

Abstract
Epilepsy of the temporal lobe (TLE) is the most common form of focal epilepsy, and in adults, it most frequently develops after injury. However, the mechanisms by which a normal functioning brain turns into an epileptic one still remain obscure. Recent studies point to vascular involvement and particularly blood-brain barrier (BBB) dysfunction in the development of epilepsy. The BBB is a specialized structure which functions to control the neuronal extracellular milieu. BBB dysfunction is found in many diseases of the central nervous system, including stroke, traumatic injuries, tumors and infections. Interestingly, all these insults may initiate an epileptogenic process which eventually leads to spontaneous, recurrent seizures. This epileptogenic time frame usually lasts weeks, months, or even years in man, and days to weeks in rodents and may serve as a “window of opportunity” for the prevention of epilepsy. However, no prevention strategy exists, stressing the importance of research into the mechanisms of epileptogenesis. Here, we will underscore recent experiments suggesting that BBB dysfunction directly induces epileptogenesis. We will provide new evidence to support the hypothesis that BBB breakdown and specifically exposure of temporal lobe structures to the most common serum protein, albumin, is sufficient to induce epileptogenesis.
1. Brain Insults and Temporal Lobe Epilepsy: Vascular Pathology as the Missing Link
The onset of TLE frequently follows cerebral insults. In infants, prolonged febrile seizures are considered the most common precipitating event [1], while in adults, stroke, traumatic brain injury (TBI), tumors, infections and iatrogenic artifacts are often the triggering insults [2]. Stroke or vascular occlusion are the most common form of brain insult in the adult population and the number one known cause of epilepsy [3], raising the hypothesis of a vascular-related pathology as the underlying basis for epilepsy. Importantly, the increased vascular seen after stroke is often not confined to the ruptured or clotted vessels nor to the ischemic region of the brain, rather it affects a wide area around the focus of the lesion. Moreover, the increased permeability is not limited to the acute time point of the insult and may last from days up to years [4]. The mechanisms underlying BBB dysfunction and restoration after brain insults are still not completely understood but seem to be associated with metabolic failure [5] and immune processes involving the innate brain and peripheral inflammatory responses [6, 7]. The BBB, as a dynamic transport mechanism, requires continuous supply of nutrients and metabolites to maintain proper function. Therefore, the BBB is sensitive to the metabolic failure deriving from insults [8], and in rats even a short metabolic disruption during a transient ischemic attack may be enough to cause leakage [9, 10]. The inflammatory response that ensues after an insult may be variable, but commonly involves a BBB opening [11–13]. The exact mechanisms, through which the inflammatory mediators' interactions affect the BBB, are yet unknown; however, experimental data suggest that cytokines secreted from astrocytes and microglia, including IL-6, IL-1β, and TGF-β, affect endothelial tight junctions culminating in increased BBB permeability. Cytokines interact with cellular receptors on the endothelial membrane thereby inducing a signal cascade that mediates an increase in adhesion molecules (I-Cam and V-Cam), aquaporin channels (AQP4), and cytoskeletal changes, resulting in a pathological dysfunction of the BBB, adhesion of white blood cells, diffusion of blood proteins into the brain [14] and induction of astroglial activation, thereby creating a positive feedback cycle, thus keeping the BBB open for significant time periods.
Cerebral tumors may directly damage blood vessels, compressing the venous efflux, as well as induce aberrant angiogenesis [15, 16]. These damaged or newly formed vessels are dysfunctional and allow the leakage of blood constituents into the neuropil [17, 18]. Physiological cerebral angiogenesis is initiated by a short phase of vascular permeability mediated by vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). Under pathological conditions such as tumor growth, inflammation or stroke, large amounts of VEGF are released. This overproduction modifies the balance between angiogenic and stabilizing factors in favor of vascular permeability [16, 19, 20]. Signaling via VEGF and bFGF receptors induces the production of collagenase, heparinase, plasminogen activators (uPA, tPA), and matrix metalloproteinases (mainly MMP-2 and MMP-9), which contribute to the proteolysis of the basement membrane and to the disassembly of interendothelial junctions [21–23].
