

Abstract
Metabolic syndrome and its components are associated with chronic kidney disease (CKD) development. Insulin resistance (IR) plays a central role in the metabolic syndrome and is associated with increased risk for CKD in nondiabetic patients. IR is common in patients with mild-to-moderate stage CKD, even when the glomerular filtration rate is within the normal range. IR, along with oxidative stress and inflammation, also promotes kidney disease. In patients with end stage renal disease, IR is an independent predictor of cardiovascular disease and is linked to protein energy wasting and malnutrition. Systemic inflammation, oxidative stress, elevated serum adipokines and fetuin-A, metabolic acidosis, vitamin D deficiency, depressed serum erythropoietin, endoplasmic reticulum stress, and suppressors of cytokine signaling all cause IR by suppressing insulin receptor-PI3K-Akt pathways in CKD. In addition to adequate renal replacement therapy and correction of uremia-associated factors, thiazolidinedione, ghrelin, protein restriction, and keto-acid supplementation are therapeutic options. Weight control, reduced daily prednisolone dosage, and the use of cyclosporin decrease the risk of developing new-onset diabetes after kidney transplantation. Improved understanding of the pathogenic mechanisms underlying IR in CKD may lead to more effective therapeutic strategies to reduce uremia-associated morbidity and mortality.
1. Introduction
Despite recent advances in the treatment of chronic kidney disease (CKD), it remains an important public health challenge [1]. CKD is a major risk factor for end stage renal disease (ESRD) and cardiovascular disease [2], and cardiovascular disease is the leading cause of mortality in uremic patients. Cardiac mortality in dialysis patients is more than 10-fold greater than that of the general population [2]. The excess cardiovascular mortality in patients with CKD is not explained by traditional risk factors such as hypertension and hypercholesterolemia. Identifying and treating risk factors for early CKD may be the best approach to prevent and delay adverse outcome [3].
The metabolic syndrome (MetS), characterized by abdominal obesity, hypertriglyceridemia, depressed serum high-density lipoprotein cholesterol (HDL-C), high blood pressure, and high fasting glucose level [4], is commonly seen in patients with CKD. Insulin resistance (IR) plays a central role in the MetS and is associated with increased risk for CKD in nondiabetic patients [5, 6]. The MetS and its components are associated with the development of CKD, microalbuminuria, and overt proteinuria [7, 8]. There is a strong association between HDL-C, inflammatory surrogates, and IR in this nondiabetic, nonobese hemodialysis (HD) patient group. In addition, serum HDL-C level is a good screening parameter in high-risk patients [9].
Rodents are widely used to mimic human diseases to improve understanding of the causes and progression of disease symptoms and to test possible therapeutic interventions. For instance, Diabetic Zucker fatty rats (ZDFs), a model of early onset obesity, have a mutation in the leptin receptor gene. Also, Goto-Kakizaki (GK) rodents, which are nonobese to begin with and genetically engineered to have malfunctioned β-cells, can mimic noninsulin-dependent disorder [10]. These models can utilize urine concentration of energy metabolites to evaluate the metabolic syndrome in prediabetic insulin-resistant and hyperglycemia stage. Chronic consumption of a high-carbohydrate, high-fat diet by normal rodents may provide an adequate rodent model to mimic the human metabolic syndrome and for testing potential therapeutic interventions [10, 11].
IR is common in patients with mild-to-moderate CKD [12]. IR is also an independent predictor of cardiovascular mortality in ESRD [13] and is increasing recognized as a nontraditional risk factor [14, 15]. IR is common in patients with ESRD [16] and is linked to protein energy wasting and malnutrition [17, 18]. Nutritional, metabolic, and cardiovascular complications of renal disease may be a consequence of abnormal insulin action [19]. Therefore, IR may be an important therapeutic target for reduction of cardiovascular mortality in patients with CKD.
2. Measurement of IR
There are a number of methods to assess insulin sensitivity in the dynamic versus static state. Estimates of IR based on fasting insulin concentration may not be adequate in patients with CKD as it largely reflects hepatic defects, whereas CKD impairs insulin catabolism [20]. Hyperinsulinemic euglycemic clamp is the gold standard for IR determination because it provides a direct measure of whole-body sensitivity to insulin, primarily skeletal muscle. At lower doses of insulin infusion, the addition of labeled glucose can allow for specific assessment of the ability of insulin to suppress endogenous glucose production. This method can differentiate between peripheral IR and hepatic IR and provides a direct and precise IR measurement [20, 21]. The oral glucose tolerance test (OGTT) primarily measures glucose tolerance, which reflects both IR and beta-cell function; IR can be calculated from OGTT using validated formulae. On the other hand, the hyperglycemic clamp is the gold standard for the measurement of beta-cell function. Due to the complexity of this methodology, more practical methods such as homeostatic model assessment (HOMA) and the quantitative insulin sensitivity check index (QUICKI) are widely used in clinical studies. The HOMA test estimates IR from fasting glucose and insulin concentrations, but primarily reflects hepatic IR rather than peripheral IR. There is an inverse linear correlation between IR by euglycemic hyperinsulinemic clamp and log-transformed IR by HOMA [22]. Therefore, IR by HOMA can be used as an alternative method to assess IR in patients with renal failure.
3. IR in CKD
IR is a well-known complication of ESRD based on hyperinsulinemic euglycemic clamp testing. IR is also present in early stage CKD, even when the glomerular filtration rate (GFR) is within the normal range. Glucose disposal rate (GDR) correlates negatively with serum creatinine level and positively with creatinine clearance [12]. A large prospective study using data from the Atherosclerosis Risk in Communities (ARICs) cohort confirmed a step-wise increase in risk for CKD development with the number of MetS criteria met in nondiabetic adults, ever after controlling for the development of diabetes mellitus (DM) and hypertension [5]. A prospective cohort study found that MetS predicts the risk of prevalent and incident CKD. This same study found IR to be associated with prevalent CKD and rapid decline in renal function among elderly individuals in an older Asian nondiabetic cohort [23]. However, there is little evidence that preventing or treating symptoms of the MetS protects patients with renal impairment. IR, along with oxidative stress and inflammation, is suggested to play a role in the development of albuminuria and declining kidney function [23, 24]. Hyperglycemia may not only cause poor renal function but also lower expression of Klotho protein in diabetic kidney, and these defects could be reversed by correction of blood glucose levels using insulin treatment in diabetic rats [25].
3.1. IR Accentuates Kidney Injury
IR promotes kidney disease by worsening renal hemodynamics through mechanisms such as activation of the sympathetic nervous system [26], sodium retention, decreased Na+, K+-ATPase activity, and increased GFR [24]. Endoplasmic reticulum (ER) stress seems to be the factor-linking inflammation and IR at the molecular level. The suppression of insulin signaling via phosphorylation of the insulin receptor substrate (IRS-1) due to activation of c-Jun N-terminal kinase (JNK) plays an important role [27]. Renal ER stress is associated with proteinuria-induced podocyte damage and alteration of nephrin N-glycosylation in podocytes, which is the underlying factor in the pathogenesis of proteinuria, and is involved in the pathophysiology of chronic kidney injury with tubulointerstitial damage [28]. According to Sowers [29], IR and inflammatory cytokine release may be partly responsible for glomerular mesangial expansion, basement membrane thickening, podocytopathy, and the loss of slit pore diaphragm integrity ultimately leading to glomerulosclerosis and tubulointerstitial injury. Megalin, an endocytic receptor, mediates the conservation of nutrients and carrier bound vitamins in glomerular filtrates via interaction with various molecules in renal proximal tubular epithelial cells (PTECs) [30]. In metabolic syndrome or dyslipidemia, free fatty acids are delivered to PTECs with the carrier proteins such as albumin or liver-type fatty acid-binding protein [31]. Metabolically overloaded PTECs are activated to express proinflammatory cytokines, such as MCP1 and TNFα, and lead to epithelial-mesenchymal transition [32].
IR may drive the overproduction of very low-density lipoprotein cholesterol and contribute to hypertriglyceridemia [36]. Triglyceride-rich apolipoprotein B-containing lipoproteins clearly promote the progression of renal insufficiency [37]. High triglyceride levels are a risk factor for proteinuria development [38].
