

Abstract
The P2Y1 receptor (P2Y1R) is a G protein-coupled receptor naturally activated by extracellular ADP. Its stimulation is an essential requirement of ADP-induced platelet aggregation, thus making antagonists highly sought compounds for the development of antithrombotic agents. Here, through a virtual screening campaign based on a pharmacophoric representation of the common characteristics of known P2Y1R ligands and the putative shape and size of the receptor binding pocket, we have identified novel antagonist hits of µM affinity derived from a N,N’-bis-arylurea chemotype. Unlike the vast majority of known P2Y1R antagonists, these drug-like compounds do not have a nucleotidic scaffold or highly negatively charged phosphate groups. Hence, our compounds may provide a direction for the development of receptor probes with altered physicochemical properties.
Keywords: P2Y1 receptor, G protein-coupled receptor, antagonist, virtual screening, molecular modeling
1. Introduction
The P2Y receptor (P2YR) family comprises eight subtypes of G protein-coupled receptors (GPCRs) that are activated by extracellular nucleotides.1 Two subtypes, namely the Gq-coupled P2Y1R and the Gi-coupled P2Y12R, play a prominent role in platelet aggregation, since their stimulation by adenosine 5’-diphosphate (ADP) synergistically mediates a proaggregatory response.2 Since the simultaneous activation of both receptors is required to elicit aggregation, antagonists of either subtype act as effective antithrombotic agents. In particular, antagonists of the P2Y12R, such as liver-activated clopidogrel (Plavix®), are already widespread clinical tools, while antagonists of the P2Y1R are still under study as an alternative platform for the development of antiaggregatory drugs.3, 4 With the exception of a few compounds recently identified by researchers at GlaxoSmithKline5 and Pfizer,6 most of the known P2Y1R antagonists are based on the structure of adenosine-3',5'-bisphosphate (A3P5P).7, 8 For example, the selective antagonist (1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane (MRS2500), compound 1 (Figure 1), is used widely as a pharmacological probe of the P2Y1R. It contains a substituted ribose ring moiety, i.e. a methanocarba ring system (bicyclo[3.1.0]hexane) that is constrained in the P2Y1R-preferring Northern (N) conformation.9 Notably, compound 1 was shown to be effective in reducing thrombus formation in vivo and to be more resistant to degradation than related 9-riboside derivatives.10
Figure 1.

Representation of the pharmacophore at the base of the virtual screening, embedded within the homology model of the P2Y1R: the hydrogen bond donor feature is in light purple; its projection is in dark purple; the aromatic or hydrogen bond acceptor feature is in indigo; the negatively charged or hydrogen bond acceptor features are in pink. The ribbon representation of the backbone of the receptor is colored with a continuum spectrum going from red at the N-terminus to purple at the C-terminus, with TM1 in orange, TM2 in yellow/green, TM3 in green, TM4 in blue/green, TM5 in cyan, TM6 in blue and TM7 in purple. The high affinity antagonist MRS2500 (1) appears in the figure, with its carbon atoms in yellow, while a schematic 2D representation is shown in the lower left inset. Three cationic residues that, according to experimental evidence, are fundamental for ligand recognition are also shown, with their carbon atoms in grey.
Although a crystal structure of the P2Y1R in complex with its ligands has not been solved, we have extensively studied the molecular recognition at the P2Y1R using a combination of site-directed mutagenesis, structure-activity relationships (SARs), and molecular modeling based on the known structure of homologue GPCRs.1, 8, 11–13 In the course of these studies, we have docked several nucleotide antagonists to the putative ligand binding site of the P2Y1R. Here, in the quest for P2Y1R antagonists with more drug-like characteristics, we generated a three-dimensional pharmacophore that reflected the common chemical features shown by these docked known antagonists. We then performed a pharmacophore-based virtual screen, which led us to the discovery of novel antagonists derived from non-nucleotide chemical scaffolds.
2. Results and Discussion
2.1. Construction of the pharmacophore
We have recently published a docking-based QSAR analysis of 53 in-house developed nucleotide-based antagonists of the P2Y1R.8 On the basis of the docked conformations of said compounds, here we generated a pharmacophore that reflected their common chemical characteristics as well as the shape of the putative ligand binding pocket in our P2Y1R homology model (Figure 1). The number, size and position of the pharmacophoric features were then adjusted in order to maximize the number of known active compounds able to pass the pharmacophoric search, leading to a final pharmacophoric query consisting of: a) a hydrogen-bond donor feature with a radius of 1 Å corresponding to the exocyclic amino group of the known bisphosphate antagonists, supplemented by a projected feature with a radius of 1.4 Å - this was necessary to ensure that the H-bond donor feature maintained the orientation observed in the docked P2Y1antagonists; b) an aromatic or hydrogen bond acceptor feature with a radius of 2.6 Å, corresponding to the adenine moiety of the known antagonists; c) six negatively charged or hydrogen-bond acceptor features with a radius of 1.3 Å, corresponding to the phosphate moieties of the known antagonists. The criteria for the successful passing of the pharmacophore search by a given compound were defined as the essential matching of the hydrogen-bond donor feature and its projection, and the additional matching of four of the remaining features. The shape of the P2Y1R binding pocket was taken into account by adding to the pharmacophore query an excluded volume corresponding to the residues that line it in our model. These regions were treated as impenetrable by the ligands. Under these conditions, all the abovementioned 53 known inhibitors were successfully recognized as ligands through the pharmacophore search.