In infections, BBB opening may also be induced as part of the inflammatory response, granting brain access to the systemic immune system, or directly by the infecting pathogen, attacking the endothelium of the cerebral vessels [24, 25].
Status epilepticus (SE), a prolonged seizure, commonly appears in patients previously diagnosed with epilepsy. However, SE can occur spontaneously or following any one of the aforementioned insults, without a prior history of epilepsy [26]. In infancy, TLE usually accompanies genetic or developmental deficits; however, a subpopulation of infants acquires epilepsy after suffering an episode of prolonged febrile seizures (PFS) [27, 28]. Recent animal studies have shown that SE and PFS are associated with angiogenesis, BBB disruption [29, 30], and inflammatory response [31, 32] as well as metabolic dysfunction, thereby causing an extensive long-lasting vascular pathology similar to that found after other insults.
To summarize, epilepsy-initiating insults, as a rule, affect cerebral vessels and are associated with BBB dysfunction via three main mechanisms: inflammation, metabolic failure, and angiogenesis (Figure 1). This raises the question of whether BBB dysfunction plays a crucial role in epileptogenesis or whether it is a coincidental finding.
Figure 1.
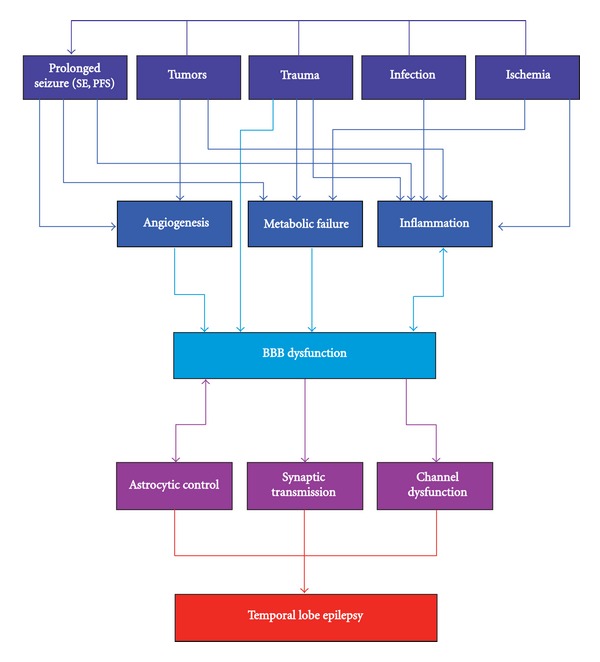
Flowchart illustrating a possible pathway leading from common brain insults to the development of epilepsy. Stroke, trauma, tumor and infections, the common insults with an epileptic sequela, usually cause BBB dysfunction by three main mechanisms: inflammation, angiogenesis and metabolic failure. The inflammatory response, inducing BBB dysfunction, is increasingly enhanced by the BBB dysfunction itself, creating a positive feedback loop that may develop into a chronic condition. BBB dysfunction induces direct signaling in astrocytes and neurons, resulting in hyperexcitability and network hypersynchronicity, which manifests as spontaneous recurrent seizures.
2. Blood-Brain Barrier Dysfunction Induces Epileptogenesis
The frequent findings of BBB dysfunction after brain insults and in epileptic patients raised the captivating hypothesis for a direct role of BBB dysfunction in epileptogenesis [33]. The hypothesis was further supported by the recent observation of a strong positive correlation between the extent of serum albumin leakage through the BBB of rats after induced SE and the number of subsequent seizures ([30, 34, 35], see Figure 2). Direct evidence for the involvement of BBB dysfunction in epileptogenesis was recently established by a series of animal studies, in which a long-lasting opening of the BBB in the rat neocortex was shown to result in the delayed appearance of hypersynchronous epileptiform activity [36–39]. Epileptogenesis following BBB injury does not seem to be mediated by a significant loss of inhibitory interneurons, but rather seems to involve early dysfunction of astrocytes (AKA “reactive astrogliosis”) and an inflammatory response [40, 41]. Importantly, BBB dysfunction does not seem to induce epileptogenesis by provoking SE or immediate neuronal death, although under some conditions (e.g., intrarterial mannitol) it may induce immediate seizures [42, 43], and, in epileptic animals, it lowers seizure threshold [30].