3.2. IR in CKD Patients with Dialysis Therapy
Adequate HD has a positive effect on IR (Table 1), but there is little clinical data regarding the effect of peritoneal dialysis (PD) on insulin sensitivity. After comparing the effect of continuous cycling PD (CCPD) to that of HD on IR in younger adolescent uremic patients [39], the percentage increase and the final insulin sensitivity were significantly higher in the CCPD group than the HD group. This may be because PD provided better clearance of middle-molecule-weight uremic toxins than HD. In contrast, in 19 nondiabetic patients with ESRD, CAPD therapy for 5.4 weeks normalized IR similar to HD. There is close correlation between changes in blood urea nitrogen (BUN) or HCO3− and IR [34]. The variations in effect on IR with PD may be due to the different modalities used.
Table 1.
Clinical studies assessing IR in healthy controls, CKD patients, and patients receiving PD, HD, or kidney transplantation (KT).
| Patients | n | GDR (mg/kg/min) | HOMA index | ||
|---|---|---|---|---|---|
| Controls [33] | 10 | 9.93 ± 1.33 | 1.85–1.95 | ||
| CKD [12] | 29a | 6.91 ± 2.46 | 2.24 ± 1.06 | ||
|
| |||||
| Before Tx | After Tx | Before KT | After KT | ||
|
| |||||
| With PD [34] | 9a | 6.35 ± 1.65 | 8.18 ± 1.76 | ||
| With HD [34] | 10a | 6.53 ± 1.84 | 9.74 ± 2.88 | ||
| 1 M after KT [35] | 34 | 3.05 ± 2.72 | 1.67 ± 1.25 | ||
| 6 M after KT [35] | 34 | 3.05 ± 2.72 | 2.32 ± 2.83 | ||
CKD: chronic kidney disease; GDR: glucose disposal rate; HOMA: homeostasis model assessment for insulin resistance; ESRD: end stage renal disease; CAPD: continuous ambulatory PD; Tx: dialysis treatment; KT: kidney transplantation.
aNondiabetic.
3.3. IR in CKD Patients with Kidney Transplantation
After renal transplantation, serum leptin level dramatically decreased immediately and correlated significantly with serum insulin level and HOMA index. Serum leptin levels at six months after transplant were significantly higher than that of controls and correlation between serum leptin levels and HOMA index persisted [35]. Five years after transplant, serum leptin, IR, body fat percentage, and serum lipids had a profile similar to those in the pretransplant period. A positive correlation between leptin and IR in the late postrenal transplantation period persisted [40]. Distribution of obesity and prednisolone treatment are predominant determinants of IR in the long term after transplantation [41]. These findings suggest that IR might play a role in increased leptin concentration in renal transplant recipients together with increased body fat mass. IR after transplantation could be managed favorably by weight control in overweight patients and prednisolone dose reduction. Pretransplant glycemia and high posttransplant BUN were associated with a greater risk of posttransplant hyperglycemia [42].
4. Factors Affecting IR in CKD
IR in CKD exists mainly at periphery. As adipose tissue accounts for the disposal of less than 2% of the glucose load, muscle tissue must represent the primary site of IR in patients with ESRD. Hepatic glucose production is not increased and is suppressed normally in response to insulin in patients with ESRD. IR may not lead to a compensatory increase in insulin secretion, as compared to patients without advanced kidney disease [43]. In contrast to animal models with high-fat intake-induced IR, IR is first manifested in the liver and then in white adipose tissue, while skeletal muscle remains insulin sensitive [44]. A postreceptor defect is recognized as the primary defect in CKD. One study demonstrated that IRS-1-associated phosphoinositol (PI) 3-kinase (K) activity is suppressed. The potential mechanism for the reduction in IRS-1-associated PI3-K activity is induction of the p85 PI3-K regulatory subunit, but another mechanism may also be involved [45]. Downstream function of the IRS-PI3 K-Akt pathways was associated with chronic inflammation, metabolic acidosis, vitamin D and parathyroid hormone status, anemia, uremic toxins, and adipokines.
4.1. IR Associated with Inflammation in CKD
A complex network of nutritional and metabolic alterations underlies CKD, including chronic inflammation, oxidative stress, IR, and protein energy wasting. Like other chronic diseases, patient with CKD demonstrates low-grade systemic inflammation marked by elevated levels of proinflammatory cytokines such as C-reactive protein (CRP), tumor necrosis factor alpha (TNF-α), inerleukin-6 (IL-6), and interleukin-1 beta (IL-1β) [46]. Inflammation and oxidative stress are evident in the early stage of CKD [47] and are also known to induce IR, primarily via increased production of proinflammatory cytokines.
In the presence of insulin, the insulin receptor phosphorylates IRS, which is linked to the activation of two main signaling pathways: the phosphatidylinositol 3-kinase (PI3 K)-AKT/protein kinase B (PKB) pathway, which is responsible for most of the metabolic action such as glucose transport, and the Ras-mitogen-activated protein kinase (MAPK) pathway, which regulates gene expression and cooperates with the PI3 K pathway to control cell growth and differentiation [48]. In animal models of CKD, PI3 K activity is reduced and both apoptosis [49], and ATP-ubiquitin-proteasome pathways are activated [50]. This mechanism could increase muscle atrophy in conditions with impaired insulin action.
TNF-α induces IR through direct or indirect mechanisms (Figure 1). TNF-α can induce IRS-1 serine phosphorylation through activation of serine kinases including IKK-β [51], c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), protein kinase C (PKC), Akt (PKB), mammalian target of rapamycin (mTOR), and glycogen synthase kinase 3 (GSK3) [49]. After infusion of TNF-α in healthy humans, IR develops in skeletal muscle that is associated with impaired phosphorylation of Akt substrate 160, leading to dysfunction of glucose transporter 4 (GLUT4) translocation and glucose uptake [52]. Excessive concentrations of TNF-α negatively regulate insulin signaling and whole-body glucose uptake in humans. Additionally, TNF-α stimulates lipolysis and release of free-fatty acids [53]. Free-fatty acids induce intracellular accumulation of diacylglycerol and long-chain acyl CoA, with subsequent protein kinase C activation and dephosphorylation of IRS-1/2 in human skeletal muscle cell. Other studies demonstrate that IL-6 can also inhibit the insulin-signaling pathway at the level of the insulin receptor and IRS-1 interaction [54]. Administration of an IL-1 receptor antagonist in patients with type II DM not only decreases serum CRP and IL-6 levels but also improves insulin sensitivity [55]. Defects in insulin signaling through the protein kinase Akt underlie the development of IR. Akt acts as a nodal point in the control of both the metabolic and pleiotropic effects of insulin. Imbalance among these effects leads to cardiac hypertrophy, fibrosis, and apoptosis, reduced angiogenesis, metabolic remodeling, and altered calcium cycling, all key features of uremic cardiomyopathy [56].
Figure 1.
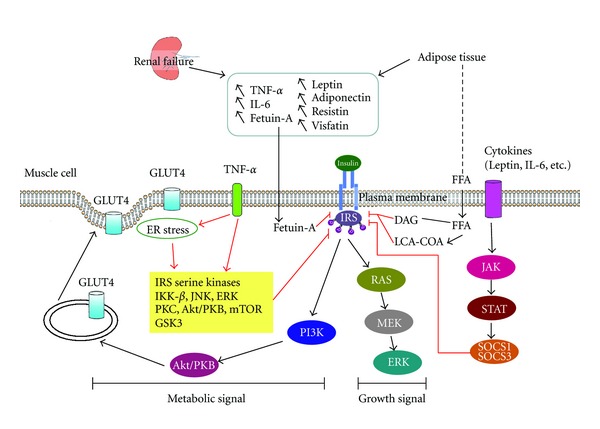
Insulin resistance in chronic kidney disease. Renal failure and adipose tissue lead to systemic inflammation and increased plasma levels of adipokines. TNF-α activates adipose tissue lipolysis, which generates free-fatty acids (FFAs). On muscle cells, FFAs activate transcription factors, such as peroxisome proliferator-activated receptor (PPAR), and generate messengers including diacylglycerol (DAG) and long-chain acyl-CoA (LCA-CoA), which will lead to protein kinase C (PKC) activation and dephosphorylation of insulin receptor substrate (IRS)-1/2. On muscle cells, TNF-α activates a series of kinases including IKK-β, c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), protein kinase C (PKC), Akt (PKB), mammalian target of rapamycin (mTOR), and glycogen synthase kinase 3 (GSK3) responsible for phosphorylation of insulin receptor (InsR) and IRS-1 on serine/threonine residues. Inhibition of IRS-1 function will block Akt leading to cytosol glucose transporter 4 (GLUT4) sequestration. Fetuin-A could also inhibit IRS and induce low-grade inflammation. IL-6 is responsible for the induction of different suppressors of cytokine signaling (SOCS) proteins through the Janus kinase/signal transducer and activator of transcription (Jak/STAT)-signaling pathway. SOCS will inhibit IRS-1/2 and protein kinase A. Endoplasmic reticulum stress seems to be another factor linking inflammation and insulin resistance at the molecular level. Red line illustrates the mechanism of CKD-related factors in IR.
