

Abstract
In Alzheimer's disease, lipid alterations point towards peroxisomal dysfunctions. Indeed, a cortical accumulation of saturated very long chain fatty acids (VLCFAs: C22:0, C24:0, C26:0), substrates for peroxisomal β-oxidation, has been found in Alzheimer patients. This study was realized to investigate the effects of VLCFAs at the mitochondrial level since mitochondrial dysfunctions play crucial roles in neurodegeneration. On human neuronal SK-NB-E cells treated with C22:0, C24:0, or C26:0 (0.1–20 μM; 48 h), an inhibition of cell growth and mitochondrial dysfunctions were observed by cell counting with trypan blue, MTT assay, and measurement of mitochondrial transmembrane potential (Δψ m) with DiOC6(3). A stimulation of oxidative stress was observed with DHE and MitoSOX used to quantify superoxide anion production on whole cells and at the mitochondrial level, respectively. With C24:0 and C26:0, by Western blotting, lower levels of mitochondrial complexes III and IV were detected. After staining with MitoTracker and by transmission electron microscopy used to study mitochondrial topography, mass and morphology, major changes were detected in VLCFAs treated-cells: modification of the cytoplasmic distribution of mitochondria, presence of large mitochondria, enhancement of the mitochondrial mass. Thus, VLCFAs can be potential risk factors contributing to neurodegeneration by inducing neuronal damages via mitochondrial dysfunctions.
1. Introduction
Alzheimer's disease (AD; OMIM #104300) is a neurodegenerative disorder. This is a clinicopathological entity regarded as progressive dementia, which includes episodic memory impairment and the involvement of other cognitive domains or skills that are associated with the reduction of daily activities and behavioral changes [1, 2]. AD is characterized by the presence of two aberrant structures in patients' brains: senile plaques and neurofibrillary tangles [3]. Senile plaques are composed of β-amyloid peptides, particular fragments of the amyloid peptide precursor protein (APP) [4], whereas the main component of neurofibrillary tangles is the cytoskeleton protein interacting with the microtubules known as the tau protein, in its hyperphosphorylated form [5, 6]. Currently, only very few treatments, which minimally impact the disease, are available [1]. Therefore, for a better understanding of the physiopathology of AD, there is a real need to identify the molecules capable of favoring the development of this disease, to determine their biological activities and the associated metabolic pathways in order to discover new therapeutic targets. Some investigations support a linkage between lipid metabolism and AD [7–9]. The most widely studied risk factors are the three common variants of the human apolipoprotein E (apoE) protein, known as E2, E3, and E4. The gene variants underlying these protein isoforms, known as ε2, ε3, and ε4 are allelic forms of the APOE gene, resulting from different haplotypes at the APOE locus (19q13.31) [10]. As many carriers of the APOE risk allele (ε4) live into their 90 s, the existence of other late-onset AD genetic and/or environmental risk factors have yet to be identified [11]. Some evidence has accumulated that cholesterol oxide derivatives, also called oxysterols, which are 27 carbon derivatives of cholesterol containing additional oxygen atoms on the steroid's nucleus or on the side chain, can be used as markers of brain atrophy in patients with various neurodegenerative diseases [12, 13]. The accumulation of oxysterols in the body may be due to dietary consumption (dried egg products, powdered milk, cheeses, and dried animal products); they can also be formed enzymatically, mostly by cytochromes P450 (CYPs) [14], and also nonenzymatically by reaction with oxygen [15]. Oxysterols have various biological activities (induction of cell death, activation of inflammation, and/or oxidative processes) and are therefore assumed to play major roles in various pathologies including AD [13, 16]. Potential roles of 7-ketocholesterol and 7β-hydroxycholesterol have been suggested [17, 18], and it was found that reduction of the plasmatic level of 24S-hydroxycholesterol correlated with the severity of dementia or the degree of brain atrophy [19]. 24S-hydroxycholesterol formed by a specific cholesterol 24-hydroxylase (CYP46A1), a cytochrome P450 enzyme expressed in neurons, is considered as a marker of metabolically active neurons [20], and under in vitro conditions, it is an efficient inhibitor of β-amyloid peptide formation, a major component of senile plaques [21, 22]. Therefore, conversion of cholesterol into 24S-hydroxycholesterol in the brain is of critical importance for central cholesterol homeostasis and 24S-hydroxycholesterol could play important roles in the development of AD [23, 24]. Moreover, regarding the decline of peroxisomal function with age [25, 26], peroxisomal dysfunctions associated with aging might favor neurodegenerative diseases, including AD [27, 28]. In agreement with this hypothesis, some lipid alterations observed in AD, concerning docosahexaenoic acid (DHA, C22:6 n-3), very long chain fatty acids (VLCFAs: C22:0, C24:0 and C26:0), and plasmalogens also point towards peroxisomal dysfunctions given that the β-oxidation or the synthesis of these lipids occurs, at least in part, in the peroxisome [29, 30]. Recently, lipid analyses of cortical regions from patients with AD revealed accumulation of VLCFAs (C22:0, C24:0, and C26:0), three substrates for peroxisomal β-oxidation, in patients with stages V and VI pathology compared with those modestly affected (stages I and II) based on the neuropathological Braak staging for AD [31]. Conversely, the level of plasmalogens, which need intact peroxisomes for their biosynthesis, was decreased in severely affected tissues, in agreement with a peroxisomal dysfunction. In addition, the peroxisomal volume density was increased in some neurons in the gyrus frontalis at advanced AD stages [31]. Based on these later observations, it is tempting to speculate that the accumulation of C22:0, C24:0, and C26:0 in the cortex of patients with AD could contribute to neuron dysfunctions. Indeed, these fatty acids are known to induce various types of cellular damages, in vitro and in vivo, especially at the mitochondrial and lysosomal level, and to trigger oxidative stress and cell death in the cells of the central nervous system [32–35].
In order to evaluate the ability of C22:0, C24:0, and C26:0 to induce neuronal damages, human neuronal cells (SK-NB-E) were cultured in the absence or in the presence of these fatty acids at various concentrations (0.1, 1, 5, 10, and/or 20 μM) for 48 h. We focused on the effects of these fatty acids at the mitochondrial level since mitochondria are assumed to play important roles in the development of AD [36–38], and we observed that C22:0, C24:0, and C26:0 were able to induce morphological and functional modifications of this organelle.
2. Materials and Methods
2.1. Cells and Cell Treatments
Human neuronal cells (SK-NB-E) were seeded at 200,000 cells per well in 12-well microplates containing 1 mL of culture medium constituted by Dulbecco's Modified Eagle Medium with L-gutamine (DMEM) (Lonza) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Pan Biotech) and 1% antibiotics (100 U/mL penicillin, 100 mg/mL streptomycin) (Pan Biotech). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and passaged twice a week. At each passage, SK-NB-E cells were trypsinized with a (0.05% trypsin-0.02% EDTA) solution (Pan Biotech).