Figure 2.
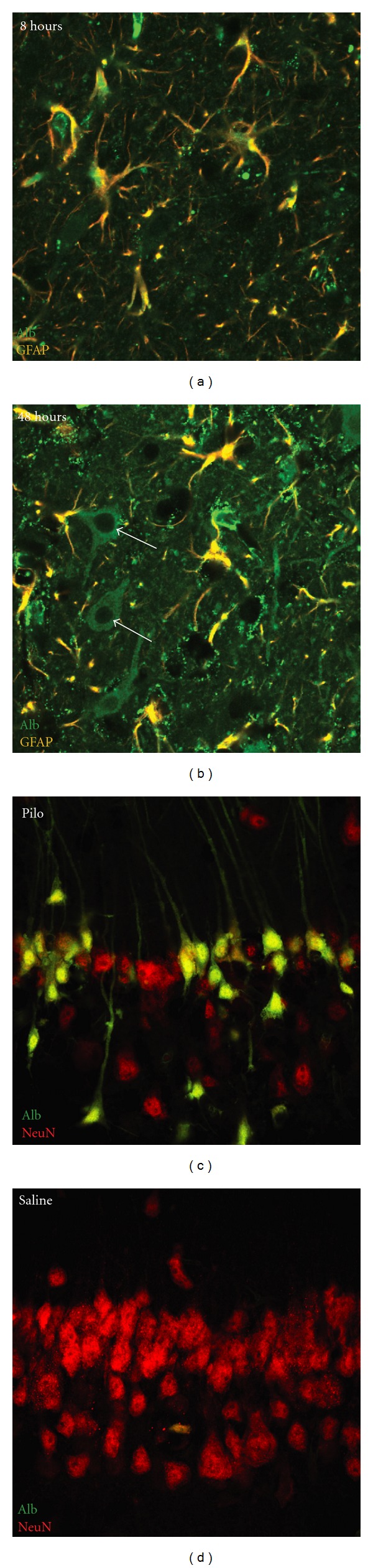
Albumin uptake in hippocampal astrocytes and neurons after SE. (a) 8 hours after SE, endogenous albumin as visualized by immunostainings with antirat albumin (green) is observed in the neuropil indicating breakdown of the BBB. Note that albumin is preferentially taken up by GFAP-positive (red) astrocytes. (b) 48 hours after SE, endogenous albumin is also transported into the cytoplasm of GFAP-negative, morphologically identified neurons. (c) Pyramidal cell layer neurons of the CA1 region of the rat hippocampus (anti-NeuN, red) accumulate FITC-labeled exogenous BSA (green) 48 h after pilocarpine-induced SE, whereas neurons of animals injected with saline do so very sparsely (d). Note the marked neuronal cell loss after SE.
