about 25% of the short domains in i had transformed into tall domains without any dissolution of S-layer proteins from the domains. (iii) and (iv) At Δt ∼ 3.2 and 19.2 h, most of the short domains had transformed into tall domains. However, a few short domains still remained, indicating that the transformation does not go to 100% completion until much later times. Series in B shows transformation of a single domain in protein solution with CP = 70 μg/mL (10 mM Tris pH 7.2, 50 mM CaCl2, 100 mM NaCl) following an incubation time of 2.5 h. The transition began at the free edge of the short domain. In between (iii*) and (iv*), the short domain continued to grow by adding new tetramers at the bottom edge. Times of image collection were (i*) 2.5, (ii*) 2.7, (iii*) 2.8, and (iv*) 2.9 h.
Fig. 3B shows sequential in situ AFM images of a single short domain neighbored by three tall domains and one short domain during its transformation from short to tall (see SI Appendix, Fig. S5 and Movie S1). The transformation starts at a domain edge with a change in the height of an individual tetramer and then gradually propagates from one side of the domain to the other, rather than occurring simultaneously throughout the entire domain.
High-resolution AFM height images (Fig. 4 A and B) of the short and tall domains reveal nearly identical lattice spacings. However, image analysis in Fig. 4 B and C show clear topological differences in the submolecular details of the tetramers in the two domains. For example, the tall domain (Fig. 4C) consists of tetrameric units that are more compact within the plane of the domain and exhibit loop-like regions that are not evident in tetramers of the short domain (Fig. 4D), which in turn expose portions of the proteins in interstitial regions that are not evident in the tall domains. The structural difference that distinguishes the short and tall domains becomes evident when the high-resolution images shown in Fig. 4 are analyzed for statistical variations. Image analysis (SI Appendix, Fig. S4) shows that the standard deviation of the S-layer structure away from the average is also significantly greater for the less stable short domains. These results imply that the transformation from short to tall is due to a slight conformational transition that amounts to folding under portions of the S-layer proteins to make them less extended and produce a more uniform structure.
Fig. 4.

High-resolution AFM images of tall (A, C) and short (B, D) domains. A and B show unprocessed AFM images. C and D show high resolution height images of four tetramers from tall and short domains that reveal distinct topological differences C and D were processed through correlation averaging (see SI Appendix for details). S-layers were initially grown in protein solution with Cp ∼ 41 μg/mL (10 mM Tris pH 7.2, 50 mM CaCl2, 100 mM NaCl) at 25 °C for 4.5 h. Both A and B were imaged in the same solution with minimal imaging forces (ca. 85 pN) (full Z color scale: 8 nm for A and B, 2.5 nm for C and D).
The presence of two domains with distinct conformations was confirmed by cryo transmission electron microscopy (cryo-TEM) analysis of S-layers grown on approximately 10–20 nm thick mica flakes as shown in SI Appendix, Figs. S6 and S7. (See Materials and Methods for details of procedure.) Cross-sectional views confirmed the presence of a single S-layer (SI Appendix, Fig. S7A). Comparison to cryo-TEM images of S-layers grown in mica free solutions (SI Appendix, Fig. S6) showed that one domain type (SI Appendix, Fig. S7D) exhibited the typical doughnut-shaped tetrameric lattice units whereas the other type (SI Appendix, Fig. S7C) did not and indeed appeared to be less ordered.
Discussion
The results presented above show that S-layer assembly on mica follows two multistage pathways as shown schematically in Fig. 5A. The crystalline phase that forms is set at the time of cluster nucleation, with the fraction that form the tall phase being greater (see Fig. 1 B–D) and increasing with temperature (Fig. 2A). After the growth process is complete, the short phase undergoes a conformational transformation to form the tall phase, exhibiting an exponential dependence of number fraction on time, whereas the tall phase was never observed to transform into the short phase. These observations are consistent with the short domain being less thermodynamically stable and lead to the energy barrier diagram shown in Fig. 5B.
Fig. 5.

(A) Proposed scheme of multiple pathways of S-layer assembly on mica. S-layer assembly on mica starts from the condensation of monomer S-layers and then follow two different pathways. One leads to low energy, final ordered state known as tall phase (T phase); however, the other leads to kinetically trapped, more disordered state known as short phase (S phase). Early crystal clusters are defined as crystalline domains of S-layers that are still in the process of the growth whereas mature crystal grains are defined as fully grown, closely neighbored crystalline domains of which their growth is limited due to no available space for monomers. (B) Proposed energy diagram of S-layer assembly from the monomer in solution to the crystal on mica. Energy barriers to formation of two states only differ by 1.6 kJ/mol; however, the energy barrier to transformation from one to the other is approximately 38 times larger. These energy barriers are qualitatively presented along with two pathways during the assembly.