2.2. Virtual screening
A database of approximately 250,000 commercially available compounds was then virtually screened according to the scheme represented in Figure 2. Prior to the pharmacophore search, the database was subjected to a descriptor-based filtering to rapidly eliminate the compounds devoid of the characteristics necessary to match the pharmacophore. In particular, we eliminated all the compounds endowed with less than one hydrogen bond donor and less than four hydrogen bond acceptors (or one aromatic ring and three hydrogen bond acceptors). The resulting filtered database, amounting to about half of the original size, was then subjected to the pharmacophore search and yielded 362 virtual hits. Aiming at testing about one hundred compounds, we subjected these virtual hits to a molecular fingerprint-based clustering, which yielded 51 clusters of molecules with similar structures and 59 unique singletons. We then selected a total of 110 molecules, i.e. all 59 singletons and a representative compound for each cluster, choosing the member that in the pharmacophore search yielded the lowest root mean square deviation (RMSD) with the query.
Figure 2.

Schematic representation of the virtual screening process.
2.3. Biological evaluation of the virtual hits and identification of novel ligands
The selected compounds were evaluated for their ability to inhibit the binding of a P2Y1 antagonist radioligand in membranes of human (h) P2Y1R-expressing Sf9 insect cells. The experimental screening, which was initially conducted studying the inhibition of radioligand binding by a 20 µM concentration of the tested compounds, yielded the identification of several novel ligand hits with ≥30% inhibition of [125I]MRS2500 binding (2 – 4).16 Among them, N,N’-bis-arylurea 2a was found to have an affinity in the low µM range, thus constituting a very good starting basis for hit optimization. In a subsequent search after the chemical modification of 2a was initiated, we tested commercially available analogs of compound 2a and found two structurally related compounds, 2b and 2c, of comparable affinity. The structure of other tentative library hits (5 – 10) that inhibited only 20% of radioligand binding are shown in Supporting Information (Table S1).
2.4. Synthesis and biological evaluation of analogs of compound 2a
As mentioned, before the identification of the activity of compounds 2b and 2c we conducted a preliminary exploration of the SAR of the novel antagonists by synthesizing analogs of compound 2a. Compound 2a was also resynthesized in order to confirm the structure provided. In particular, taking advantage of the commercial availability of several aryl isocyanates, we replaced the dichloro-substituted phenyl ring of 2a with six different disubstituted analogs. The central phenylsulfonamido or isoxazole rings remained unmodified. The synthetic route for the preparation of various urea derivatives 2a and 15a-i is shown in Scheme 1. A coupling reaction between 5-amino-3,4-dimethylisoxazole 11 and p-nitrobenzenesulfonyl chloride 12 generated the nitro compound 13 in 41% yield. Our efforts to reduce the nitro group under Pd/C catalyzed hydrogenation reaction conditions resulted in the nitro reduction along with reduction of the less substituted double bond of the 3,4-dimethylisoxazol-5-yl moiety (results not shown). However, the nitro group was chemo-selectively reduced using Zn dust in acetic acid to obtain the amine 14 in 69% yield. The amine 14 was then used as a common intermediate to generate the various urea derivatives 2a and 15a-i, by reacting with the corresponding isocyanates.
Scheme 1.

The compounds were evaluated for their ability to inhibit P2Y1 antagonist radioligand binding and to increase intracellular Ca2+ concentration. Selected compounds were found to inhibit 2-MeSADP-stimulated phospholipase C (PLC) signaling. The binding was measured in membranes of hP2Y1R-expressing Sf9 insect cells, and the functional assays were performed in hP2Y1R-expressing 1321N1 human astrocytoma cells. The results, shown in Table 2, indicated that several of the in-house synthesized analogs of 2a bind to the P2Y1R with an affinity in the low µM range, although none of the compounds appeared to be a better binder than the parent compound. Moreover, the data show that antagonism of P2Y1R-induced effects was present. In particular, when administered at a concentration of 10 µM, the compounds endowed with higher affinity for the receptor inhibited about 30 % of the P2Y1R-mediated calcium mobilization induced by the agonist 2-MeSADP. The only exception appears to be compound 15b that, despite a relatively high binding affinity, seemed to be a weaker antagonist.