Docosanoic acid (C22:0), tetracosanoic acid (C24:0), and hexacosanoic acid (C26:0) (Sigma-Aldrich) were solubilized in α-cyclodextrin (Sigma-Aldrich) as previously described [39]. The final maximum concentration of α-cyclodextrin (vehicle) in the culture medium was 1 mg/mL [33].
The conditions of treatment with very long chain fatty acids (VLCFAs) (C22:0, C24:0, or C26:0) were the following: after plating SK-NB-E cells for 24 h, the cells were further treated for 48 h with various VLCFA concentrations (0.1, 1, 5, 10, and/or 20 μM) in HAM's F-10 medium (Pan Biotech). The concentrations of VLCFAs were chosen according to measurements made in similar experimental conditions on the plasma of healthy subjects (C22:0, C24:0, or C26:0: less than 1 μM), of patients with X-ALD (C22:0, C24:0, or C26:0: from 1 to 5 μM) (used as positive controls) [34, 40], and of patients with AD. Our preliminary data concern 15 Alzheimer patients from the department of Neurology (University Hospital, Monastir, Tunisia): C22:0 (mean, 4.2 μM; range, 0.8–8.4 μM) and C24:0 (mean, 4.9 μM; range, 0.8–10.6 μM).
2.2. Colorimetric MTT Assay
The MTT assay was carried out as previously described [41] on SK-NB-E cells plated in 12-well flat-bottom culture plates after 48 h of treatment with C22:0, C24:0, or C26:0 (0.1, 1, 5, 10, and 20 μM). The MTT assay was used to evaluate the effects of C22:0, C24:0, and C26:0 on cell proliferation and/or viability. MTT salt is reduced to formazan in the metabolic active cells by dehydrogenase to form NADH and NADPH [42]. The plates were read at 570 nm with a microplate reader.
2.3. Cell Counting with Trypan Blue
After trypsinization, cells were centrifuged and resuspended in culture medium. The total number of viable cells was determined in the presence of trypan blue, which stains dead cells blue, after 48 h of treatment with C22:0, C24:0, and C26:0 (0.1, 1, 5, 10, and 20 μM) under an inverted-phase contrast microscope Diaphot (Nikon).
2.4. Study of Cell Morphology Using Phase Contrast Microscopy
Cell morphology of SK-NB-E cells was observed after 48 h of culture in the absence or the presence of fatty acids (C22:0, C24:0, and C26:0: 0.1, 1, 5, 10, and 20 μM) under an inverted-phase contrast microscope (Axiovert 40 CFL, Zeiss). Digitalized images were obtained with a camera (Axiocam ICm1, Zeiss).
2.5. Flow Cytometric Measurement of Transmembrane Mitochondrial Potential with DiOC6(3)
Variations of the mitochondrial transmembrane potential (ΔΨm) were measured with 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3) (Invitrogen), which allows the percentage of cells with low ΔΨm to be determined.With DiOC6(3), mitochondrial depolarization is indicated by a decrease in green fluorescence collected through a 520/10-nm band pass filter. DiOC6(3) was used at a 40 nM [43]. Flow cytometric analyses were performed on a Galaxy flow cytometer (Partec). Ten thousand cells were acquired for each sample. Data were analyzed with Flomax software (Partec) or FlowJo software (Tree Star Inc.).
2.6. Flow Cytometric Measurement of the Whole Intracellular Production of Superoxide Anion with Dihydroethidium
The whole intracellular production of superoxide anion (O2 ∙−) was detected with dihydroethidium (DHE; Invitrogen/Life Technologies), which is a nonfluorescent compound that can diffuse through cell membranes, rapidly oxidized in hydroethidine (HE) under the action of O2 ∙− [44]. DHE was initially prepared at 10 mM in dimethyl sulfoxide (DMSO) and used at a 2 μM final concentration on cell samples of 1 × 106 cells/mL of HAM'S F10 medium. Cells were incubated for 15 min at 37°C before flow cytometric analysis. The fluorescent signals were collected through a 580 ± 20 nm band pass filter on a logarithmic scale of four decades of log on a GALAXY flow cytometer (Partec). For each sample, 10,000 cells were acquired. Data were analyzed with Flomax software (Partec).
2.7. Flow Cytometric Measurement of Mitochondrial Superoxide Anions with MitoSOX
The mitochondrial production of superoxide anion (O2 ∙−) was detected with MitoSOX (Invitrogen/Life Technologies). MitoSOX Red reagent is a live-cell permeant and is rapidly and selectively targeted to the mitochondria [45]. Once in the mitochondria, MitoSOX Red reagent is oxidized by superoxide and exhibits orange/red fluorescence (λEx = 510 nm; λEm = 580 nm). The oxidation product becomes highly fluorescent upon binding to nucleic acids. MitoSOX was initially prepared at 5 mM in PBS and used at 5 μM on cell samples of 1 × 106 cells/mL of PBS. Cells were incubated for 15 min at 37°C before flow cytometry analysis. The fluorescent signals were collected through a 580 ± 20-nm band pass filter on a Galaxy flow cytometer (Partec). For each sample, 10,000 cells were acquired. Data were analyzed with Flomax software (Partec).
2.8. Labeling of Mitochondria with MitoTracker for the Microscopical Evaluation of Mitochondrial Distribution and Flow Cytometric Evaluation of Mitochondrial Mass
The mitochondrial mass and the cellular distribution of mitochondria were evaluated after staining with MitoTracker Red (Invitrogen) [45].
To evaluate mitochondrial distribution, SK-NB-E cells were cultured on glass coverslips, and after 48 h of culture in the absence or in the presence of fatty acids, they were stained with MitoTracker Red at 1 μM (15 min, 37°C). After labeling, the coverslips were washed in PBS, mounted in Dako fluorescent mounting medium (Dako), and stored in the dark at 4°C until observation under blue light. Observations were made under an Axioskop fluorescent microscope (Zeiss) using green excitation, and digitalized images were obtained with an Axiocam Zeiss camera.
To determine the mitochondrial mass, SK-NB-E cells were cultured in 12-well microplates, and after 48 h of culture in the absence or in the presence of fatty acids, they were stained with MitoTracker Red (1 μM, 15 min, 37°C). After trypsinization, the cells were washed and resuspended in PBS and immediately analyzed using flow cytometry. The fluorescent signals were collected through a 580 ± 20 nm band pass filter on a Galaxy flow cytometer (Partec). For each sample, 10,000 cells were acquired and the data were analyzed with Flomax software (Partec).