The most frequently used animal model for TLE is the pilocarpine-induced SE in rodents. Indeed, in this model, similar to the human disease, a significant cell loss can be found within the hippocampus; most of this damage, however, is caused during the initiating SE [44] linking the neuronal necrosis to excitotoxicity [45]. Consistent with the hypothesis that BBB opening is required to initiate epileptogenesis, van Vliet and colleagues [30] showed the accumulation of serum-derived albumin after SE in glia and neurons. Importantly, albumin positive neurons were positively costained by fluoro jade; while this may indicate that intracellular albumin induces neuronal damage, a more likely explanation is that the neuronal uptake of albumin is limited to injured neurons. Indeed, immunostaining against serum albumin reveals BBB dysfunction a few hours after SE. While during the first 24 hours after SE albumin uptake is mostly observed in GFAP (+) astrocytes (Figure 2(a)), at 48 hours, many GFAP (−) cells, with a typical neuronal morphology are also stained to albumin (Figure 2(b)). Moreover, while exposure of acute brain slices from healthy rats to FITC-labeled albumin reveals a prominent astrocytic uptake [37], exposure of slices from SE-experiences rats results in prominent neuronal uptake, mostly in regions that are subject to extensive damage and neuronal cell loss following SE (hippocampal regions CA1, CA3, and hilus; Figures 2(c) and 2(d)). The mechanism of albumin uptake is yet unknown. TGF-β receptor-mediated albumin uptake has been repeatedly shown in different nonneuroglial cell populations [37, 46, 47]. Evidence for a similar mechanism in astroglia has been recently shown by our group [48]. Furthermore, the expression of TGF-β receptor 1 and 2 is increased in neurons during ischemic insult, highlighting the potential role of these receptors in neurons challenged by noxious stimuli [49]. Thus, it is conceivable, that upregulation of TGF-β receptors in hippocampal neurons after SE underlies the uptake of serum albumin extravagated via the disrupted BBB. It has also been proposed that albumin could itself convey a neurotoxic effect, which leads to neuronal death. Studies using injection of hemolysate into the brain parenchyma have shown that presence of serum proteins in the brain parenchyma is associated with accumulation of the albumin-binding dye Evans blue in the cytoplasm of injured neurons and with cytochrome c release and DNA fragmentation [50]. Furthermore, in vitro experiments in cell cultures demonstrated that microglial cells produce neurotoxic agents in response to albumin exposure [51]. However, a direct intraventricular injection of labeled albumin alone does not result in SE or a significant neuronal uptake of albumin, making the direct neurotoxic effects of albumin less likely, but rather suggesting that injured neurons after SE uptake albumin. To summarize, while the mechanisms underlying albumin uptake are not entirely clear, it seems that after SE, albumin is first transported into astrocytes, and only at a later point into damaged neurons (Figure 2). While experimental data show that the rapid transcriptional brain response to serum albumin involves TGF-β signaling, the connection between this signaling pathway to the delayed albumin uptake into neurons is not clear. Other serum-derived proteins (e.g., thrombin, see [52]) may also take a role in signaling within the neurovascular unit after injury.
To challenge the hypothesis that serum albumin is sufficient to induce epileptogenesis within the temporal lobe, we introduced albumin directly into the ventricles, which are anatomically related to temporal lobe structures. In 2–3 month anesthetized FVB-N mice, we implanted a 200 μl osmotic pump (ALZET osmotic pump 2001) filled with 0.4 mM bovine serum albumin (BSA) in artificial cerebral spinal fluid (ACSF; for ACSF composition see [53]). The pump was connected to a brain infusion kit (ALZET brain infusion kit 3) which was stereotactically inserted to the right lateral ventricle, frontal to the hippocampus (0.5 mm posterior, 1 mm lateral, and 3 mm ventral to Bregma), through a 0.7 mm diameter hole drilled in the skull. Control animals were perfused with ACSF alone (sham controls). The implanted pump continuously infused 1 μl/hour and was surgically removed after 7 days. Animal's electroencephalographic (EEG) activity was telemetrically continuously monitored from moment of implant, throughout the duration of the experiment. Single-channel electrographic signals were acquired using a telemetric EEG monitoring system (Data Science International), as previously described [54]. Briefly, a transmitter (TA10EA-F20) was placed subcutaneously on the right dorsal side of the animal. Two holes were drilled through the skull using a surgical drill and stainless steel screws, serving as electrodes, were inserted through the drilled holes to the epidural surface. The epicranial side of each screw was connected to a transmitter lead and secured with bone cement. Signals were transmitted to a receiver placed below the animal's cage and saved on a personal computer (PC), with a sampling rate of 1 kHz. Using fluorescent-labeled albumin (FITC-albumin), we confirmed a preferential accumulation of the infused albumin in temporal lobe structures, specifically the hippocampus and the entorhinal cortex, as well as in the corpus callosum. While no instant changes in EEG were observed immediately following pump implantation (i.e., no SE), 3–5 days after, spontaneous seizures were recorded in treated animals and persisted for the remaining duration of recording (4–6 weeks, Figure 3). While many of the seizures were behaviorally expressed as staring behavior, occasional stage 5 generalized seizures were observed. No seizures were recorded in sham-treated controls. The induction of chronic spontaneous seizures by the exposure of the temporal lobe structures to albumin, emphasizes the important role of BBB integrity for normal brain functions and the potential epileptogenic role for BBB dysfunction and exposure of temporal lobe structures to serum-derived albumin.