Fetuin-A, also called a2-Heremans-Schmid glycoprotein (Ahsg), is a powerful circulating inhibitor of vascular calcification [57]. CKD patients have significantly lower levels of fetuin-A than healthy controls. Short-term treatment with sevelamer increases serum fetuin-A concentration in stage 4 CKD patients [58]. In dialysis patients, calcimimetic therapy for secondary hyperparathyroidism significantly decreases fetuin-A levels [59]. However, serum fetuin-A levels increased obviously following parathyroidectomy in patients with refractory renal hyperparathyroidism [60]. Fetuin-A inhibits insulin receptor tyrosine kinase activity by blocking autophosphorylation of tyrosine kinase and IRS-1 and induces lower-grade inflammation [61, 62], resulting in IR [63, 64]. Recently, epidemiological studies showed that serum fetuin-A was associated with IR [65] and its comorbidities, such as MetS [66] and type 2 DM [67, 68]. Higher fetuin-A concentrations were associated with type 2 DM and IR in middle-aged and elderly Chinese patients [69]. However, the possible effects of fetuin-A on IR in CKD deserve further elucidation.
Suppressors of cytokine signaling (SOCS) are a family of intracellular proteins, several of which have emerged as key physiological regulators of cytokine-mediated homeostasis, including innate and adaptive immunity. In CKD or ESRD patients, increased expression of SOCS in monocytes and lymphocytes, accompanied by increased plasma levels of the inflammatory cytokines IL-6, CRP, and TNFα, was reported [70, 71]. Recent observations also suggest that SOCS expression levels are also profoundly altered in kidney disease [72].
SOCS proteins act as a classical feedback loop to ensure fine regulation of cytokine-signaling pathways, and the classical target of the SOCS proteins is inhibition of the JAK-STAT-signaling pathway. SOCS proteins can also inhibit insulin signaling; SOCS-1 knock-out mice show hypoglycemia and increased insulin signaling. Recent studies showed that SOCS proteins not only exacerbate IR but also act as an important link between elevated cytokine levels and IR [73]. SOCS-3 can bind phosphorylated Tyr960 on the insulin receptor, which is important for IRS-1 binding [74]. Inhibition of SOCS-1 is likely mediated through binding to the kinase domain of the insulin receptor, preventing further phosphorylation [75]. Targeting of IRS-1 and -2 for ubiquitin-dependent degradation via the proteasomal machinery may be another inhibitory mechanism [76].
Elevated IL-6 induced SOCS-3 expression and consequently inhibited insulin signaling in human differentiated myotubes grown in vitro [77]. SOCS-3 expression was not elevated in muscle from nondiabetic obese and type 1 DM patient. Increased SOCS-3 expression in patients with type 2 DM may be explained by the combination of high glucose and IL-6 levels in the blood. High levels of inflammatory cytokines lead to increased SOCS-1 and -3 expression in insulin sensitive tissues, which induce IR via inhibition of the insulin-signaling pathway.
4.2. Role of Adipokines
Adipokines are markedly elevated in the plasma of uremic patients, mainly due to reduced renal excretion. Obesity superimposed on uremia may further aggravate hyperadipokinemia. Abnormal adipocytokine levels, including leptin and adiponectin, may further promote IR and a proinflammatory state in CKD [78]. The exact pathophysiological role of adipokines [79, 80] in the pathogenesis of IR in CKD is still unclear. Leptin may be considered as a uremia toxin and is associated with reduced energy intake and protein-energy wasting in uremic patients [81]. Leptin signaling in the central nervous system was recently shown to be an important cause of anorexia [82]. Leptin also significantly interferes with neutrophil chemotaxis [83] and produces vascular damage through proatherogenic and proinflammatory effects [84]. Adiponectin could mediate insulin-sensitizing [85, 86], antiatherogenic, and anti-inflammatory actions [87]. However, whether the removal of leptin by dialysis may translate into prospective improvements in appetite, nutrition status, neutrophil chemotaxis, and other clinical benefits in uremic patients still needs to be clarified [88]. Experiments conducted on animal models and in vitro on cell culture demonstrated that adiponectin signaling via MAPK regulates oxidative stress, segmental fusion of podocyte foot processes, and albuminuria [89, 90]. Adiponectin treatment decreased albuminuria, glomerular hypertrophy, and tubulointerstitial fibrosis due to the restoration of VCAM-1, monocyte chemoattractant protein-1, TNF-α, TGF-β1, collagen type I/III, and Nox to the levels found in wild-type mice [91]. In patients on HD, plasma adiponectin levels were inversely related to body mass index values, insulin levels, and HOMA index [92]. Plasma adiponectin is an inverse predictor of cardiovascular outcome. However, the paradoxical unfavorable effect of high adiponectin could be a consequence of a concurrent process of wasting and salt and volume overload.
In humans, resistin is expressed by macrophages residing in adipose tissue. Resistin appears to disturb the immune response and contribute to increased atherosclerotic risk by modulating the activity of endothelial [93]. Circulating resistin levels are strongly associated with both GFR and inflammatory biomarkers in CKD, but not with IR [94]. Visfatin is mainly synthesized in visceral fat and is linked to impaired endothelial function and angiogenesis [95]. In PD patients, serum visfatin might be a sensitive marker for cardiac performance [96]. Chemerin is another novel adipokine linked to obesity and is also increased in uremic patients. In incident dialysis patients, elevated chemerin is associated with a survival advantage despite its significant positive correlation with markers of inflammation and dyslipidemia [97].
Previous study showed that adipose-derived proinflammatory cytokines exert direct inhibitory effects on muscle insulin signaling, accounting for the close link between IR and systemic inflammation [98]. Obesity or abdominal fat accumulation is associated with oxidative stress and impaired insulin action in patients with CKD. There is a plausible relationship between inflammatory biomarkers and truncal fat mass in patients with ESRD. Weight loss may decrease the inflammatory and oxidative burden in CKD and attenuate cardiovascular risk in this population [99].
4.3. Role of Vitamin D and Secondary Hyperparathyroidism
There are evidences that the vitamin D and or parathyroid hormone (PTH) axis is important in the pathogenesis of glucose intolerance and IR in patients with CKD. IR is present in the early stages of CKD and has an inverse association with 25-hydroxyvitamin D levels [100]. Vitamin D metabolism is profoundly disturbed in CKD; abnormalities begin during early CKD stages, that is, stage 3 or earlier, and progress as renal function declines [101]. Vitamin D is now recognized as having pleiotropic roles beyond bone and mineral homeostasis, with vitamin D receptor and metabolizing machinery identified in multiple tissues. Circulating 25-hydroxyvitamin D concentration correlates with glucose tolerance, beta-cell function, and insulin sensitivity, measured using oral glucose tolerance tests and hyperglycemia clamps [102, 103]. Intravenous vitamin D3 corrects glucose intolerance and stimulates insulin secretion in response to a glucose challenge in 1,25-dihydroxyvitamin D3 (1,25(OH)2D3)-deficient animals [104]. After four weeks of intravenous 1,25(OH)2D3 therapy, glucose intolerance, IR, hypoinsulinemia, and hypertriglyceridemia were corrected in patients on HD, in the absence of PTH suppression [105]. A single dose of 1,25(OH)2D3 also improved insulin sensitivity [106].