2.9. Western Blot Analysis of Mitochondrial Proteins
SK-NB-E cells were treated with C26:0, C24:0, and C22:0 at 5 and 10 μM for 24 and 48 h. After treatment, cells were trypsinized and incubated with Ripa lysis buffer (0.1% (w/v) SDS, 1% (w/v) NP40, 0.5% (w/v) Na-desoxycholate, 150 mM NaCl, 2 mM EDTA, and 50 mM TrisHCl pH 8) containing a mixture of protease and phosphatase inhibitors (NaF 50 mM, Protease Inhibitor Cocktail Tablets (Roche)). After 30 min incubation at 4°C in the lysis buffer, the cell debris were eliminated by centrifugation (20 min, 10,000 g), and the supernatant was collected. The protein concentrations were measured using bicinchoninic acid reagent (Sigma) [46]. Fifty micrograms of protein per slot were diluted in loading buffer (125 mmol/L Tris/HCl, pH 6.8, 10% (w/v) mercaptoethanol, 4.6% (w/v) SDS, 20% (v/v) glycerol, and 0.003% (w/v) Bromophenol blue), separated by SDS-PAGE and transferred to nitrocellulose membrane. After blocking nonspecific binding sites overnight by 5% nonfat milk in TPBS (PBS, 0.1% Tween20), the membranes were incubated for 1 h at room temperature with the anti-OXPHOS mixture (MS601, Abcam/Mitosciences) against structural components of all five oxidative phosphorylation complexes (Complex I subunit NDUFB8 (20 kDa), Complex II subunit (30 kDa), Complex III subunit Core 2 (47 kDa), Complex IV subunit II (24 kDa), and Complex V ATP synthase subunit alpha (53 kDa)) at a 1 : 500 dilution (TPBS with 1% nonfat milk). After three 10-min washes with TPBS, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) at a 1 : 5000 dilution (TPBS with 5% nonfat milk) for 1 h at room temperature and washed three times in TPBS for 10 min. Autoradiography of the immunoblots was performed using an enhanced chemoluminescence detection kit (Santa Cruz Biotechnology) and they were quantified with Image J Software. The results were expressed as a fold change to untreated SK-NB-E cells. Actin was used as a loading control. The monoclonal antibodies in the cocktail were chosen because they are against subunits that are labile when the corresponding complex is not assembled. Moreover, the combination is readily resolved in SDS-PAGE when the appropriate gel conditions are used.
2.10. Transmission Electron Microscopy
Transmission electron microscopy is a valuable tool to evaluate the morphological changes occurring under the action of various physical or chemical agents [47]. It was used to visualize the potential ultrastructural modifications induced especially at the mitochondrial level by fatty acids (C22:0, C24:0, and C26:0 used at 10 μM; 24 h) and α-cyclodextrin (1 mg/mL for 24 h, used as vehicle to dissolve fatty acids) on SK-NB-E cells, and was conducted as previously described [48].
2.11. Statistical Analysis
Statistical analyses were performed using WinSTAT software (Microsoft). The Mann-Whitney U test was used to compare the different groups, and data were considered to be statistically different at a P value of 0.05 or less.
3. Results
3.1. Evaluation of the Effects of C22:0, C24:0, and C26:0 on SK-NB-E Cells with the MTT Assay
When SK-NB-E cells were incubated for 48 h with C22:0, C24:0, and C26:0 (0.1–20 μM) a dose-dependent decrease of MTT reduction was observed. The absorbance began to decrease significantly with 0.1 μM of C22:0, and with 5 μM of C24:0 and C26:0, and similar absorbencies were obtained in control and α-cyclodextrin (vehicle)-treated cells (Figure 1). As MTT is cleaved into a blue color product (formazan) by the mitochondrial enzyme succinate dehydrogenase, this assay gives information on cell proliferation and mitochondrial metabolism. In order to determine whether the decrease in formazan production under treatment with C22:0, C24:0, and C26:0 is simply a function of the reduction of the number of viable cells, and/or an induction of mitochondrial dysfunctions, the effects of these VLCFAs on the number of viable cells and the mitochondrial transmembrane potential (ΔΨm) were measured [49].
Figure 1.

Evaluation of the effects of C22:0, C24:0, and C26:0 on cell proliferation and mitochondrial metabolism with the MTT assay. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 0.1–20 μM. Data shown are mean ± SD from two independent experiments conducted in triplicates. Significance of the difference between vehicle- and fatty acid-treated cells is indicated by *(Mann Whitney test; *P < 0.05). No significant difference was observed between control and vehicle-treated cells.
3.2. Effects of C22:0, C24:0, and C26:0 on SK-NB-E Cells in Terms of Cell Growth and Cell Morphology
Incubation of SK-NB-E cells with C22:0, C24:0, and C26:0 (0.1–20 μM) resulted in a marked and dose-dependent reduction in the total number of viable cells after 48 h. The decrease in the total number of viable cells determined in the presence of trypan blue became statistically significant with 0.1 μM of C22:0 and 5 μM of C24:0 and C26:0; a similar number of cells were obtained in untreated cells (control) and α-cyclodextrin (vehicle)-treated cells (Figure 2(a)). With the different fatty acids used, compared to control cells and vehicle-treated cells, the decreased number of viable cells was associated with an increase in the presence of some round cells (recalling less adherent dying cells [50]) in the culture dishes, especially under treatment with C22:0 at 20 μM (Figure 2(b)). The shape index based on the observations performed by phase contrast microscopy (Figure 2(b)) was calculated according to the following formula (number of round cells/total number of cells/mm2). This index shows a dose-dependent increase of round cells under treatment with the different VLCFAs used, especially with C22:0 (20 μM) (Figure 3).
Figure 2.
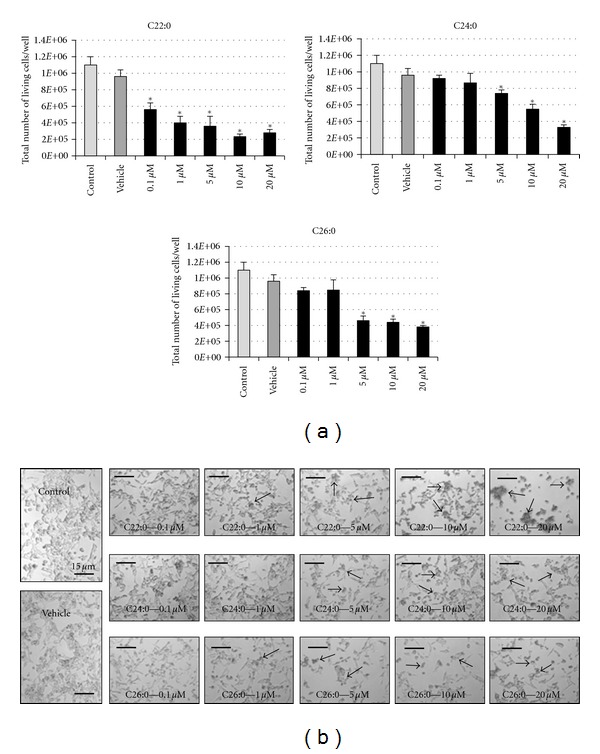
Evaluation of the effects of C22:0, C24:0, and C26:0 on SK-NB-E cells in terms of cell growth and cell morphology. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 0.1–20 μM. (a) Number of viable cells per well of 12 well plates determined in the presence of trypan blue; (b) observation with phase contrast microscopy. The arrows point towards aggregates of round cells. Data shown are mean ± SD from at least three independent experiments conducted in duplicates. Significance of the difference between vehicle- and fatty acid-treated cells is indicated by *(Mann-Whitney test; *P < 0.05). No significant difference was observed between control and vehicle-treated cells.
Figure 3.