Table 2.
In vitro pharmacological data for the synthesized analogs of compound 2a at the hP2Y1R in binding, determined as in Table 1, and in the antagonism increase of intracellular calcium ion concentration induced by 30 nM 2-MeSADP in 1321N1 astrocytoma cells expressing the hP2Y1R.
 | |||
|---|---|---|---|
| Compound No. | R | Binding Ki, µM |
% inhibition in Ca2+ assay at 10 µM |
| 2a (F3099-0019) |  |
13 ± 3 | 36.8 ± 2.2a |
| 15 a | NE | < 5 | |
| 15b |  |
27 ± 10 | 17.6 ± 1 |
| 15c |  |
≥ 100 | 21.7 ± 3.5 |
| 15d |  |
42 ± 15 | 31.3 ± 2.5 |
| 15e |  |
NE | 11.3 ± 2.3 |
| 15f | 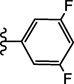 |
≥ 100 | 10.2 ± 2.6 |
| 15g | 45 ± 8 | 32.7 ± 3.7 | |
| 15h | 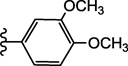 |
NE | 9.1 ± 0.4 |
| 15i | 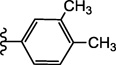 |
NE | 9.4 ± 0.2b |
IC50 in the IP Assay: 0.14 ± 0.04 µM;
IC50 in the IP Assay: 0.13 ± 0.01 µM; NE – less than 10% inhibition at 20 µM.
2.5. Analysis of the SAR data
The SAR data indicate that the presence of chlorine substituents at the meta and para positions of the phenyl ring is crucial for the affinity and the antagonistic activity of compound 2a. The removal of both chlorine substituents (compound 15a) from compound 2a leads to a complete loss of affinity. Even the sole removal of the chlorine at the para position (compound 15c) leads to a substantial loss of affinity, which however is partially regained when a second chlorine atom is reintroduced at the other meta position (compound 15b). The substitution of the chlorine atoms with an electron withdrawing trifluoromethyl group (compound 15g) is tolerated by the receptor, while their substitution with electron donating methyl or methoxy groups is not (compounds 15h and 15i). Moreover, the substitution of the chlorine in para with a fluorine (compound 15d), although somewhat detrimental, is tolerated. However, the substitution of both chlorines with fluorines leads to a complete loss of affinity (compound 15e and 15f). Taken together, these data suggest that the region of the receptor that surrounds the phenyl ring is not inclined to the recognition of electron rich moieties, thus supporting the structural results of our pharmacophore search that placed the disubstituted phenyl ring of compound 2a distantly from the negatively charged phosphate groups of 1. In particular, our pharmacophore search suggests that the ureic nitrogen of 2a closer the dichlorophenyl ring mimics the exocyclic amino group of 1, while the 5-sulfonamido-isoxazole moiety of 2a substitutes the two phosphates groups of 1 (Figure 3). According to our published models of the P2Y1R in complex with 1, the phosphates of this nucleotide antagonist bind to a portion of the binding cavity lined by cationic residues, which are notoriously electrophilic and fluorophilic residues.14 Compound 2a was more drug-like in its physicochemical properties (cLog p = 3.79) than antagonists having negatively-charged phosphate groups.
Figure 3.

Superimposition of MRS2500 (1) and compound 2, as resulting from the pharmacophore search. MRS2500 is shown in ball and stick representation, with the carbon atoms colored in green. Compound 2 is shown in stick representation, with the carbon atoms colored in grey.
3. Conclusions
In conclusion, our pharmacophore-based virtual screening led to the identification of novel P2Y1R antagonists endowed with drug-like structures. One of the more promising hits, a N,N’-bis-phenylurea 2a, from the in silico screen was validated by de novo synthesis and the preparation of closely related analogues to generate family of non-nucleotide P2Y1 antagonists, some of which displayed affinities and antagonistic potencies in the low µM range. Thus, these molecules represent good candidates for a follow-up hit optimization study. Our initial attempts to explore the SAR of the phenyl ring, did not improve the potency as antagonists, but shed light onto the requirements of ligand recognition. Moreover, there are additional sites on the molecules, such as the isoxazole ring, that can be explored, with additional information on other heterocycles from library hit compounds 2b and 2c.