The relative magnitudes of the barriers to nucleation of the tall and short phases can be determined directly from the relative number of clusters that transform into the two phases by using ϕS/ϕT = exp(-(ES - ET)/kT), where ϕi and Ei are the fractions of clusters and activation barriers, respectively, and the subscripts S and T refer to short and tall. From analysis of the data we find that the difference between the two barriers (Δ = ES - ET) is 1.6 kJ/mol or 0.7 kT (see SI Appendix for details). For comparison, we previously found that the kinetic barrier to creating new tetramers at S-layer boundaries was about 50 kJ/mol and the estimated free energy to folding of individual S-layer proteins based on known correlations between protein size and folding time is over 100 kJ/mol (18). Consequently, this barrier difference (Δ) is much smaller by comparison. Thus the notion that the kinetic trap associated with the short domains can be viewed as a subtle perturbation in the folding funnel to S-layer tetramer formation is sensible.
Although the barrier to formation of the transient short phase is a little bit larger than the barrier to direct formation of the tall state, the barrier to transformation from short to tall is quite large and thus the characteristic relaxation time at room temperature is long (approximately 10 h). To estimate the barrier to the transformation we can write NS(t) = NS(t = 0) exp(-t/τ), where 1/τ is the characteristic rate coefficient and NS(t = 0) is initial number of short domains before transformation. Because the transformation of an entire domain is triggered by transformation of a single tetramer (Fig. 3B), 1/τ and can be estimated from 1/τ = f exp(-EB/kT), where f is the characteristic attempt frequency associated with conformational fluctuations and EB is the barrier to transformation. As Fig. 3D shows, the fraction of short domains does indeed follow a simple exponential dependence. From the dependence of protein folding rates on protein size (19), the lower limit of f should be approximately 1.0 × 106 (Hz) (20), which leads to an estimate for the barrier EB of 61 kJ/mol or 25 kT (see SI Appendix). Thus, once the high-energy domains form, the barrier to their relaxation into low-energy domains is nearly as great as the barrier to folding of individual S-layer proteins.
These findings raise the obvious question of why mica reveals a kinetic trap that is not seen on lipid bilayers or in bulk solution. If there was a sufficient population of distinctive sites on the mica surface that exhibited stronger binding to the S-layer protein they could inhibit the conformational transformation from the extended monomeric state to the compact tetrameric state. As a consequence nucleation might be biased toward one of the two phases, presumably the short phase. There are two obvious sources for such sites, both of which are related to the chemistry of the mica surface. The first is impurities in the mica. Mica (muscovite phase) is a potassium alumino silicate [KAl2(AlSi3O10)(F,OH)2] in which divalent cations such as Mg2+, Mn2+, and Fe2+ commonly substitute for K+ ions (21). Due to the different size and valency between these impurities and K+ ions, these substitutions create structurally distinct defect sites at the surface. However, typical impurity levels are less than approximately 10% and the nominal level of divalent impurities in the mica used for this study was < 6%; hence, they are at insufficient concentrations to account for a nearly 50/50 mix of short and tall clusters.
The second potential source of distinct sites is Ca2+ substitution for K+, which occurs upon exposure to Ca2+-bearing solutions, such as those used in the experiments reported here. However, this exchange process is rapid and leads to 100% substitution on timescales that are short compared to the timescale for the X-ray photoelectron spectroscopy (XPS) measurements used to probe this exchange (22). Nonetheless, to be certain this could not be a source of short domains, we incubated mica for overnight (i.e., approximately 21 h) in CaCl2 solution before exposure to S-layer proteins solutions for 4 h but found no difference in the occurrence of short and tall domains when compared to samples that were not preincubated in CaCl2 solution. Apparently, even when the surface cation sites are 100% occupied by Ca2+ ions, both short and tall domains still form.
The second potential reason for the effect of mica is intrinsic to the S-layer/mica system. The interaction with the rigid mica surface, pure or impure, is simply strong enough to hinder the folding process so that the proteins get trapped in a poorly folded state. This same state may well be accessible in the bulk solution or on fluidic lipid bilayer surfaces, but is much more transient because the barrier to the conformational transformation into the well-ordered state is smaller in the absence of the S-layer/mica interaction.
Conclusions
The findings reported here demonstrate the importance of kinetic traps in determining the pathway of S-layer crystallization and suggest that the concept of the folding funnel for individual proteins can be equally applied to assembly of extended ordered protein structures. Beginning with rapid collapse into clusters of monomers, the S-layer system explores many configurations before transforming into the final ordered state. However, a large percentage of the trajectories render the system temporarily trapped in a high-energy, less ordered structure, from which it slowly relaxes into the low-energy equilibrium structure. Indeed, the language used to describe the protein folding funnel is well suited to this picture of S-layer assembly, with the amorphous liquid-like clusters investigated in our previous study representing the multiprotein equivalent of the molten globules and the high-energy domains constituting the partially ordered intermediates. Because extended protein architectures with long-range order commonly require conformational changes from the monomeric structures, this picture may be generally applicable to the phenomenon of protein self-assembly.