The mechanisms through which excess PTH blunts insulin sensitivity are uncertain. Medical treatment of hyperparathyroidism in patients with CKD led to correction of glucose intolerance and increased insulin secretion [107]. In a CKD animal model, PTH did not affect tissue resistance to insulin, and the normalization of glucose metabolism in the absence of PTH was due to increased insulin secretion [108]. One study indicates that PTH could stimulate calcium influx into cells and that increased cellular calcium concentration enhances insulin secretion [109]. Contrary to some published data, peripheral blood mononuclear cells from patients with CKD had higher cytosolic calcium concentration and intracellular calcium stores, and long-term vitamin D3 supplement can only normalize the cytosolic calcium concentration without any effect on intracellular calcium store in the absence of PTH [110]. Calcitriol treatment of patients on HD with secondary hyperparathyroidism is associated with increased insulin secretion; this is linked to decreased intracellular free calcium [111]. It is possible that the effect of altered calcium content in beta cells on insulin secretion depends on the magnitude and duration of the change. Recent evidence indicates that the ER mediates cytosolic calcium concentration, and ER stress is a central feature of IR. Cytosolic calcium levels or calcium modulation of the signal transduction of the insulin receptor might play a central role in the mechanisms of IR [112].
4.4. Other Clinical Conditions in CKD
Accumulation of uremic toxins may contribute to IR in ESRD. The middle-molecular range peptides in uremic serum induce IR in adipose cells [113]. Pseudouridine [65], which accumulates in the circulation, and asymmetric dimethyl arginine (ADMA) [114] are recognized as an important uremic toxins associated with CKD and IR. Urea, which was previously considered to have negligible toxicity, induced reactive oxygen species generation and caused IR in an animal model [115].
Metabolic acidosis may also play a significant role in IR in CKD. It is linked to protein-energy malnutrition and inflammation. Evidence suggests that the catabolic effects of metabolic acidosis may be due to increased activity of the adenosine triphosphate (ATP)-dependent ubiquitin-proteasome and branched-chain ketoacid dehydrogenase [116]. Treatment of metabolic acidosis with oral bicarbonate increased both insulin sensitivity and insulin secretion in patients with uremia measured by hyperinsulinemic euglycemic clamp [117].
Patients with ESRD generally have low-exercise tolerance, and this may contribute to IR and metabolic abnormalities [118]. Moderate endurance training programs improved some metabolic abnormalities and insulin sensitivity. Correction of anemia by erythropoietin (EPO) therapy in HD reportedly improves exercise tolerance. Significant improvements in glucose tolerance and insulin sensitivity have been found in CKD patients after six [65] or nine months [119] of EPO therapy for anemia. Improvement of exercise tolerance following EPO treatment may be the important factor in the improvement in glucose metabolism in patients with CKD.
5. Treatment of IR in CKD
IR is highly prevalent in CKD patients and is associated with a complex network of nutritional and metabolic alterations including chronic systemic inflammation, oxidative stress, and protein-energy wasting. Whether IR is antecedent to CKD or a consequence of impaired kidney function has been the subject of debate. The goal of IR treatment is traditionally aimed at etiologies including uremic toxins, protein catabolism, vitamin D deficiency, metabolic acidosis, anemia, poor physical fitness, and cachexia.
Thiazolidinediones (TZDs) are a class of oral diabetic medications that function via binding peroxisome proliferator-activated receptor γ, thereby increasing insulin sensitivity in peripheral tissue [120]. Evidence demonstrates that TZD therapy can alter cardiovascular risk factors leading to increased HDL-C and circulating adiponectin, decreased triglycerides, visceral adiposity, circulating inflammatory mediators, albuminuria, improved flow-mediated vasodilation, and blunted carotid intima-media thickening. Observations suggest that improvement in glycemic control is not the dominant operative mechanism for the effects of TZD. In patients with noninsulin-dependent DM on HD, TZD treatment was associated with significantly lower all-cause mortality among insulin-free but not insulin-requiring patients [121]. Although a meta-analysis suggested that TZD was associated with increased cardiovascular mortality [122], no conclusions can be drawn regarding the association between TZD and nonfatal cardiovascular events in dialysis patients.
Vitamin D supplementation for the general population may lead to a small but significant improvement in mortality but did not appear to prevent the development of DM in the largest clinical trial to date [123]. Data from the HD population consistently suggest that calcitriol improves short-term insulin secretion and insulin sensitivity in ESRD [105, 111]. This may be due to the fact that patients with CKD have profound vitamin D deficiency at baseline, and vitamin D therapy may be most effective when intrinsic 1-a hydroxylase activity is most impaired. Further long-term clinical trials in the setting of CKD are warranted to test this hypothesis.
IR was at least partially correctable by maintenance HD after EPO therapy. Treatment of malnutrition by intravenous nutrition [124] or low-protein diet supplement with amino acid-keto acid supplements improves IR. Recent studies show that dairy proteins and meat stimulate insulin/insulin-like growth factor-1 signaling and provide high amounts of leucine, a primary and independent stimulator for mTORC1 activation. The downstream target of mTORC1, kinase S6 K1, induces IR by phosphorylation of IRS-1. Attenuation of leucine-mediated mTORC1 signaling by defining appropriate upper limits of the daily intake of leucine-rich proteins were advocated for the prevention of type II DM and obesity [125]. However, this cannot apply to the advanced CKD patients as leucine, and essential amino acids have a critically important role in muscle protein turnover. Although there is some resistance to leucine in CKD, leucine-rich supplements in the management of uremic muscle wasting were suggested [126]. IR in children with CKD improved during treatment with dietary protein restriction and essential amino acid and keto-acid supplement. These metabolic changes were not related to hyperparathyroidism and were probably due to a reduction in nitrogen toxicity [127].
Early detection of MetS or high triglyceride levels might be beneficial if accompanied by early intervention such as fibrate therapy to lower triglyceride levels and suppress pathways for renal injury [128]. However, studies showed a controversial role of hypertriglyceridemia in CKD. Patients with hypertriglyceridemia had a relative risk of 2.01 for nephropathy, and higher risk of CKD was found in patients with triglyceride levels greater than 200 mg/dl [129]. The Helsinki Heart Study, a randomized clinical trial of gemfibrozil for the prevention of coronary heart disease, found no association between triglyceride levels and CKD [130].
Sympathetic hyperactivity, activation of the renin-angiotensin-aldosterone system (RAAS), and the related development of hypertension are implicated in CKD; this may be further aggravated by excess dietary salt intake and effects of IR and hyperinsulinemia on sodium retention [78]. IR in patients on maintenance HD was ameliorated after 12 weeks of treatment with angiotensin II receptor blockers [131]. However, insulin per se may have a modest anti-inflammatory effect as evidenced by a reduction in serum CRP that appears to have a persistent effect over the 24 h after dialysis [132].
Ghrelin is a gastric hormone [133] discovered in its acylated form and extensively studies for its appetite-stimulating effect. Evidence shows that total circulating ghrelin is positively associated with insulin sensitivity in general [134] and in patients with CKD. Nonobese, nondiabetic HD patients may present with higher circulating total ghrelin [135]. In a nephrectomy animal model, two weeks of subcutaneous ghrelin administration reduced loss of muscle mass, associated with enhanced muscle transcription of PPAR-γ coactivator 1-α and anti-inflammatory effects [136]. Ghrelin may be considered a novel potential therapeutic agent to effectively counteract CKD-related loss of lean body mass.
Lowering daily prednisolone toward 5 mg/day has beneficial effects on insulin action after renal transplantation, but withdrawal of 5 mg prednisolone may not influence IS significantly [137]. New onset DM after renal transplantation increases the risk of morbidity and mortality. The use of cyclosporine rather than tacrolimus decreases the risk of developing new-onset DM after transplantation, while the potential long-term benefits of this strategy require further study [138].