The novel P2Y1 antagonists identified through this work lack negatively charged phosphate groups, thus providing more suitable scaffolds for the development of development of receptor probes with different physicochemical properties from the canonical A3P5P-based antagonists. However, being devoid of ionized groups, these novel antagonists demonstrate that the ionic interactions that the nucleotide antagonists establish with the P2Y1R are not essential for ligand recognition. Our pharmacophore model suggests that the phosphates of nucleotide antagonists are replaced by 5-sulfonamido-isoxazole moiety of the novel antagonists, which most likely establish hydrogen-bonds and cation-aromatic interactions with the receptor. The confirmation that these substances bind as suggested by the pharacophore model will depend on the identification of analogues with enhanced affinity. These molecules may now be subjected to further structural optimization and more extensive pharmacological characterization in platelet aggregation and other models.
4. Materials and methods
4.1. Molecular Modeling
The molecular modeling study was performed with the Molecular Operating Environment (MOE), Chemical Computing Group, Inc (www.chemcomp.com). The molecular database subjected the virtual screening process was the catalogue of compounds commercially available from Life Chemicals, Inc. (Burlington, ON, Canada, www.lifechemicals.com).
4.1.1. Construction of the pharmacophore
The pharmacophore query was generated with the "pharmacophore query editor" of MOE. A set of 53 in house-developed A3P5P-based P2Y1 antagonists8 were loaded into MOE, and an initial query was generated using the "Consensus" function, according to the PCHD scheme. Only the phosphate groups, the purine ring, and the exocyclic amino group were taken into account. The resulting query was then simplified by unifying the aromatic/hydrogen bond acceptor feature relative to the purine ring and deleting all the projected features. However, we preserved the projected donor feature relative to the exocyclic amine, in order to ensure its directionality. Moreover, an excluded volume was added on the basis of all the atoms of the residues lining the binding pocket in our rhodopsin-based model of the P2Y1R,8 thus accounting for its shape and size. Specifically, the generation of the excluded volume was based on the following residues, indicated through their sequence number as well as their GPCR residue index: L54(1.35), V57(1.38), Y58(1.39), V61(1.42), Y100(2.53), L105(2.57), L108(2.61), R128(3.29), F131(3.32), H132(3.33), L135(3.36), K196(EL2.44), N197(EL2.45), I200(EL2.48), T201(EL2.49), Y203(EL2.51), D204(EL2.52), F276(6.51), H277(6.52), K280(6.55), N283(6.58), Q307(7.36), R310(7.39), G311(7.40), S314(7.43) – for more information on the GPCR residue index, see Ballesteros and Weinstein15 and Costanzi and coworkers.1 Finally, we tested the generated pharmacophore query for its ability to correctly recognize the 53 known antagonists and adjusted the size and the position of the features in order to ensure the correct recognition of the entire set of the known ligands. For this test, the 53 known antagonists where sketched from scratch in MOE and subjected to the same conformational search as the Life Chemicals database, thus recreating the condition used in the actual pharmacophore search.
4.1.2. Descriptor-based filtering of the database
The number of hydrogen bond acceptors, hydrogen bond donors and aromatic atoms was calculated for all compounds in the Life Chemicals database with the "calculate descriptors" function of MOE. To expedite the screening, we then deleted all the compounds that did not the have the necessary features to match the pharmacophore query.
4.1.3. Conformational explosion
The resulting filtered Life Chemicals database was then subjected to a conformational search with the "Conformation Import" function of MOE, in order to generate for each compound multiple conformers to be evaluated in the pharmacophore search. The maximum number of conformations per compound was set to 250, and the maximum strain energy for a conformation to be accepted was set to 100 kcal/mol. A screenshot with the complete settings is provided in Figure S1 of the Supplementary Data.
4.1.4. Fingerprint-based clustering
The compounds that passed the pharmacophore search were clustered on the basis of their molecular similarity according to two different fingerprints: the MACCS structural keys and the Typed Graph Triangle (TGT) fingerprint. Both fingerprints are conformation-independent and can be calculated from a two-dimensional representation of the molecules. The molecular similarity was calculated according to the Tanimoto coefficient, and the clustering was done setting the threshold for similarity and overlap to 80% for the MACCS and 88% for the TGT fingerprints. From the clusters independently calculated with the two fingerprints, a consensus cluster was then generated with a union operation via an ad hoc written script.
4.2. Chemical synthesis
The synthetic procedures for intermediates are described here, while analytical and spectroscopic data for each compound tested biologically are given below. Compounds subjected to biological testing were of >95% purity. For structural confirmation, 1H NMR spectra were recorded at 300 Mz. Chemical shifts are reported in parts per million (ppm) relative to deuterated solvent as the internal standard. ESI-High resolution mass spectroscopic measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. cLog p was calculated using ChemBioDraw Ultra v. 12.03 (PerkinElmer, Boston, MA).