Materials and Methods
Preparation of S-layer Crystals on Mica for in Situ AFM.
Disassembled S-layer proteins, SbpA from Lysinibacillus sphaericus, were prepared in water following previously described protocols (17). Freshly peeled mica (Grade V-1 Muscovite, Structural Probe, Inc.) was incubated in 5 mL growth solution (10 mM Tris pH 7.2, 50 mM CaCl2, 100 mM NaCl) containing the protein at concentrations of 15–40 μg/mL. After incubation, the substrate was placed in an AFM fluid cell that was filled with the same growth solution and the surface was imaged in situ. In some cases noted below, samples were first incubated for a fixed amount of time over a range of incubation temperatures between 4 and 37 °C and then examined by AFM in fluid (see SI Appendix for details).
In Situ AFM and AFM Image Analysis.
In situ AFM was performed using an AFM fluid cell within a multimode AFM (Nanoscope VIII controller, Bruker Nano: Bruker AXS) equipped both with liquid-resistant, vertical engagement 125-μm “JV” (AS-130VLR) and 10-μm “EV” (AS-12VLR) scanners. The AFM probe consisted of a sharp silicon tip on a silicon nitride cantilever (HYDRA probe, length: 200 μm, spring constant k = ca. 0.035 N/m, average tip diameter < 10 nm, AppNano). Freshly cleaved mica discs (Grade V-1 Muscovite, Structural Probe, Inc.) glued on stainless steel metal discs using 60-min two parts Double/Bubble urethane glue (Hardman, Inc.) were used as substrates. For typical imaging conditions, AFM images were collected at scan frequencies of 1–8 Hz while applying a minimum loading force of approximately 120 pN or less using optimized feedback and setpoint parameters for stable imaging conditions. Ex situ and in situ AFM images of crystalline domains of T and S phase S-layer on mica were collected and processed by correlation averaging using scanning probe image processor (SPIP, Image Metrology A/S). More than 100 tetramers from T and S phase were separately sampled and averaged by a proprietary protocol of SPIP and typically a set of output images such as correlation average (CA), cross correlation (CC), and standard deviation (SD) images were calculated from the process. The later contains information regarding the structural uniformity of the tetramers (see SI Appendix for details).
Preparation of S-layer Crystals on Mica for Cryo-TEM.
Disassembled S-layer proteins, SbpA from Lysinibacillus sphaericus, were prepared in water following previously described protocols (17). Freshly peeled mica (Grade V-1 Muscovite, Structural Probe, Inc.) was mechanically ground using a mortar and pestle and then dispersed in water via ultrasonication for approximately 1 h. The solution was then left for approximately 3 h to allow for sedimentation of large pieces. Afterwards, the supernatant of the solution containing the dispersed mica flakes with sizes ranging from a few hundred nanometers to a few micrometers with a minimum thickness of approximately 10 nm was decanted (23). Disassembled S-layer proteins (70 μg/mL) were incubated with this aqueous dispersion of mica flakes in the growth buffer (10 mM Tris pH 7.2, 50 mM CaCl2, 100 mM NaCl). After incubation for approximately 4 h at 25 °C, approximately 5 μL aliquots of the dispersion solution were dropped on lacey carbon grids (Model #01881, Ted Pella, Inc.) that were pretreated by glow discharge. The Formvar supports of the grids were not removed. Finally, the grids were manually blotted and plunged into liquid ethane by a piston and stored in liquid nitrogen before being imaged by cryo-TEM.
Cryo-TEM and Image Analysis.
Cryo-TEMimages were acquired on a JEOL-3100 electron microscope equipped with a field emission gun electron source operating at 300 kV, an Omega energy filter, a Gatan 795 4 K × 4 K CCD camera mounted at the exit of an electron decelerator (24), and a cryo-transfer stage. The decelerator was operated at 200 kV resulting in images formed by electron beam accelerated with approximately 100 kV at the CCD. The stage was cooled at 80 K with liquid nitrogen for all cryogenic data sets.
In order to have a statistically relevant survey, typically greater than approximately 150 images were acquired using a magnification of × 46,000 at the CCD giving a pixel size of approximately 0.28 nm at the specimen. Underfocus values ranged from 3.4 ± 0.25 μm to 6 ± 0.25 μm, and the widths of energy filter were typically around 22 ± 0.2 eV. Structurally informative, high quality 2D projection images were acquired using electron dosages between approximately 45 e-/Å to approximately 80 e-/Å per image. ImageJ [National Institutes of Health (NIH), http://rsb.info.nih.gov/ij/] program was used to analyze 2D projection images.