Quantification of morphological changes induced by C22:0, C24:0, and C26:0 on SK-NB-E cells. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 0.1–20 μM. The induction of morphological changes associated with the presence of round cells observed under treatment with VLCFA was quantified according to the shape index (number of round cells/total number of cells/mm2). Data shown are mean ± SD from three independent experiments. Significance of the difference between vehicle- and fatty-acid-treated cells is indicated by *(Mann-Whitney test; *P < 0.05). No significant difference was observed between control and vehicle-treated cells.
3.3. Effects of C22:0, C24:0, and C26:0 on SK-NB-E Cells in Terms of Mitochondrial Transmembrane Potential
Incubation of SK-NB-E cells with C22:0, C24:0, and C26:0 (0.1–20 μM) for 48 h resulted in a marked and dose-dependent increase in the percentage of cells with depolarized mitochondria measured by flow cytometry after staining with DiOC6(3). The increase in cells with depolarized mitochondria became statistically significant with 1 μM of C22:0 and C24:0, and with 0.1 μM of C26:0. Similar percentages of cells with depolarized mitochondria were obtained between control and α-cyclodextrin (vehicle)-treated cells (Figure 4).
Figure 4.

Flow cytometric evaluation of the effects of C22:0, C24:0, and C26:0 on mitochondrial transmembrane potential. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 0.1–20 μM. The mitochondrial transmembrane potential was determined after staining with DiOC6(3) in order to quantify the cells with depolarized mitochondria. Data shown are mean ± SD from three independent experiments conducted in duplicate. Significance of the difference between vehicle- and fatty acid-treated cells is indicated by *(Mann-Whitney test; *P < 0.05). No significant difference was observed between control and vehicle-treated cells.
3.4. Effects of C22:0, C24:0, and C26:0 on Superoxide Anion Production in Whole Cells and at the Mitochondrial Level
The effects of C22:0, C24:0, and C26:0 on oxidative stress, mainly on superoxide anion production, were investigated at concentrations inducing modifications on cell growth and on mitochondrial transmembrane potential. To this end, SK-NB-E cells were incubated with C22:0, C24:0, and C26:0 (5–20 μM) for 48 h and the production on superoxide anions on whole cells and at the mitochondrial level was determined after staining with DHE and MitoSOX, respectively. After staining with DHE, whatever the fatty acid considered, a marked increase of cells overproducing superoxide anions (HE-positive cells) was observed at 10 and 20 μM (Figure 5(a)). With MitoSOX, whatever the fatty acid considered, our data show an increase of superoxide anion production at the mitochondrial level at 10 and 20 μM with C22:0 and C24:0, and at 20 μM with C26:0 (Figure 5(b)). As the increase of MitoSOX positive cells was lower than those of HE-positive cells, these data suggest that the mitochondria contribute in part to the increase of superoxide anion production on whole cells.
Figure 5.
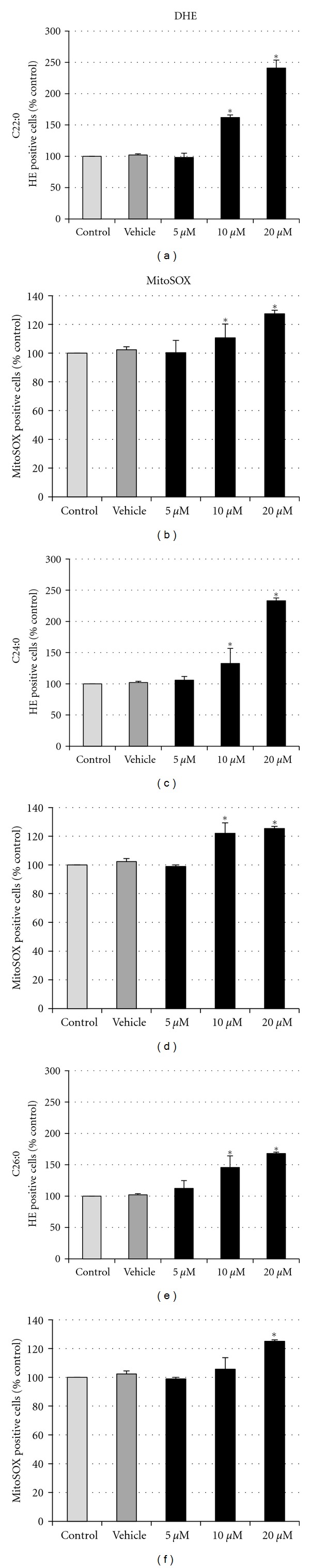
Flow cytometric evaluation of the effects of C22:0, C24:0, and C26:0 on superoxide anion production in whole cells and at the mitochondrial level. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle) or a VLCFA (C22:0, C24:0, or C26:0) used at 5, 10, and 20 μM. The production of superoxide anion on whole cells (HE-positive cells) (a) and at the mitochondrial level (MitoSOX-positive cells) (b) was determined after staining with DHE and MitoSOX, respectively. Data shown are mean ± SD from three independent experiments conducted in duplicates. Significance of the difference between vehicle- and fatty acid-treated cells is indicated by *(Mann-Whitney test; *P < 0.05). No significant difference was observed between control and vehicle-treated cells.
3.5. Effects of C22:0, C24:0, and C26:0 on the Expression of Mitochondrial Complex Subunits
The effect of C22:0, C24:0, and C26:0 (5 and 10 μM; 48 h) was studied on SK-NB-E cells in terms of the expression of proteins belonging to the different mitochondrial Complexes (Complex I subunit NDUFB8 (20 kDa), Complex II subunit (30 kDa), Complex III subunit Core 2 (47 kDa), Complex IV subunit II (24 kDa), and Complex V ATP synthase subunit alpha (53 kDa)) using Western blotting (Figure 6(a)) associated with a quantitative densitometry analysis (Figure 6(b)). In these conditions, when compared to untreated cells (control), similar values of the different complexes were observed in α-cyclodextrin (vehicle)-treated cells, and under treatment with C22:0. However, some changes were observed especially under treatment with C24:0 and C26:0, mainly on Complexes III and IV, which are proton pumps contributing to the proton gradient by an asymmetric absorption/release of protons. Thus, C24:0 (5 μM) reduced the level of Complex III subunit core 2, whereas C24:0 (10 μM) increased it, and the opposite was shown for C26:0. However, both C24:0 and C26:0 (5 and 10 μM) reduced the level of Complex IV subunit II (Figure 5(b)).
Figure 6.

Analysis of the effects of C22:0, C24:0, and C26:0 on the expression of mitochondrial complex subunits. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 10 μM. The Complex I subunit NDUFB8 (20 kDa), Complex II subunit (30 kDa), Complex III subunit Core 2 (47 kDa), Complex IV subunit II (24 kDa), and Complex V ATP synthase subunit alpha (53 kDa) involved in oxidative phosphorylation obtained from whole cellular extracts were analyzed with specific antibodies (MitoProfile Total OXPHOS Human WB Antibody Cocktail, Abcam/Mitosciences) by Western blotting (a). Quantification was carried out with Image J Software; each complex was normalized versus actin, and for each complex data were expressed as [(normalized value of the assay)/(normalized value of the control)]. Similar values were obtained between control, vehicle, and C22:0-treated cells, and differences were observed especially under treatments with C24:0 and C26:0, mainly for Complex III subunit Core 2 and Complex IV subunit II (b). Data shown are representative of two independent experiments.