N-(3,4-Dimethylisoxazol-5-yl)-4-nitrobenzenesulfonamide (13)
5-Amino-3,4-dimethylisoxazole 11 (0.20 g, 1.78 mmol) and p-nitrobenzenesulfonyl chloride 12 (0.39 g, 1.78 mmol) were dissolved in anhydrous CH2Cl2 (20 mL). Et3N (0.75 mL, 5.36 mmol) and N,N-dimethylaminopyridine (44 mg, 0.36 mmol) were then added. After stirring the reaction mixture for 42 h, it was diluted with EtOAc (50 mL) and washed with sat. aq. NaHCO3 (1 × 20 mL) and sat. aq. NaCl (1 × 20 mL). The aqueous phase was back extracted with EtOAc (3 × 20 mL), and the combined organic phases were evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in EtOAc, v/v) to afford compound 13 (0.22 g, 41%) as a solid.
4.2.1. 4-Amino-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (14)
Nitro compound 13 (0.71 g, 2.40 mmol) was dissolved in a mixture of AcOH:CH3CN (20 mL, 1:1, v/v) and cooled to 0 °C. Zn powder (1.5 g) was added in small portions over a period of 10 min. After stirring the reaction for 3 h at rt, reaction mixture was filtered off and filtrate was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in EtOAc, v/v) to afford amine 14 (0.65 g, 69%) as a solid.
4.2.2. General experimental procedure for compounds 2a and 15a-i
Method A
Amine 14 (25 mg, 0.094 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous THF (2 mL). Diisopropylethylamine (66 µL, 0.38 mmol) and the corresponding isocyanate (0.140 mmol) were added. After stirring for 2 h at 70 °C, the reaction mixture was diluted with EtOAc (25 mL) and washed with saturated aq. NaCl (1 × 15 mL). The aqueous phase was back extracted with EtOAc (3 × 20 mL), and the combined organic phases were evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in EtOAc, v/v) to afford target compounds 2a and 15b, 15d-f, and 15h-i, as solid materials.
Method B
Amine 14 (15 mg, 0.056 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous THF (2 mL). Diisoproylethylamine (40 µL, 0.224 mmol) and corresponding isocyanate (0.224 mmol) were added. After stirring for 16 h at room temperature, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–6% MeOH in EtOAc, v/v) to afford target compounds 15a, 15c and 15g as solid materials.
4.2.2.1. 4-(3-(3,4-Dichlorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (2a)
Rf = 0.5 (10% MeOH in EtOAc, v/v); ESI-HRMS m/z 455.0331 ([M + H]+, C18H16N4O4SCl2·H+: Calcd. 455.0348); 1H NMR (CD3OD) δ 7.81 (d, J = 2.8 Hz, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.63 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.8 Hz, 1H), 7.31 (dd, J = 2.8, 8.8 Hz, 1H), 2.14 (s, 3H), 1.78 (s, 3H).
4.2.2.2. N-(3,4-Dimethylisoxazol-5-yl)-4-nitrobenzenesulfonamide (13)
Rf = 0.2 (10% MeOH in EtOAc, v/v); ESI-HRMS m/z 298.0492 ([M + H]+, C11H11N3O5S·H+: Calcd. 298.0498); 1H NMR (CDCl3) δ 8.37 (d, J = 8.8 Hz, 2H), 8.01 (d, J = 8.8 Hz, 2H), 2.22 (s, 3H), 1.98 (s, 3H), 1.57 (s, 1H).
4.2.2.3. 4-Amino-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (14)
Rf = 0.4 (7% MeOH in EtOAc, v/v); ESI-HRMS m/z 268.0767 ([M + H]+, C11H13N3O3S·H+: Calcd. 268.0756); 1H NMR (CDCl3) δ 7.56 (d, J = 8.8 Hz, 2H), 6.64 (d, J = 8.8 Hz, 2H), 4.19 (s, 2H), 2.18 (s, 3H), 1.91 (s, 3H), 1.59 (s, 1H).
4.2.2.3.4. N-(3,4-Dimethylisoxazol-5-yl)-4-(3-phenylureido)benzenesulfonamide (15a)
Rf = 0.2 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 387.1118 ([M + H]+, C18H18N4O4S·H+: Calcd. 387.1127); 1H NMR (CD3OD) δ, 7.72 (d, J = 9.1 Hz, 2H), 7.64 (d, J = 9.1 Hz, 2H), 7.40–7.48 (m, 2H), 7.27–7.34 (m, 2H), 7.02–7.07 (m, 1H), 2.16 (s, 3H), 1.79 (s, 3H).
4.2.2.3.5. 4-(3-(3,5-Dichlorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15b)
Rf = 0.2 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 455.0348 ([M + H]+, C18H16N4O4SCl2·H+: Calcd. 455.0348); 1H NMR (CD3OD) δ 7.93 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 1.7 Hz, 2H), 7.06–7.08 (m, 1H), 2.07 (s, 3H), 1.19 (s, 3H).