6. Conclusion
IR is prevalent in CKD patients and plays a role in declining renal function. The etiology of IR is multifactorial and associated with a complex network including chronic inflammation, oxidative stress, vitamin D deficiency, anemia, and malnutrition. These factors are associated with elevated inflammatory cytokines, adipokines, ER stress, and SOCS, leading to an acquired defect of the insulin receptor-signaling pathway. TZD treatment is associated with significantly reduced all-cause mortality and higher serum albumin among an insulin-free population but is used sparingly due to possible side effects. Proper nutritional supplementation and prevention of uremia-associated complications including dyslipidemia, vitamin D and EPO deficiency, and anemia may also improve insulin sensitivity among the CKD population. Ghrelin may be a novel intervention to enhance appetite in malnourished CKD patients. Further studies with larger patient populations are necessary to clarify long-term effects of these treatments. More novel approaches to improving IR in the CKD population might lead to potential strategies for preventing excess mortality.
Authors' Contributions
M.-T. Liao and C.-C. Sung contributed equally to this work.
Acknowledgments
The Research Fund from Cardinal-Tien Hospital (CTH-99-1-2A06) and Teh-Tzer Study Group for Human Medical Research Foundation (B-99-10-53) supported this study.
References
- 1.Kasiske BL, Chavers B, Foley R, et al. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. American Journal of Kidney Diseases. 2002;39(2) supplement 1:S1–S266. [PubMed] [Google Scholar]
- 2.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. American Journal of Kidney Diseases. 1998;32(5):S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 3.Muntner P, He J, Hamm L, Loria C, Whelton PK. Renal insufficiency and subsequent death resulting from cardiovascular disease in the United States. Journal of the American Society of Nephrology. 2002;13(3):745–753. doi: 10.1681/ASN.V133745. [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM, Becker D, Clark LT, et al. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) Journal of the American Medical Association. 2001;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 5.Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. Journal of the American Society of Nephrology. 2005;16(7):2134–2140. doi: 10.1681/ASN.2005010106. [DOI] [PubMed] [Google Scholar]
- 6.Ryu S, Chang Y, Woo HY, et al. Time-dependent association between metabolic syndrome and risk of CKD in Korean men without hypertension or diabetes. American Journal of Kidney Diseases. 2009;53(1):59–69. doi: 10.1053/j.ajkd.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Gu D, Chen CS, et al. Association between the metabolic syndrome and chronic kidney disease in Chinese adults. Nephrology Dialysis Transplantation. 2007;22(4):1100–1106. doi: 10.1093/ndt/gfl759. [DOI] [PubMed] [Google Scholar]
- 8.Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD. Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clinical Journal of the American Society of Nephrology. 2011;6(10):2364–2373. doi: 10.2215/CJN.02180311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng YS, Chiu YL, Chen HY, et al. Decreased high-density lipoprotein cholesterol is associated with inflammation and insulin resistance in non-diabetic haemodialysis patients. Nephrology. 2010;15(7):692–699. doi: 10.1111/j.1440-1797.2010.01295.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhao LC, Zhang XD, Liao SX, Wang HY, Lin DH, Gao HC. A metabonomic comparison of urinary changes in Zucker and GK rats. Journal of Biomedicine and Biotechnology. 2010;2010:6 pages. doi: 10.1155/2010/431894.431894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panchal SK, Brown L. Rodent models for metabolic syndrome research. Journal of Biomedicine and Biotechnology. 2011;2011:14 pages. doi: 10.1155/2011/351982.351982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi S, Maesato K, Moriya H, Ohtake T, Ikeda T. Insulin resistance in patients with chronic kidney disease. American Journal of Kidney Diseases. 2005;45(2):275–280. doi: 10.1053/j.ajkd.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 13.Shinohara K, Shoji T, Emoto M, et al. Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. Journal of the American Society of Nephrology. 2002;13(7):1894–1900. doi: 10.1097/01.asn.0000019900.87535.43. [DOI] [PubMed] [Google Scholar]
- 14.Becker BN, Himmelfarb J, Henrich WL, Hakim RM. Reassessing the cardiac risk profile in chronic hemodialysis patients: a hypothesis on the role of oxidant stress and other non-traditional cardiac risk factors. Journal of the American Society of Nephrology. 1997;8(3):475–486. doi: 10.1681/ASN.V83475. [DOI] [PubMed] [Google Scholar]
- 15.Cheung AK, Sarnak MJ, Yan G, et al. Atherosclerotic cardiovascular disease risks in chronic hemodialysis patients. Kidney International. 2000;58(1):353–362. doi: 10.1046/j.1523-1755.2000.00173.x. [DOI] [PubMed] [Google Scholar]
- 16.DeFronzo RA, Alvestrand A, Smith D. Insulin resistance in uremia. Journal of Clinical Investigation. 1981;67(2):563–568. doi: 10.1172/JCI110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fouque D, Kalantar-Zadeh K, Kopple J, et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney International. 2008;73(4):391–398. doi: 10.1038/sj.ki.5002585. [DOI] [PubMed] [Google Scholar]
- 18.Barazzoni R, Gortan Cappellari G, Zanetti M, Guarnieri G. Ghrelin and muscle metabolism in chronic uremia. Journal of Renal Nutrition. 2012;22(1):171–175. doi: 10.1053/j.jrn.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Mak RHK, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron. 1992;61(4):377–382. doi: 10.1159/000186953. [DOI] [PubMed] [Google Scholar]
- 20.Pham H, Utzschneider KM, De Boer IH. Measurement of insulin resistance in chronic kidney disease. Current Opinion in Nephrology and Hypertension. 2011;20(6):640–646. doi: 10.1097/MNH.0b013e32834b23c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. The American Journal of Physiology. 1979;237(3):E214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 22.Shoji T, Emoto M, Nishizawa Y. HOMA index to assess insulin resistance in renal failure patients. Nephron. 2001;89(3):348–349. doi: 10.1159/000046098. [DOI] [PubMed] [Google Scholar]
- 23.Cheng H-T, Huang J-W, Chiang C-K, Yen C-J, Hung K-Y, Wu K-D. Metabolic syndrome and insulin resistance as risk factors for development of chronic kidney disease and rapid decline in renal function in elderly. Journal of Clinical Endocrinology and Metabolism. 2012;97(4):1268–1276. doi: 10.1210/jc.2011-2658. [DOI] [PubMed] [Google Scholar]
- 24.Gluba A, Mikhailidis DP, Lip GY, et al. Metabolic syndrome and renal disease. doi: 10.1016/j.ijcard.2012.01.013. International Journal of Cardiology. In press. [DOI] [PubMed] [Google Scholar]
- 25.Cheng M-F, Chen L-J, Cheng J-T. Decrease of Klotho in the kidney of streptozotocin-induced diabetic rats. Journal of Biomedicine and Biotechnology. 2010;2010:7 pages. doi: 10.1155/2010/513853.513853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowe JW, Young JB, Minaker KL. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes. 1981;30(3):219–225. doi: 10.2337/diab.30.3.219. [DOI] [PubMed] [Google Scholar]
- 27.Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 28.Cybulsky AV. Endoplasmic reticulum stressin proteinuric kidney disease. Kidney International. 2010;77(3):187–193. doi: 10.1038/ki.2009.389. [DOI] [PubMed] [Google Scholar]
- 29.Sowers JR. Metabolic risk factors and renal disease. Kidney International. 2007;71(8):719–720. doi: 10.1038/sj.ki.5002006. [DOI] [PubMed] [Google Scholar]
- 30.Saito A, Sato H, Iino N, Takeda T. Molecular mechanisms of receptor-mediated endocytosis in the renal proximal tubular epithelium. Journal of Biomedicine & Biotechnology. 2010;2010:7 pages. doi: 10.1155/2010/403272.403272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oyama Y, Takeda T, Hama H, et al. Evidence for megalin-mediated proximal tubular uptake of L-FABP, a carrier of potentially nephrotoxic molecules. Laboratory Investigation. 2005;85(4):522–531. doi: 10.1038/labinvest.3700240. [DOI] [PubMed] [Google Scholar]