3.6. Effects of C22:0, C24:0, and C26:0 on Mitochondrial Topography, Mitochondrial Mass, and Ultrastructural Mitochondrial Characteristics
The effects of C22:0, C24:0, and C26:0 on mitochondrial topography, mitochondrial mass, and ultrastructural mitochondrial characteristics were investigated at concentrations inducing mitochondrial transmembrane potential modifications and an overproduction of superoxide anions at the mitochondrial level. To this end, SK-NB-E cells were incubated with C22:0, C24:0, and C26:0 (5, 10 and 20 μM) for 48 h and the cells were stained with MitoTracker Red. Whereas a few fluorescent dots were most often observed in untreated cells (control) and in α-cyclodextrin (vehicle)-treated cells, showing a regular cytoplasmic distribution of mitochondria, a diffuse staining pattern was frequently detected in fatty acid-treated cells, suggesting either modifications of the mitochondrial distribution and/or of the mitochondrial membrane characteristics (Figure 7(a)). It is noteworthy that these mitochondrial changes revealed by fluorescence microscopic observations were associated with an enhancement of the mitochondrial mass measured by flow cytometry (Figure 7(b)). In agreement with the data obtained with MitoTracker Red, the observations made using transmission electron microscopy also showed substantial mitochondrial changes. Whereas the distribution (Figures 8(a) and 8(b) and the aspects (size and shape) (Figures 8(f) and 8(g)) of mitochondria were similar in control and α-cyclodextrin (vehicle)-treated cells, major changes were found in fatty acid-treated cells. Thus, in C22:0-, C24:0-, and C26:0-treated cells, clusters of mitochondria were often detected in various areas of the cytoplasm (Figures 8(c), 8(d) and 8(e)), suggesting mitochondrial biogenesis. Moreover, some of these mitochondria have different sizes and shapes than in the control and α-cyclodextrin (vehicle)-treated cells: indeed, some elongated (Figure 8(h), 8(i)) and round (Figure 8(j)) mitochondria were very often observed in VLCFA-treated cells.
Figure 7.

Analysis by fluorescence microscopy and flow cytometry of the effects of C22:0, C24:0, and C26:0 on mitochondrial topography and mitochondrial mass. SK-NB-E cells were cultured for 48 h in the absence (control) or presence of α-cyclodextrin (1 mg/mL) (vehicle), or a VLCFA (C22:0, C24:0, or C26:0) used at 10 μM. Mitochondria were identified by staining with MitoTracker Red. (a) Evaluation of mitochondrial topography by fluorescence microscopy; whereas some fluorescent dots are often observed in the cytoplasm of untreated (control) and α-cyclodextrin (vehicle)-treated cells, a more diffuse and irregular staining pattern is detected under treatment with VLCFAs. (b) Quantification of mitochondrial mass by flow cytometry. Data shown are representative of at least two independent experiments.
Figure 8.

Evaluation by transmission electron microscopy of the ultrastructural characteristics of human neuronal cells (SK-NB-E) treated with C22:0, C24:0, or C26:0 and of their mitochondrial characteristics. Transmission electron microscopy of SK-NB-E cells cultured for 48 h in the absence (control cells) (a, f) or presence of α-cyclodextrin (1 mg/mL) (vehicle) (b, g), C22:0 (10 μM) (c, h), C24:0 (10 μM) (d, i), or C26:0 (10 μM) (e, j). The insets in Figures (a, b, c, d, and e) correspond to Figures (f, g, h, i, and j), respectively. No differences were observed between control (a, f) and vehicle-treated cells (b, g). In C22:0-, C24:0- and C26:0-treated cells, no signs of apoptosis (cells with perinuclear chromatin, with condensed and/or fragmented nuclei) were detected [47, 48], but some mitochondrial modifications were observed. In C22:0- (h), C24:0- (i), and C26:0- (j) treated SK-NB-E cells, when compared to control and vehicle-treated cells, higher numbers of mitochondria, often larger in size, were found.
4. Discussion
Brain aging leading to cognitive decline, including AD, is an incompletely understood process that different hypotheses attempt to explain. Currently, the agents contributing to brain aging-associated dysfunctions are not well known; nor are the possible underlying mechanisms involved in cognitive decline [38]. The increased accumulation of VLCFAs (C22:0, C24:0, and C26:0) found in cortical regions from patients with AD [31], the important roles played by various lipids, including different types of fatty acids, in the physiopathology of AD [7], and the mitochondrial changes occurring with advancing age [51, 52] led us to attempt to characterize the effects of C22:0, C24:0, and C26:0 at the mitochondrial level on human neuronal cells (SK-NB-E). We observed that these different fatty acids induced important morphological and functional changes at the mitochondrial level.
Since mitochondria are unlikely to play a significant role in cellular MTT reduction, the MTT assay, which gives information on mitochondrial activity and cell growth, was used to evaluate the sensitivity of SK-NB-E cells to C22:0, C24:0, and C26:0. With the MTT assay, the lower ability of SK-NB-E cells to reduce MTT to formazan blue in the presence of these fatty acids revealed the cytotoxic effects of these compounds in concentrations 5 μM and higher, which was similar to those previously described on glial cells [34]. Compared to untreated and vehicle-treated cells, the higher MTT value observed under treatment with C24:0 (0.1 μM) suggests mitochondrial hyperactivity (previously reported with cytotoxic compounds [53]), which could take place to counteract early mitochondrial dysfunctions. We cannot exclude that a similar effect occurs with C22:0 and C26:0 used at 0.1 μM before or after 48 h of treatment. Moreover, in agreement with data obtained on glial cells (oligodendrocytes, astrocytes), C22:0 and VLCFAs (C24:0, C26:0) also inhibit cell growth in a dose-dependent manner and favor cell detachment, as suggested by the presence of an increased number of clusters of round cells detected under treatment with these fatty acids [34]. The neurotoxic effects of C22:0, C24:0, and C26:0, via alterations of mitochondrial activity suggested by the MTT assay, were confirmed with the use of DiOC6(3), which is a reliable fluorescent probe used to measure the mitochondrial transmembrane potential (ΔΨm) [54]. With this probe, the higher percentages of cells with depolarized mitochondria found under treatment with C22:0, C24:0, and C26:0 demonstrate that there is a significant impact of these fatty acids on mitochondrial activity, which can contribute to triggering cell death [34, 48]. This mitochondrial disturbance was also observed on rat C6 glioma cells, murine oligodendrocytes 158 N [34], normal fibroblasts, and fibroblasts of X-ALD patients [55] as well as oligodendrocytes and astrocytes of rat brain [33] treated with C24:0 or C26:0 used at 20 μM and higher, indicating that a decrease of ΔΨm is common to neural cells and fibroblasts and provides evidence that mitochondrial-dependent cellular dysfunctions are triggered by elevated VLCFA concentrations. Therefore, when the effects of C22:0, C24:0, and C26:0 were evaluated with the MTT assay by cell counting in the presence of trypan blue or after staining with DiOC6(3), since similar activities of these fatty acids were found on glial cells and neural cells, these different data suggest that these compounds may have dramatic consequences on various brain functions and that they can trigger neurodegeneration.