4.2.2.3.6. 4-(3-(3-Chlorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15c)
Rf = 0.3 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 421.0725 ([M + H]+, C18H17N4O4SCl·H+: Calcd. 421.0737); 1H NMR (CD3OD) δ, 7.74 (d, J = 8.1 Hz, 2H), 7.60–7.69 (m, 3H), 7.23–7.32 (m, 2H), 7.02–7.07 (m, 1H), 2.16 (s, 3H), 1.79 (s, 3H).
4.2.2.3.7. 4-(3-(3-Chloro-4-fluorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15d)
Rf = 0.2 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 439.0641 ([M + H]+, C18H16N4O4FClS·H+: Calcd. 439.0643); 1H NMR (CD3OD) δ 7.70–7.80 (m, 3H), 7.65 (d, J = 8.8 Hz, 2H), 7.20–7.30 (m, 1H), 7.05–7.15 (m, 1H), 2.13 (s, 3H), 1.79 (s, 3H).
4.2.2.3.8. 4-(3-(3,4-Difluorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15e)
Rf = 0.2 (7% MeOH in EtOAc, v/v); ESI-HRMS m/z 423.0924 ([M + H]+, C18H16N4O4SF2·H+: Calcd. 423.0939); 1H NMR (CD3OD) δ 7.76 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.18–7.21 (m, 2H), 7.09–7.12 (m, 1H), 2.16 (s, 3H), 1.77 (s, 3H).
4.2.2.3.9. 4-(3-(3,5-Difluorophenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15f)
Rf = 0.2 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 423.0934 ([M + H]+, C18H16N4O4F2S·H+: Calcd. 423.0939); 1H NMR (CD3OD) δ 7.75 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.2 Hz, 2H), 6.54–6.61 (m, 1H), 2.13 (s, 3H), 1.78 (s, 3H).
4.2.2.3.10. N-(3,4-Dimethylisoxazol-5-yl)-4-(3-(4-(trifluoromethyl)phenyl)ureido)-benzenesulfonamide (15g)
Rf = 0.2 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 387.1118 ([M + H]+, C19H17N4O4S·H+: Calcd. 387.1127); 1H NMR (CD3OD) δ, 7.73 (d, J = 8.6 Hz, 2H), 7.60–7.68 (m, 4H), 7.58 (d, J = 8.7 Hz, 2H), 2.15 (s, 3H), 1.79 (s, 3H).
4.2.2.3.11. 4-(3-(3,4-Dimethoxyphenyl)ureido)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (15h)
Rf = 0.2 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 447.1343 ([M + H]+, C20H23N4O6S·H+: Calcd. 447.1338); 1H NMR (CD3OD) δ 7.72 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.22 (d, J = 2.2 Hz, 1H), 6.83–6.92 (m, 2H), 3.84 (s, 3H), 3.81 (s, 3H), 2.13 (s, 3H), 1.77 (s, 3H).
4.2.2.3.12. N-(3,4-Dimethylisoxazol-5-yl)-4-(3-(3,4-dimethylphenyl)ureido)benzenesulfonamide (15i)
Rf = 0.2 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 415.1447([M + H]+, C20H22N4O4S·H+: Calcd. 415.1440); 1H NMR (CD3OD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.11–7.21 (m, 2H), 7.04 (d, J = 8.2 Hz, 1H), 2.24 (s, 3H), 2.21 (s, 3H), 2.14 (s, 3H), 1.77 (s, 3H).
4.3. Pharmacological evaluation
4.3.1. Radioligand binding assay
Binding of [125I]MRS2500, a potent, selective, antagonist of the P2Y1R was performed as reported elsewhere.16, 17 2-MeSADP was purchased from Sigma (St. Louis, MO). Myo-[3H]inositol (20 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO).
4.3.2. Assay of inositol phosphates
Selected compounds were examined for inhibition of P2Y1R-promoted stimulation of inositol phosphate formation measured at the hP2Y1R stably expressed in 1321N1 human astrocytoma cells as previously described.8, 18, 19 The IC50 values were averaged from 3 independently determined concentration-effect curves for each compound. Briefly, cells plated in 24-well dishes were labeled in inositol-free medium (DMEM; Gibco, Gaithersburg, MD) containing 1.0 µCi of 2-[3H]myo-inositol (20 Ci/mmol; American Radiolabeled Chemicals, Inc., St. Louis, MO) for 18–24 h in a humidified atmosphere of 95% air / 5% CO2 at 37°C. PLC activity was measured the following day by quantitating [3H]inositol phosphate accumulation after a 10 min incubation at 37°C in the presence of 10 mM LiCl. Total [3H]inositol phosphates were quantified by anion exchange chromatography as previously described.