- 32.Winbanks CE, Darby IA, Kelynack KJ, Pouniotis D, Becker GJ, Hewitson TD. Explanting is an ex vivo model of renal epithelial-mesenchymal transition. Journal of Biomedicine and Biotechnology. 2011;2011:7 pages. doi: 10.1155/2011/212819.212819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esteghamati A, Ashraf H, Esteghamati AR, et al. Optimal threshold of homeostasis model assessment for insulin resistance in an Iranian population: the implication of metabolic syndrome to detect insulin resistance. Diabetes Research and Clinical Practice. 2009;84(3):279–287. doi: 10.1016/j.diabres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi S, Maejima S, Ikeda T, Nagase M. Impact of dialysis therapy on insulin resistance in end-stage renal disease: comparison of haemodialysis and continuous ambulatory peritoneal dialysis. Nephrology Dialysis Transplantation. 2000;15(1):65–70. doi: 10.1093/ndt/15.1.65. [DOI] [PubMed] [Google Scholar]
- 35.Kayacan SM, Yildiz A, Kazancioglu R, Sahin S, Sever MS, Ark E. The changes in serum leptin, body fat mass and insulin resistance after renal transplantation. Clinical Transplantation. 2003;17(1):63–68. doi: 10.1034/j.1399-0012.2003.02078.x. [DOI] [PubMed] [Google Scholar]
- 36.Tsimihodimos V, Dounousi E, Siamopoulos KC. Dyslipidemia in chronic kidney disease: an approach to pathogenesis and treatment. American Journal of Nephrology. 2008;28(6):958–973. doi: 10.1159/000144024. [DOI] [PubMed] [Google Scholar]
- 37.Samuelsson O, Attman PO, Knight-Gibson C, et al. Complex apolipoprotein B-containing lipoprotein particles are associated with a higher rate of progression of human chronic renal insufficiency. Journal of the American Society of Nephrology. 1998;9(8):1482–1488. doi: 10.1681/ASN.V981482. [DOI] [PubMed] [Google Scholar]
- 38.Tozawa M, Iseki K, Iseki C, Oshiro S, Ikemiya Y, Takishita S. Triglyceride, but not total cholesterol or low-density lipoprotein cholesterol levels, predict development of proteinuria. Kidney International. 2002;62(5):1743–1749. doi: 10.1046/j.1523-1755.2002.00626.x. [DOI] [PubMed] [Google Scholar]
- 39.Mak RHK. Insulin resistance in uremia: effect of dialysis modality. Pediatric Research. 1996;40(2):304–308. doi: 10.1203/00006450-199608000-00019. [DOI] [PubMed] [Google Scholar]
- 40.Nicoletto BB, Souza GC, Gonçalves LF, Costa C, Perry IS, Manfro RC. Leptin, insulin resistance, and metabolic changes 5 years after renal transplantation. Journal of Renal Nutrition. 2012;22(4):440–449. doi: 10.1053/j.jrn.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Oterdoom LH, De Vries APJ, Gansevoort RT, et al. Determinants of insulin resistance in renal transplant recipients. Transplantation. 2007;83(1):29–35. doi: 10.1097/01.tp.0000245844.27683.48. [DOI] [PubMed] [Google Scholar]
- 42.Bergrem HA, Valderhaug TG, Hartmann A, Bergrem H, Hjelmesæth J, Jenssen T. Glucose tolerance before and after renal transplantation. Nephrology Dialysis Transplantation. 2010;25(3):985–992. doi: 10.1093/ndt/gfp566. [DOI] [PubMed] [Google Scholar]
- 43.Zheng CM, Ma WY, Wu CC, et al. Glycated albumin in diabetic patients with chronic kidney disease. Clinica Chimica Acta. 2012;413(19-20):1555–1561. doi: 10.1016/j.cca.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 44.Kleemann R, Van Erk M, Verschuren L, et al. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PLoS One. 2010;5(1) doi: 10.1371/journal.pone.0008817.e8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE. Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. Journal of the American Society of Nephrology. 2006;17(5):1388–1394. doi: 10.1681/ASN.2004100842. [DOI] [PubMed] [Google Scholar]
- 46.Ikizler TA. Nutrition, inflammation and chronic kidney disease. Current Opinion in Nephrology and Hypertension. 2008;17(2):162–167. doi: 10.1097/MNH.0b013e3282f5dbce. [DOI] [PubMed] [Google Scholar]
- 47.Menon V, Rudym D, Chandra P, Miskulin D, Perrone R, Sarnak M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clinical Journal of the American Society of Nephrology. 2011;6(1):7–13. doi: 10.2215/CJN.04140510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews Molecular Cell Biology. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 49.Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. Journal of Biological Chemistry. 2003;278(27):24944–24950. doi: 10.1074/jbc.M300423200. [DOI] [PubMed] [Google Scholar]
- 50.Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. Journal of the American Society of Nephrology. 2004;15(6):1537–1545. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 51.Lee DF, Kuo HP, Chen CT, et al. IKKβ suppression of TSC1 function links the mTOR pathway with insulin resistance. International Journal of Molecular Medicine. 2008;22(5):633–638. doi: 10.3892/ijmm_00000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-α induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54(10):2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- 53.Plomgaard P, Fischer CP, Ibfelt T, Pedersen BK, Van Hall G. Tumor necrosis factor-α modulates human in vivo lipolysis. Journal of Clinical Endocrinology and Metabolism. 2008;93(2):543–549. doi: 10.1210/jc.2007-1761. [DOI] [PubMed] [Google Scholar]
- 54.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51(12):3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 55.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New England Journal of Medicine. 2007;356(15):1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 56.Semple D, Smith K, Bhandari S, Seymour AML. Uremic cardiomyopathy and insulin resistance: a critical role for Akt? Journal of the American Society of Nephrology. 2011;22(2):207–215. doi: 10.1681/ASN.2009090900. [DOI] [PubMed] [Google Scholar]
- 57.Heiss A, DuChesne A, Denecke B, et al. Structural basis of calcification inhibition by α2-HS glycoprotein/fetuin-A: formation of colloidal calciprotein particles. Journal of Biological Chemistry. 2003;278(15):13333–13341. doi: 10.1074/jbc.M210868200. [DOI] [PubMed] [Google Scholar]
- 58.Caglar K, Yilmaz MI, Saglam M, et al. Short-term treatment with sevelamer increases serum fetuin-A concentration and improves endothelial dysfunction in chronic kidney disease stage 4 patients. Clinical Journal of the American Society of Nephrology. 2008;3(1):61–68. doi: 10.2215/CJN.02810707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Messa P, Alberti L, Como G, et al. Calcimimetic increases osteoprotegerin and decreases fetuin-A levels in dialysis patients. Nephrology Dialysis Transplantation. 2007;22(9):2724–2725. doi: 10.1093/ndt/gfm312. [DOI] [PubMed] [Google Scholar]
- 60.Wang CC, Hsu YJ, Wu CC, et al. Serum fetuin-A levels increased following parathyroidectomy in uremic hyperparathyroidism. Clinical Nephrology. 2012;77(2):89–96. [PubMed] [Google Scholar]
- 61.Hennige AM, Staiger H, Wicke C, et al. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS One. 2008;3(3) doi: 10.1371/journal.pone.0001765.e1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang AYM, Woo J, Lam CWK, et al. Associations of serum fetuin-A with malnutrition, inflammation, atherosclerosis and valvular calcification syndrome and outcome in peritoneal dialysis patients. Nephrology Dialysis Transplantation. 2005;20(8):1676–1685. doi: 10.1093/ndt/gfh891. [DOI] [PubMed] [Google Scholar]
- 63.Mathews ST, Chellam N, Srinivas PR, et al. α2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Molecular and Cellular Endocrinology. 2000;164(1-2):87–98. doi: 10.1016/s0303-7207(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 64.Auberger P, Falquerho L, Contreres JO, et al. Characterization of a natural inhibitor of the insulin receptor tyrosine kinase: cDNA cloning, purification, and anti-mitogenic activity. Cell. 1989;58(4):631–640. doi: 10.1016/0092-8674(89)90098-6. [DOI] [PubMed] [Google Scholar]