Currently, some investigations support that oxidative damage can favor brain senescence and neurodegeneration via mitochondrial dysfunctions [36, 37, 52] and that oxidative-stress-mediated energetic failure may be at the core of multifactorial neurodegenerative diseases [56]. There is also some evidence that accumulation of VLCFAs in different tissues and biological fluids simultaneously stimulates oxidative stress leading to lipid peroxidation and decreases the antioxidant defenses [57–60]. Moreover, VLCFAs are known to favor overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS) on various types of cells, including neuronal cells [35, 55], and VLCFA-induced oxidative damage could further compromise energy metabolism [61]. It was therefore useful to specify the ability of C22:0, C24:0, and C26:0 to favor the overproduction of ROS on whole SK-NB-E cells and at the mitochondrial level. Data obtained on SK-NB-E cells confirm the potential prooxidative activity of VLCFAs and show similar effects with C22:0. Indeed, when ROS overproduction, especially superoxide anion production, was measured with DHE [44], higher percentages of cells overproducing superoxide anions were found under treatment with C22:0, C24:0, and C26:0 compared to untreated cells and α-cyclodextrin-treated cells. As it has been postulated that oxidative stress in mitochondria can reduce their activities and contribute to decreasing the mitochondrial transmembrane potential (ΔΨm) [52, 61], the overproduction of superoxide anions at the mitochondrial level was investigated with the fluorescent probe MitoSOX [45]. In these conditions, our data show that mitochondrial dysfunctions were associated with an increase of superoxide anion production at the mitochondrial level. However, as the increase of MitoSOX-positive cells was lower than those of HE-positive cells, we can assume that C22:0, C24:0, and C26:0 have other targets than mitochondria to stimulate superoxide anion production. It is therefore tempting to speculate that extramitochondrial sources of ROS production, especially superoxide anion, could be activated by C22:0, C24:0, and C26:0 and could in turn contribute to inducing mitochondrial damage [62]. Indeed, the accumulation of VLCFAs in X-ALD lymphoblasts has been described to increase the level of NADPH oxidase gp91PHOX observed in total cell homogenate and membrane fractions of those cells [63, 64]. Therefore, C24:0 and C26:0 might favor the activation of NADPH oxidase activity, leading to an overproduction of ROS, which might further contribute to modifying mitochondrial activity and to triggering cell death [65]. Since it has been demonstrated that excess C26:0 generates ROS, which results in oxidation of mitochondrial enzymes (glycolysis and tricarboxylic acid cycle enzymes) and reduces their activity, contributing in turn to enhancing mitochondrial ROS production [60, 61], C22:0 and C24:0 may activate similar mechanisms. Although by Western blotting, under treatment with C22:0, we did not observe quantitative modifications of the Complex I subunit NDUFB8 (20 kDa), Complex II subunit (30 kDa), Complex III subunit Core 2 (47 kDa), Complex IV subunit II (24 kDa), and Complex V ATP synthase subunit alpha (53 kDa) involved in oxidative phosphorylation, whose defect may result in nonfunctional synapses, axonal degeneration, and cell death [66], this does not exclude oxidative modifications of these complexes, which may not affect the reactivity with the antibodies used. This also does not exclude oxidative modifications on other proteins of these complexes as well as decreased activities of these complexes as observed in hepatocytes of PEX5-deficient mice (a model of peroxisome biogenesis disorders), where a peroxisomal defect leads to lower β-oxidation and accumulation of C24:0 and C26:0 [67]. It is noteworthy that under treatment with C24:0 and C26:0 the decrease of Complex III subunit Core 2 (only with C24:0 (5 μM) and C26:0 (10 μM)) and Complex IV subunit II (both with C24:0 and C26:0 (5 and 10 μM)) observed on SK-NB-E cells was in agreement with observations on fibroblast primary cultures of PEX5-deficient mice where a reduction of the core I subunit of Complex III and subunit I of Complex IV was found [68]. It remains to be determined whether the different effects between C24:0 and C26:0 on Complex III subunit Core 2 are characteristic of these fatty acids or whether they depend not only on their concentrations, but also on the time of treatment considered.
In addition to these functional mitochondrial dysfunctions, some topographical, quantitative, and morphological modifications of the mitochondria were also observed under treatment of SK-NB-E cells with C22:0, C24:0, and C26:0. Thus, using MitoTracker Red, which identifies the cytoplasmic distribution of mitochondria by fluorescence microscopy [45], important changes were found under treatment with C22:0, C24:0, and C26:0. In contrast to the fluorescent dots often observed in control and α-cyclodextrin-treated cells, a more diffuse and irregular staining pattern was detected in the presence of C22:0, C24:0, and C26:0. Moreover, with flow cytometry the higher fluorescence intensity of MitoTracker Red observed under treatment with C22:0, C24:0, and C26:0, compared to control and α-cyclodextrin-treated cells, favors the hypothesis of an increase of the mitochondrial mass, recently reported with the use of this fluorochrome on cells submitted to chemical or physical stresses [69, 70], which could correspond to either a higher number of mitochondria and/or the presence of mitochondria of larger sizes. Interestingly, these two possibilities are supported by the observations from transmission electron microscopy. Since fusion and fission of mitochondria are strictly controlled in neurons, malfunction in this process triggered by VLCFAs may contribute to activating neuronal death [48, 71]. In addition, as described on cardiac myocytes in stress conditions [72], the biogenesis of mitochondria under treatment with C22:0, C24:0, and C26:0 could occur to compensate the loss of mitochondrial activity and to maintain the stability of mitochondrial metabolism. Moreover, in agreement with this hypothesis, on eicosapentaenoic acid-treated rat glioma C6 cells, it has been suggested that enhancement of mitochondrial biogenesis through enhanced PGC1-α and Tfam transcriptional activities may provide partial protection against cytotoxicity [73].
Altogether, our data indicate that VLCFAs (C22:0, C24:0, and C26:0) could constitute potential risk factors in AD. They could favor neurodegeneration by inducing neuronal damages via their ability to induce mitochondrial dysfunctions. Therefore, as C22:0, C24:0, and C26:0 have been identified in the brain of patients with AD, they could contribute to the development of this pathology, and the mitochondria may constitute a rational target for therapeutic intervention [74]. Therefore, the ability to counteract the associated mitochondrial dysfunctions might help identify new therapeutic targets and develop efficient treatments in AD.
Acknowledgments
This work was supported by grants from the INSERM, the Université de Bourgogne (Dijon, France), the Université de Monastir (Monastir, Tunisia), and the Conseil Régional de Bourgogne.