4.3.3. Calcium mobilization assay
Changes in intracellular calcium ion concentration induced by the hP2Y1R stably expressed in 1321N1 human astrocytoma cells were measured as reported.20 Specifically, 1321N1 human astrocytoma cells stably expressing the hP2Y1R were cultured in DMEM (JRH Biosciences, Inc., Lenexa, KS) supplemented with 5% fetal bovine serum, 100 units penicillin mL−1, 100 µg streptomycin mL−1, 2 µmol glutamine mL−1, and 800 µg geneticin mL−1. For the assay, cells were grown overnight in 100 µL of media in 96-well flat-bottom plates at 37 °C at 5% CO2 or until approximately 60–80% confluency. The calcium 4 assay kit (Molecular Devices, Sunnyvale, CA) was used as directed without washing of the cells. Cells were loaded with 30 µL of dye to each well and incubated for 1 h at room temperature. All the tested compounds were diluted to the appropriate concentration in Hank’s buffer. Dose-response curves for the agonist 2-MeSADP were obtained treating the cells with six different concentration of the compound in the range between (10−9 M to 10−5 M). For the antagonist studies, the same dose-response curves were registered in the presence of the newly identified P2Y1 ligands, pretreating the cells for 20 min at room temperature with a 10 µM concentration of each candidate antagonist before application of the agonist 2-MeSADP. Samples were run in triplicates using a Molecular Devices FLIPRTETRA at room temperature. Cell fluorescence (excitation = 485 nm, emission = 525 nm) was monitored following exposure to the compound. Increases in intracellular calcium are reported as the maximum fluorescence value after exposure minus the basal fluorescence value before exposure.
Binding and functional parameters were estimated using GraphPAD Prism software (GraphPAD, San Diego, CA).
Supplementary Material
Table 1.
In vitro pharmacological data for compounds selected through an in silico screen (Life Chemicals code shown) for inhibition of radioligand binding at the hP2Y1R.
| Compound No. | Structure | Binding Ki, µM, or % inhibitiona |
|---|---|---|
| 2a, F3099-0019 | 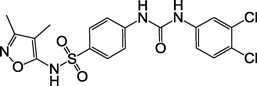 |
13 ± 3 |
| 2b, F0348-3162 | 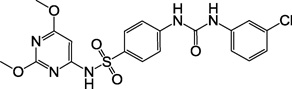 |
6 |
| 2c, F1766-0041 | 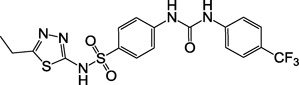 |
10 |
| 3, F2019-1559 | 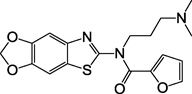 |
40 %b |
| 4, F1795-0008 | 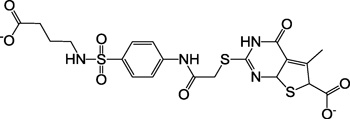 |
30 %b |
Assays were with membranes from Sf9 insect cells expressing the hP2Y1R and performed with a radioligand ([125I]MRS2500) concentration of approximately 0.2 nM.
Percent inhibition of specific binding of [125I]MRS2500 by a 20 µM concentration of test compound.
Acknowledgements
This research was supported in part by the Intramural Research Program of NIDDK and by the National Institutes of General Medical Sciences (GM38213).
Abbreviations
- ADP
adenosine 5’-diphosphate
- A3P5P
adenosine-3',5'-bisphosphate
- DMEM
Dulbecco’s modified Eagle medium
- ESI-HRMS
electrospray ionization-high resolution mass spectroscopy
- GPCR
G protein-coupled receptor
- 2-MeSADP
2-methylthio-adenosine 5’-diphosphate
- MOE
Molecular Operating Environment
- MRS2500
(1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane
- PLC
phospholipase C
- QSAR
quantitative structure activity relationship
- RMSD
root mean square deviation
- THF
tetrahydrofuran
- TGT
Typed Graph Triangle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
A screenshot with the settings for the conformational explosion of the screened database of compounds and additional data concerning tentative hits can be found in the online version.