- 65.Xu Y, Xu M, Bi Y, et al. Serum fetuin-A is correlated with metabolic syndrome in middle-aged and elderly Chinese. Atherosclerosis. 2011;216(1):180–186. doi: 10.1016/j.atherosclerosis.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 66.Reinehr T, Roth CL. Fetuin-A and its relation to metabolic syndrome and fatty liver disease in obese children before and after weight loss. Journal of Clinical Endocrinology and Metabolism. 2008;93(11):4479–4485. doi: 10.1210/jc.2008-1505. [DOI] [PubMed] [Google Scholar]
- 67.Ix JH, Wassel CL, Kanaya AM, et al. Fetuin-A and incident diabetes mellitus in older persons. Journal of the American Medical Association. 2008;300(2):182–188. doi: 10.1001/jama.300.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stefan N, Fritsche A, Weikert C, et al. Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes. 2008;57(10):2762–2767. doi: 10.2337/db08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song A, Xu M, Bi Y, et al. Serum fetuin-A associates with type 2 diabetes and insulin resistance in Chinese adults. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0019228.e19228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rastmanesh MM, Bluyssen HAR, Joles JA, Boer P, Willekes N, Braam B. Increased expression of SOCS3 in monocytes and SOCS1 in lymphocytes correlates with progressive loss of renal function and cardiovascular risk factors in chronic kidney disease. European Journal of Pharmacology. 2008;593(1-3):99–104. doi: 10.1016/j.ejphar.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 71.Rastmanesh MM, Braam B, Joles JA, Boer P, Bluyssen HAR. Increased SOCS expression in peripheral blood mononuclear cells of end stage renal disease patients is related to inflammation and dialysis modality. European Journal of Pharmacology. 2009;602(1):163–167. doi: 10.1016/j.ejphar.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 72.Wesoly J, Sikorski K, Lee CK, Bluyssen HAR. Suppressor of cytokine signaling and accelerated atherosclerosis in kidney disease. Acta Biochimica Polonica. 2010;57(3):251–260. [PubMed] [Google Scholar]
- 73.Rønn SG, Billestrup N, Mandrup-Poulsen T. Diabetes and suppressors of cytokine signaling proteins. Diabetes. 2007;56(2):541–548. doi: 10.2337/db06-1068. [DOI] [PubMed] [Google Scholar]
- 74.Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. Journal of Biological Chemistry. 2000;275(21):15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 75.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Molecular and Cellular Biology. 2004;24(12):5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. Journal of Biological Chemistry. 2002;277(44):42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 77.Rieusset J, Bouzakri K, Chevillotte E, et al. Suppressor of cytokine signaling 3 expression and insulin resistance in skeletal muscle of obese and type 2 diabetic patients. Diabetes. 2004;53(9):2232–2241. doi: 10.2337/diabetes.53.9.2232. [DOI] [PubMed] [Google Scholar]
- 78.Slee AD. Exploring metabolic dysfunction in chronic kidney disease. Nutrition & Metabolism. 2012;9(article 36) doi: 10.1186/1743-7075-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte Biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. Journal of Lipid Research. 2007;48(6):1253–1262. doi: 10.1194/jlr.R700005-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. Journal of Clinical Investigation. 2006;116(7):1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Odamaki M, Furuya R, Yoneyama T, et al. Association of the serum leptin concentration with weight loss in chronic hemodialysis patients. American Journal of Kidney Diseases. 1999;33(2):361–368. doi: 10.1016/s0272-6386(99)70313-6. [DOI] [PubMed] [Google Scholar]
- 82.Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. Role of leptin and melanocortin signaling in uremia-associated cachexia. Journal of Clinical Investigation. 2005;115(6):1659–1665. doi: 10.1172/JCI22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ottonello L, Gnerre P, Bertolotto M, et al. Leptin as a uremic toxin interferes with neutrophil chemotaxis. Journal of the American Society of Nephrology. 2004;15(9):2366–2372. doi: 10.1097/01.ASN.0000139321.98029.40. [DOI] [PubMed] [Google Scholar]
- 84.Stenvinkel P. Leptin—a new hormone of definite interest for the nephrologist. Nephrology Dialysis Transplantation. 1998;13(5):1099–1101. doi: 10.1093/ndt/13.5.1099. [DOI] [PubMed] [Google Scholar]
- 85.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nature Medicine. 2001;7(8):947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 86.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature Medicine. 2001;7(8):941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 87.Adamczak M, Chudek J, Wiȩcek A. Adiponectin in patients with chronic kidney disease. Seminars in Dialysis. 2009;22(4):391–395. doi: 10.1111/j.1525-139X.2009.00587.x. [DOI] [PubMed] [Google Scholar]
- 88.Teta D. Adipokines as Uremic Toxins. Journal of Renal Nutrition. 2012;22(1):81–85. doi: 10.1053/j.jrn.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 89.Ohashi K, Iwatani H, Kihara S, et al. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(9):1910–1917. doi: 10.1161/ATVBAHA.107.147645. [DOI] [PubMed] [Google Scholar]
- 90.Ahima RS. Linking adiponectin to proteinuria. Journal of Clinical Investigation. 2008;118(5):1619–1622. doi: 10.1172/JCI35655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ouedraogo R, Gong Y, Berzins B, et al. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. Journal of Clinical Investigation. 2007;117(6):1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zoccali C, Mallamaci F, Tripepi G, et al. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. Journal of the American Society of Nephrology. 2002;13(1):134–141. doi: 10.1681/ASN.V131134. [DOI] [PubMed] [Google Scholar]
- 93.Cohen G, Hörl WH. Resistin as a cardiovascular and atherosclerotic risk factor and uremic toxin. Seminars in Dialysis. 2009;22(4):373–377. doi: 10.1111/j.1525-139X.2009.00583.x. [DOI] [PubMed] [Google Scholar]
- 94.Axelsson J, Bergsten A, Qureshi AR, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney International. 2006;69(3):596–604. doi: 10.1038/sj.ki.5000089. [DOI] [PubMed] [Google Scholar]
- 95.Yilmaz MI, Saglam M, Carrero JJ, et al. Serum visfatin concentration and endothelial dysfunction in chronic kidney disease. Nephrology Dialysis Transplantation. 2008;23(3):959–965. doi: 10.1093/ndt/gfm727. [DOI] [PubMed] [Google Scholar]
- 96.Karakan S, Sezer S, Acar FNÖ, Haberal M. The relationship of visfatin levels with insulin resistance and left ventricular hypertrophy in peritoneal dialysis patients. Renal Failure. 2012;34(6):732–737. doi: 10.3109/0886022X.2012.676493. [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto T, Qureshi AR, Anderstam B, et al. Clinical importance of an elevated circulating chemerin level in incident dialysis patients. Nephrology Dialysis Transplantation. 2010;25(12):4017–4023. doi: 10.1093/ndt/gfq329. [DOI] [PubMed] [Google Scholar]
- 98.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramos LF, Shintani A, Ikizler TA, Himmelfarb J. Oxidative stress and inflammation are associated with adiposity in moderate to severe CKD. Journal of the American Society of Nephrology. 2008;19(3):593–599. doi: 10.1681/ASN.2007030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chonchol M, Scragg R. 25-Hydroxyvitamin D, insulin resistance, and kidney function in the Third National Health and Nutrition Examination Survey. Kidney International. 2007;71(2):134–139. doi: 10.1038/sj.ki.5002002. [DOI] [PubMed] [Google Scholar]
- 101.Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney International. 2007;71(1):31–38. doi: 10.1038/sj.ki.5002009. [DOI] [PubMed] [Google Scholar]
- 102.Baynes KCR, Boucher BJ, Feskens EJM, Kromhout D. Vitamin D, glucose tolerance anal insulinaemia in elderly men. Diabetologia. 1997;40(3):344–347. doi: 10.1007/s001250050685. [DOI] [PubMed] [Google Scholar]
- 103.Chiu KC, Chu A, Go VLW, Saad MF. Hypovitaminosis D is associated with insulin resistance and β cell dysfunction. American Journal of Clinical Nutrition. 2004;79(5):820–825. doi: 10.1093/ajcn/79.5.820. [DOI] [PubMed] [Google Scholar]
- 104.Cade C, Norman AW. Vitamin D3 improves impaired glucose tolerance and insulin secretion in the vitamin D-deficient rat in vivo. Endocrinology. 1986;119(1):84–90. doi: 10.1210/endo-119-1-84. [DOI] [PubMed] [Google Scholar]