References
- 1.Burns A, Iliffe S. Alzheimer’s disease. British Medical Journal. 2009;338, article b158 doi: 10.1136/bmj.b158. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England Journal of Medicine. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52(1):3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annual Review of Neuroscience. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. Journal of Neurochemistry. 2008;104(6):1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Experimental Neurology. 2010;223(2):322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Hartmann T, Kuchenbecker J, Grimm MO. Alzheimer’s disease: the lipid connection. Journal of Neurochemistry. 2007;103:159–170. doi: 10.1111/j.1471-4159.2007.04715.x. [DOI] [PubMed] [Google Scholar]
- 8.Grösgen S, Grimm MOW, Frieß P, Hartmann T. Role of amyloid beta in lipid homeostasis. Biochimica et Biophysica Acta. 2010;1801(8):966–974. doi: 10.1016/j.bbalip.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi H. Lipid metabolism and glial lipoproteins in the central nervous system. Biological and Pharmaceutical Bulletin. 2011;34(4):453–461. doi: 10.1248/bpb.34.453. [DOI] [PubMed] [Google Scholar]
- 10.- Seripa D, D’Onofrio G, Panza F, Cascavilla L, Masullo C, Pilotto A. The genetics of the human APOE polymorphism. Rejuvenation Research. 2011;14(5):491–500. doi: 10.1089/rej.2011.1169. [DOI] [PubMed] [Google Scholar]
- 11.Bekris LM, Yu CE, Bird TD, Tsuang DW. Review article: genetics of Alzheimer disease. Journal of Geriatric Psychiatry and Neurology. 2010;23(4):213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Javitt NB. Oxysterols: novel biologic roles for the 21st century. Steroids. 2008;73(2):149–157. doi: 10.1016/j.steroids.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Leoni V, Caccia C. Oxysterols as biomarkers in neurodegenerative diseases. Chemistry and Physics of Lipids. 2011;164(6):515–524. doi: 10.1016/j.chemphyslip.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Russell DW. Oxysterol biosynthetic enzymes. Biochimica et Biophysica Acta. 2000;1529(1–3):126–135. doi: 10.1016/s1388-1981(00)00142-6. [DOI] [PubMed] [Google Scholar]
- 15.Schroepfer GJ., Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiological Reviews. 2000;80(1):361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- 16.Vejux A, Lizard G. Cytotoxic effects of oxysterols associated with human diseases: induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Molecular Aspects of Medicine. 2009;30(3):153–170. doi: 10.1016/j.mam.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochemical Society Transactions. 2005;33(5):1033–1036. doi: 10.1042/BST20051033. [DOI] [PubMed] [Google Scholar]
- 18.Vaya J, Schipper HM. Oxysterols, cholesterol homeostasis, and Alzheimer disease. Journal of Neurochemistry. 2007;102(6):1727–1737. doi: 10.1111/j.1471-4159.2007.04689.x. [DOI] [PubMed] [Google Scholar]
- 19.Solomon A, Leoni V, Kivipelto M, et al. Plasma levels of 24S-hydroxycholesterol reflect brain volumes in patients without objective cognitive impairment but not in those with Alzheimer’s disease. Neuroscience Letters. 2009;462(1):89–93. doi: 10.1016/j.neulet.2009.06.073. [DOI] [PubMed] [Google Scholar]
- 20.Russell DW, Halford RW, Ramirez DMO, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annual Review of Biochemistry. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown J, III, Theisler C, Silberman S, et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. The Journal of Biological Chemistry. 2004;279(33):34674–34681. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- 22.Prasanthi JRP, Huls A, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on β-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Molecular Neurodegeneration. 2009;4(1, article 1) doi: 10.1186/1750-1326-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. Journal of Internal Medicine. 2006;260(6):493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- 24.Björkhem I, Cedazo-Minguez A, Leoni V, Meaney S. Oxysterols and neurodegenerative diseases. Molecular Aspects of Medicine. 2009;30(3):171–179. doi: 10.1016/j.mam.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Terlecky SR, Koepke JI, Walton PA. Peroxisomes and aging. Biochimica et Biophysica Acta. 2006;1763(12):1749–1754. doi: 10.1016/j.bbamcr.2006.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Titorenko VI, Terlecky SR. Peroxisome metabolism and cellular aging. Traffic. 2011;12(3):252–259. doi: 10.1111/j.1600-0854.2010.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santos MJ, Quintanilla RA, Toro A, et al. Peroxisomal proliferation protects from β-amyloid neurodegeneration. The Journal of Biological Chemistry. 2005;280(49):41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- 28.Cimini A, Moreno S, D’Amelio M, et al. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: a role for peroxisomes. Journal of Alzheimer’s Disease. 2009;18(4):935–952. doi: 10.3233/JAD-2009-1199. [DOI] [PubMed] [Google Scholar]
- 29.Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annual Review of Biochemistry. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 30.Lizard G, Rouaud O, Demarquoy J, Cherkaoui-Malki M, Iuliano L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. Journal of Alzheimer’s disease. 2012;29(2):241–254. doi: 10.3233/JAD-2011-111163. [DOI] [PubMed] [Google Scholar]
- 31.Kou J, Kovacs GG, Höftberger R, et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathologica. 2011;122(3):271–283. doi: 10.1007/s00401-011-0836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eichler FS, Ren JQ, Cossoy M, et al. Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Annals of Neurology. 2008;63(6):729–742. doi: 10.1002/ana.21391. [DOI] [PubMed] [Google Scholar]
- 33.Hein S, Schönfeld P, Kahlert S, Reiser G. Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Human Molecular Genetics. 2008;17(12):1750–1761. doi: 10.1093/hmg/ddn066. [DOI] [PubMed] [Google Scholar]
- 34.Baarine M, Ragot K, Athias A, et al. Incidence of Abcd1 level on the induction of cell death and organelle dysfunctions triggered by very long chain fatty acids and TNF-α on oligodendrocytes and astrocytes. Neurotoxicology. 2012;33(2):212–228. doi: 10.1016/j.neuro.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Baarine M, Andréoletti P, Athias A, et al. Evidence of oxidative stress in very long chain fatty acid—treated oligodendrocytes and potentialization of ROS production using RNA interference-directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience. 2012;213:1–18. doi: 10.1016/j.neuroscience.2012.03.058. [DOI] [PubMed] [Google Scholar]
- 36.Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radical Biology and Medicine. 2011;51(5):1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swerdlow RH. Brain aging, Alzheimer’s disease, and mitochondria. Biochimica et Biophysica Acta. 2011;1812(12):1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lezi E, Swerdlow RH. Mitochondria in neurodegeneration. Advances in Experimental Medicine and Biology. 2012;942(2):269–286. doi: 10.1007/978-94-007-2869-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh I, Kishimoto Y. Effect of cyclodextrins on the solubilization of lignoceric acid, ceramide, and cerebroside, and on the enzymatic reactions involving these compounds. Journal of Lipid Research. 1983;24(5):662–665. [PubMed] [Google Scholar]