References
- 1.Costanzi S, Mamedova L, Gao Z, Jacobson K. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J. Med. Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gachet C, Hechler B. The platelet P2 receptors in thrombosis. Semin. Thromb. Hemost. 2005;31:162–167. doi: 10.1055/s-2005-869521. [DOI] [PubMed] [Google Scholar]
- 3.Shankar H, Kunapuli SP. Is the P2Y1 receptor a better target for antithrombotic drugs? Drug Discov. Today. 2005;2:285–290. [Google Scholar]
- 4.Jacobson K, Boeynaems J. P2Y nucleotide receptors: promise of therapeutic applications. Drug Discov. Today. 2010;15:570–578. doi: 10.1016/j.drudis.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morales-Ramos A, Mecom J, Kiesow T, Graybill T, Brown G, Aiyar N, Davenport E, Kallal L, Knapp-Reed B, Li P, Londregan A, Morrow D, Senadhi S, Thalji R, Zhao S, Burns-Kurtis C, Marino JJ. Tetrahydro-4-quinolinamines identified as novel P2Y1 receptor antagonists. Bioorg. Med. Chem. Lett. 2008;18:6222–6226. doi: 10.1016/j.bmcl.2008.09.102. [DOI] [PubMed] [Google Scholar]
- 6.Pfefferkorn J, Choi C, Winters T, Kennedy R, Chi L, Perrin L, Lu G, Ping Y, McClanahan T, Schroeder R, Leininger M, Geyer A, Schefzick S, Atherton J. P2Y1 receptor antagonists as novel antithrombotic agents. Bioorg. Med. Chem. Lett. 2008;18:3338–3343. doi: 10.1016/j.bmcl.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 7.Boyer J, Romero-Avila T, Schachter J, Harden T. Identification of competitive antagonists of the P2Y1 receptor. Mol. Pharmacol. 1996;50:1323–1329. [PubMed] [Google Scholar]
- 8.Costanzi S, Tikhonova I, Ohno M, Roh E, Joshi B, Colson A, Houston D, Maddileti S, Harden T, Jacobson K. P2Y1 antagonists: combining receptor-based modeling and QSAR for a quantitative prediction of the biological activity based on consensus scoring. J. Med. Chem. 2007;50:3229–3241. doi: 10.1021/jm0700971. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, Ohno M, Xu B, Kim H, Choi Y, Ji X, Maddileti S, Marquez V, Harden T, Jacobson K. 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a northern conformation: enhanced potency as P2Y1 receptor antagonists. J. Med. Chem. 2003;46:4974–4987. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hechler B, Nonne C, Roh E, Cattaneo M, Cazenave J, Lanza F, Jacobson K, Gachet C. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba 2'-deoxyadenosine 3',5'-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J. Pharmacol. Exp. Ther. 2006;316:556–563. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov A, Costanzi S, Jacobson K. Defining the nucleotide binding sites of P2Y receptors using rhodopsin-based homology modeling. J. Comput. Aided Mol. Des. 2006;20:417–426. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- 12.Ohno M, Costanzi S, Kim H, Kempeneers V, Vastmans K, Herdewijn P, Maddileti S, Gao Z, Harden T, Jacobson K. Nucleotide analogues containing 2-oxa-bicyclo[2.2.1]heptane and l-alpha-threofuranosyl ring systems: interactions with P2Y receptors. Bioorg. Med. Chem. 2004;12:5619–5630. doi: 10.1016/j.bmc.2004.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Castro S, Maruoka H, Hong K, Kilbey SM, 2nd, Costanzi S, Hechler B, Brown GG, Jr, Gachet C, Harden TK, Jacobson KA. Functionalized congeners of P2Y1 receptor antagonists: 2-alkynyl (N)-methanocarba 2'-deoxyadenosine 3',5'-bisphosphate analogues and conjugation to a polyamidoamine (PAMAM) dendrimer carrier. Bioconjug. Chem. 2010;21:1190–1205. doi: 10.1021/bc900569u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 15.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Stuart CS, editor. Methods in Neurosciences. Vol. Volume 25. Academic Press; 1995. pp. 366–428. [Google Scholar]
- 16.Ohlmann P, de Castro S, Brown GG, Jr, Gachet C, Jacobson KA, Harden TK. Quantification of recombinant and platelet P2Y1 receptors utilizing a [125I]-labeled high-affinity antagonist 2-iodo-N6-methyl-(N)-methanocarba-2'-deoxyadenosine-3',5'-bisphosphate ([125I]MRS2500) Pharmacol. Res. 2010;62:344–351. doi: 10.1016/j.phrs.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waldo GL, Corbitt J, Boyer JL, Ravi G, Kim HS, Ji XD, Lacy J, Jacobson KA, Harden TK. Quantitation of the P2Y1 receptor with a high affinity radiolabeled antagonist. Mol. Pharmacol. 2002;62:1249–1257. doi: 10.1124/mol.62.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes CP. Phosphoinositide hydrolysis by guanosine 5'-[gamma-thio]triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem. J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyer JL, Downes CP, Harden TK. Kinetics of activation of phospholipase C by P2Y purinergic receptor agonists and guanine nucleotides. J. Biol. Chem. 1989;264:884–890. [PubMed] [Google Scholar]
- 20.Balasubramanian R, Ruiz de Azua I, Wess J, Jacobson KA. Activation of distinct P2Y receptor subtypes stimulates insulin secretion in MIN6 mouse pancreatic beta cells. Biochem. Pharmacol. 2010;79:1317–1326. doi: 10.1016/j.bcp.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.