- 105.Mak RHK. 1,25-Dihydroxyvitamin D3 corrects insulin and lipid abnormalities in uremia. Kidney International. 1998;53(5):1353–1357. doi: 10.1046/j.1523-1755.1998.00865.x. [DOI] [PubMed] [Google Scholar]
- 106.Mak RHK. Amelioration of hypertension and insulin resistance by 1,25-dihydroxycholecalciferol in hemodialysis patients. Pediatric Nephrology. 1992;6(4):345–348. doi: 10.1007/BF00869730. [DOI] [PubMed] [Google Scholar]
- 107.Mak RHK, Turner C, Haycock GB, Chantler C. Secondary hyperparathyroidism and glucose intolerance in children with uremia. Kidney International. 1983;24(16):S-128–S-133. [PubMed] [Google Scholar]
- 108.Akmal M, Massry SG, Goldstein DA. Role of parathyroid hormone in the glucose intolerance of chronic renal failure. Journal of Clinical Investigation. 1985;75(3):1037–1044. doi: 10.1172/JCI111765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.De Boer IH. Vitamin D and glucose metabolism in chronic kidney disease. Current Opinion in Nephrology and Hypertension. 2008;17(6):566–572. doi: 10.1097/MNH.0b013e32830fe377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lajdova I, Spustova V, Oksa A, Chorvatova A, Chorvat D, Dzurik R. Intracellular calcium homeostasis in patients with early stagesof chronic kidney disease: effects of vitamin D3 supplementation. Nephrology Dialysis Transplantation. 2009;24(11):3376–3381. doi: 10.1093/ndt/gfp292. [DOI] [PubMed] [Google Scholar]
- 111.Lu KC, Shieh SD, Lin SH, et al. Hyperparathyroidism, glucose tolerance and platelet intracellular free calcium in chronic renal failure. Quarterly Journal of Medicine. 1994;87(6):359–365. [PubMed] [Google Scholar]
- 112.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McCaleb ML, Izzo MS, Lockwood DH. Characterization and partial purification of a factor from uremic human serum that induces insulin resistance. Journal of Clinical Investigation. 1985;75(2):391–396. doi: 10.1172/JCI111712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kielstein JT, Zoccali C. Asymmetric dimethylarginine: a cardiovascular risk factor and a uremic toxin coming of age? American Journal of Kidney Diseases. 2005;46(2):186–202. doi: 10.1053/j.ajkd.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 115.D'Apolito M, Du X, Zong H, et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. Journal of Clinical Investigation. 2010;120(1):203–213. doi: 10.1172/JCI37672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiu YW, Kopple JD, Mehrotra R. Correction of metabolic acidosis to ameliorate wasting in chronic kidney disease: goals and strategies. Seminars in Nephrology. 2009;29(1):67–74. doi: 10.1016/j.semnephrol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 117.Kalantar-Zadeh K, Mehrotra R, Fouque D, Kopple JD. Metabolic acidosis and malnutrition-inflammation complex syndrome in chronic renal failure. Seminars in Dialysis. 2004;17(6):455–465. doi: 10.1111/j.0894-0959.2004.17606.x. [DOI] [PubMed] [Google Scholar]
- 118.Goldberg AP, Hagberg JM, Delmez JA. Metabolic effects of exercise training in hemodialysis patients. Kidney International. 1980;18(6):754–761. doi: 10.1038/ki.1980.194. [DOI] [PubMed] [Google Scholar]
- 119.Mak RHK. Effect of recombinant human erythropoietin on insulin, amino acid, and lipid metabolism in uremia. Journal of Pediatrics. 1996;129(1):97–104. doi: 10.1016/s0022-3476(96)70195-6. [DOI] [PubMed] [Google Scholar]
- 120.Yki-Jarvinen H. Thiazolidinediones. The New England Journal of Medicine. 2004;351(11):1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 121.Brunelli SM, Thadhani R, Ikizler TA, Feldman HI. Thiazolidinedione use is associated with better survival in hemodialysis patients with non-insulin dependent diabetes. Kidney International. 2009;75(9):961–968. doi: 10.1038/ki.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007;356(24):2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 123.De Boer IH, Tinker LF, Connelly S, et al. Calcium plus vitamin D supplementation and the risk of incident diabetes in the women’s health initiative. Diabetes Care. 2008;31(4):701–707. doi: 10.2337/dc07-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Church JM, Hill GL. Impaired glucose metabolism in surgical patients improved by intravenous nutrition: assessment by the euglycemic-hyperinsulinemic clamp. Metabolism: Clinical and Experimental. 1988;37(6):505–509. doi: 10.1016/0026-0495(88)90162-x. [DOI] [PubMed] [Google Scholar]
- 125.Melnik BC. Leucine signaling in the pathogenesis of type 2 diabetes and obesity. World Journal of Diabetes. 2012;3(3):38–53. doi: 10.4239/wjd.v3.i3.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen Y, Sood S, McIntire K, Roth R, Rabkin R. Leucine-stimulated mTOR signaling is partly attenuated in skeletal muscle of chronically uremic rats. American Journal of Physiology - Endocrinology and Metabolism. 2011;301(5):E873–E881. doi: 10.1152/ajpendo.00068.2011. [DOI] [PubMed] [Google Scholar]
- 127.Mak RHK, Turner C, Thompson T. The effect of a low protein diet with amino acid/keto acid supplements on glucose metabolism in children with uremia. Journal of Clinical Endocrinology and Metabolism. 1986;63(4):985–989. doi: 10.1210/jcem-63-4-985. [DOI] [PubMed] [Google Scholar]
- 128.Hadjadj S, Duly-Bouhanick B, Bekherraz A, et al. Serum triglycerides are a predictive factor for the development and the progression of renal and retinal complications in patients with type 1 diabetes. Diabetes and Metabolism. 2004;30(1):43–51. doi: 10.1016/s1262-3636(07)70088-5. [DOI] [PubMed] [Google Scholar]
- 129.Lee PH, Chang HY, Tung CW, et al. Hypertriglyceridemia: an independent risk factor of chronic kidney disease in Taiwanese adults. American Journal of the Medical Sciences. 2009;338(3):185–189. doi: 10.1097/MAJ.0b013e3181a92804. [DOI] [PubMed] [Google Scholar]
- 130.Manninen V, Tenkanen L, Koskinen P, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation. 1992;85(1):37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- 131.Satirapoj B, Yingwatanadej P, Chaichayanon S, Patumanond J. Effect of angiotensin II receptor blockers on insulin resistance in maintenance haemodialysis patients. Nephrology. 2007;12(4):342–347. doi: 10.1111/j.1440-1797.2007.00770.x. [DOI] [PubMed] [Google Scholar]
- 132.Vos FE, Manning PJ, Sutherland WH, Schollum JB, Walker RJ. Anti-inflammatory effect of an insulin infusion in patients on maintenance haemodialysis: a randomized controlled pilot study. Nephrology. 2011;16(1):68–75. doi: 10.1111/j.1440-1797.2010.01329.x. [DOI] [PubMed] [Google Scholar]
- 133.Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409(6817):194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- 134.Pöykkö SM, Kellokoski E, Hörkkö S, Kauma H, Kesäniemi YA, Ukkola O. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes. 2003;52(10):2546–2553. doi: 10.2337/diabetes.52.10.2546. [DOI] [PubMed] [Google Scholar]
- 135.Barazzoni R, Zanetti M, Stulle M, et al. Higher total ghrelin levels are associated with higher insulin-mediated glucose disposal in non-diabetic maintenance hemodialysis patients. Clinical Nutrition. 2008;27(1):142–149. doi: 10.1016/j.clnu.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 136.Barazzoni R, Zhu X, Deboer M, et al. Combined effects of ghrelin and higher food intake enhance skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rats with chronic kidney disease. Kidney International. 2010;77(1):23–28. doi: 10.1038/ki.2009.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Midtvedt K, Hjelmesæth J, Hartmann A, et al. Insulin resistance after renal transplantation: the effect of steroid dose reduction and withdrawal. Journal of the American Society of Nephrology. 2004;15(12):3233–3239. doi: 10.1097/01.ASN.0000145435.80005.1E. [DOI] [PubMed] [Google Scholar]
- 138.Chadban S. New-onset diabetes after transplantation - Should it be a factor in choosing an immunosuppressant regimen for kidney transplant recipients. Nephrology Dialysis Transplantation. 2008;23(6):1816–1818. doi: 10.1093/ndt/gfn052. [DOI] [PubMed] [Google Scholar]