- 40.Takemoto Y, Suzuki Y, Horibe R, et al. Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain and Development. 2003;25(7):481–487. doi: 10.1016/s0387-7604(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 41.Lizard G, Gueldry S, Deckert V, Gambert P, Lagrost L. Evaluation of the cytotoxic effects of some oxysterols and of cholesterol on endothelial cell growth: methodological aspects. Pathologie Biologie. 1997;45(4):281–290. [PubMed] [Google Scholar]
- 42.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 43.Lemaire-Ewing S, Prunet C, Montange T, et al. Comparison of the cytotoxic, pro-oxidant and pro-inflammatory characteristics of different oxysterols. Cell Biology and Toxicology. 2005;21(2):97–114. doi: 10.1007/s10565-005-0141-2. [DOI] [PubMed] [Google Scholar]
- 44.Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′-dichlorofluorescin. Journal of Leukocyte Biology. 1990;47(5):440–448. [PubMed] [Google Scholar]
- 45.Haugland RP. Molecular Probes Handbook, a Guide to Fluorescent Probes and Labeling Technologies. 11th edition. Molecular Probes, Invitrogen, Life Technologies; 2010. edited by I. Johnson and M. T. Z. Spence. [Google Scholar]
- 46.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Analytical Biochemistry. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 47.Lizard G, Fournel S, Genestier L, et al. Kinetics of plasma membrane and mitochondrial alterations in cells undergoing apoptosis. Cytometry. 1995;21(3):275–283. doi: 10.1002/cyto.990210308. [DOI] [PubMed] [Google Scholar]
- 48.Kahn E, Baarine M, Dauphin A, et al. Impact of 7-ketocholesterol and very long chain fatty acids on oligodendrocyte lipid membrane organization: evaluation via LAURDAN and FAMIS spectral image analysis. Cytometry A. 2011;79(4):293–305. doi: 10.1002/cyto.a.21017. [DOI] [PubMed] [Google Scholar]
- 49.Marshall NJ, Goodwin CJ, Holt SJ. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation. 1995;5(2):69–84. [PubMed] [Google Scholar]
- 50.Lizard G, Deckert V, Dubrez L, Moisant M, Gambert P, Lagrost L. Induction of apoptosis in endothelial cells treated with cholesterol oxides. American Journal of Pathology. 1996;148(5):1625–1638. [PMC free article] [PubMed] [Google Scholar]
- 51.Lenaz G, D’Aurelio M, Merlo Pich M, et al. Mitochondrial bioenergetics in aging. Biochimica et Biophysica Acta. 2000;1459(2-3):397–404. doi: 10.1016/s0005-2728(00)00177-8. [DOI] [PubMed] [Google Scholar]
- 52.Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Frontiers in Aging Neuroscience. 2010;2, article 34 doi: 10.3389/fnagi.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zahm JM, Baconnais S, Monier S, et al. Chronology of cellular alterations during 7-ketocholesterol-induced cell death on A7R5 rat smooth muscle cells: analysis by time lapse-video microscopy and conventional fluorescence microscopy. Cytometry A. 2003;52(2):57–69. doi: 10.1002/cyto.a.10027. [DOI] [PubMed] [Google Scholar]
- 54.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A. 2011;79(6):405–425. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 55.Fourcade S, López-Erauskin J, Galino J, et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Human Molecular Genetics. 2008;17(12):1762–1773. doi: 10.1093/hmg/ddn085. [DOI] [PubMed] [Google Scholar]
- 56.Galea E, Launay N, Portero-Otin M, et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochimica et Biophysica Acta. 2012;1822(9):1475–1488. doi: 10.1016/j.bbadis.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Vargas CR, Wajner M, Sirtori LR, et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochimica et Biophysica Acta. 2004;1688(1):26–32. doi: 10.1016/j.bbadis.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Deon M, Sitta A, Barschak AG, et al. Oxidative stress is induced in female carriers of X-linked adrenoleukodystrophy. Journal of the Neurological Sciences. 2008;266(1-2):79–83. doi: 10.1016/j.jns.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 59.Deon M, Sitta A, Barschak AG, et al. Induction of lipid peroxidation and decrease of antioxidant defenses in symptomatic and asymptomatic patients with X-linked adrenoleukodystrophy. International Journal of Developmental Neuroscience. 2007;25(7):441–444. doi: 10.1016/j.ijdevneu.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 60.Galino J, Ruiz M, Fourcade S, et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid Redox Signal. 2011;15(8):2095–2107. doi: 10.1089/ars.2010.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. The Journal of Biological Chemistry. 2001;276(26):23357–23361. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- 62.Fourcade S, Ruiz M, Guilera C, et al. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Human Molecular Genetics. 2010;19(10):2005–2014. doi: 10.1093/hmg/ddq082. [DOI] [PubMed] [Google Scholar]
- 63.Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Human Molecular Genetics. 2002;11(5):499–505. doi: 10.1093/hmg/11.5.499. [DOI] [PubMed] [Google Scholar]
- 64.Uto T, Contreras MA, Gilg AG, Singh I. Oxidative imbalance in nonstimulated X-adrenoleukodystrophy-derived lymphoblasts. Developmental Neuroscience. 2009;30(6):410–418. doi: 10.1159/000191212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pujol A, Ferrer I, Camps C, et al. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Human Molecular Genetics. 2004;13(23):2997–3006. doi: 10.1093/hmg/ddh323. [DOI] [PubMed] [Google Scholar]
- 66.Breuer ME, Koopman WJ, Koene S, et al. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. doi: 10.1016/j.nbd.2012.03.007. Neurobiology of Disease. In press. [DOI] [PubMed] [Google Scholar]
- 67.Dirkx R, Vanhorebeek I, Martens K, et al. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology. 2005;41(4):868–878. doi: 10.1002/hep.20628. [DOI] [PubMed] [Google Scholar]
- 68.Baumgart E, Vanhorebeek I, Grabenbauer M, et al. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse) American Journal of Pathology. 2001;159(4):1477–1494. doi: 10.1016/S0002-9440(10)62534-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1α expression. The Journal of Biological Chemistry. 2010;285(1):142–152. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tornatore TF, Dalla Costa AP, Clemente CFMZ, et al. A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. American Journal of Physiology. 2011;300(3):H902–H912. doi: 10.1152/ajpheart.00319.2010. [DOI] [PubMed] [Google Scholar]
- 71.Chang DTW, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Progress in Neurobiology. 2006;80(5):241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 72.Ahuja P, Zhao P, Angelis E, et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. Journal of Clinical Investigation. 2010;120(5):1494–1505. doi: 10.1172/JCI38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeng JY, Lee WH, Tsai YH, Chen CY, Chao SY, Hsieh RH. Functional modulation of mitochondria by eicosapentaenoic acid provides protection against ceramide toxicity to C6 glioma cells. Journal of Agricultural and Food Chemistry. 2009;57(24):11455–11462. doi: 10.1021/jf902021h. [DOI] [PubMed] [Google Scholar]
- 74.Reddy PH. Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectrums. 2009;14(8, supplement 7):8–13. doi: 10.1017/s1092852900024901. [DOI] [PMC free article] [PubMed] [Google Scholar]