Supplementary Material
ACKNOWLEDGMENTS.
We gratefully acknowledge Dr. Jinhui Tao for help in preparing dispersions of mica flakes. This work was performed at Lawrence Berkeley National Laboratory with support from the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The research was supported by the Division of Materials Science and Engineering and was carried out in the Molecular Foundry, which is supported by the Scientific User Facilities Division.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201504109/-/DCSupplemental.
References
- 1.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 2.Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA. Metal-mediated self-assembly of protein superstructures: Influence of secondary interactions on protein oligomerization and aggregation. J Am Chem Soc. 2008;130:6082–6084. doi: 10.1021/ja8012177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoen AP, Schoen DT, Huggins KNL, Arunagirinathan MA, Heilshorn SC. Template engineering through epitope recognition: A modular, biomimetic strategy for inorganic nanomaterial synthesis. J Am Chem Soc. 2011;133:18202–18207. doi: 10.1021/ja204732n. [DOI] [PubMed] [Google Scholar]
- 4.Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 6.Gruebele M. How to mark off paths on the protein energy landscape. Proc Natl Acad Sci USA. 2009;106:18879–18880. doi: 10.1073/pnas.0910764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radford SE. Protein folding: Progress made and promises ahead. Trends Biochem Sci. 2000;25:611–618. doi: 10.1016/s0968-0004(00)01707-2. [DOI] [PubMed] [Google Scholar]
- 8.Guo ZY, Thirumalai D. Kinetics of protein-folding—nucleation mechanism, time scales, and pathways. Biopolymers. 1995;36:83–102. [Google Scholar]
- 9.Noé F, Schütte C, Vanden-Eijnden E, Reich L, Weikl TR. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc Natl Acad Sci USA. 2009;106:19011–19016. doi: 10.1073/pnas.0905466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orte A, et al. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc Natl Acad Sci USA. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borgia A, Williams PM, Clarke J. Single-molecule studies of protein folding. Annu Rev Biochem. 2008;77:101–125. doi: 10.1146/annurev.biochem.77.060706.093102. [DOI] [PubMed] [Google Scholar]
- 12.Stigler J, Ziegler F, Gieseke A, Gebhardt JCM, Rief M. The complex folding network of single calmodulin molecules. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 13.Weiss S. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat Struct Biol. 2000;7:724–729. doi: 10.1038/78941. [DOI] [PubMed] [Google Scholar]
- 14.Albers SV, Meyer BH. The archaeal cell envelope. Nat Rev Microbiol. 2011;9:414–426. doi: 10.1038/nrmicro2576. [DOI] [PubMed] [Google Scholar]
- 15.Sleytr UB, Messner P, Pum D, Sara M. Crystalline bacterial cell surface layers (S layers): From supramolecular cell structure to biomimetics and nanotechnology. Angew Chem Int Ed. 1999;38:1035–1054. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Stewart M, Beveridge TJ, Trust TJ. Two patterns in the aeromonas-salmonicida a-layer may reflect a structural transition that alters permeability. J Bacteriol. 1986;166:120–127. doi: 10.1128/jb.166.1.120-127.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung S, Shin SH, Bertozzi CR, De Yoreo JJ. Self-catalyzed growth of S layers via an amorphous-to-crystalline transition limited by folding kinetics. Proc Natl Acad Sci USA. 2010;107:16536–16541. doi: 10.1073/pnas.1008280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naganathan AN, Munoz V. Scaling of folding times with protein size. J Am Chem Soc. 2005;127:480–481. doi: 10.1021/ja044449u. [DOI] [PubMed] [Google Scholar]
- 19.Thirumalai D. From minimal models to real proteins—time scales for protein-folding kinetics. J Phys I. 1995;5:1457–1467. [Google Scholar]
- 20.Li MS, Klimov DK, Thirumalai D. Thermal denaturation and folding rates of single domain proteins: size matters. Polymer. 2004;45:573–579. [Google Scholar]
- 21.Guidotti CV. Compositional variation of muscovite as a function of metamorphic Grade and Assemblage in metapelites from NW Maine. Contrib Mineral Petrol. 1973;42:33–42. [Google Scholar]
- 22.Xu L, Salmeron M. An XPS and scanning polarization force microscopy study of the exchange and mobility of surface ions on mica. Langmuir. 1998;14:5841–5844. [Google Scholar]
- 23.Heuser JE. Procedure for freeze-drying molecules adsorbed to mica flakes. J Mol Biol. 1983;169:155–195. doi: 10.1016/s0022-2836(83)80179-x. [DOI] [PubMed] [Google Scholar]
- 24.Downing KH, Mooney PE. A charge coupled device camera with electron decelerator for intermediate voltage electron microscopy. Rev Sci Instrum. 2008;79:043702. doi: 10.1063/1.2902853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.