Figure 3.
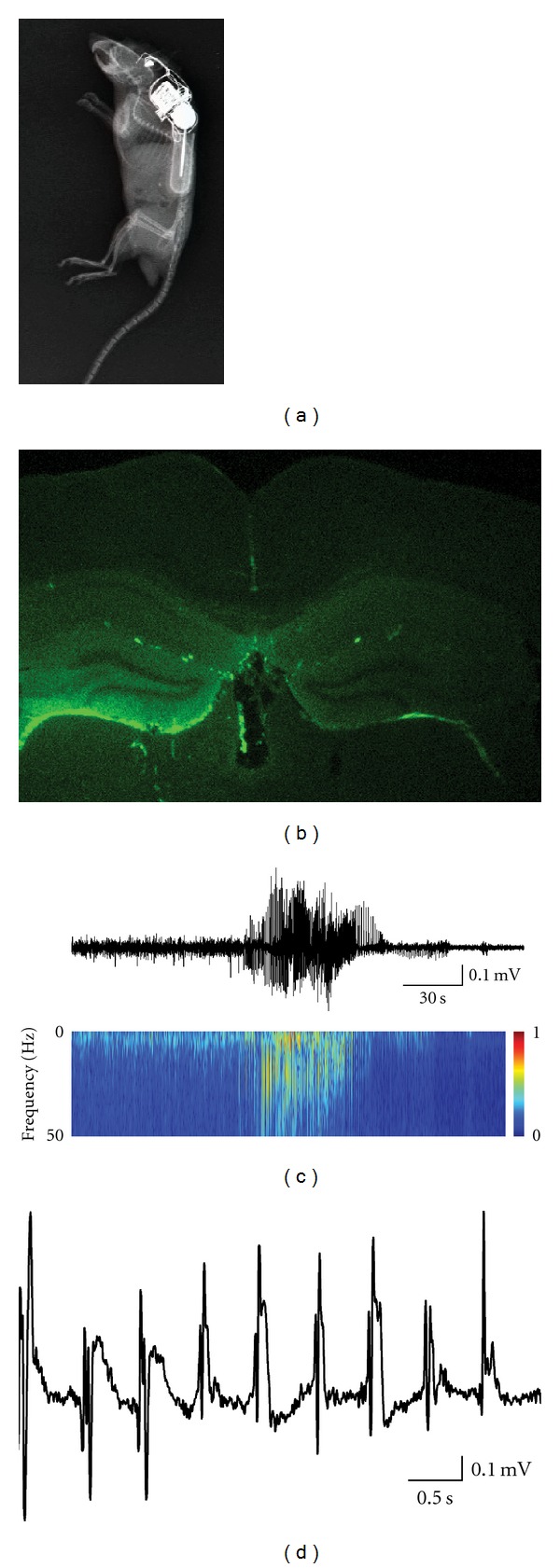
Spontaneous seizures induced by albumin. (a) An X-ray photograph of mouse implanted with FITC-BSA infusion pump and telemetric electrographic recording transmitter. (b) FITC conjugated BSA preferentially accumulates in the hippocampus after intraventricular infusion. (c) Electrographic recording (upper panel) of a spontaneous seizure 4 days after pump implant, and the corresponding spectrogram (lower panel). (d) Ictal activity (slower time scale).
3. The role of Astroglial Response in Epileptogenesis Induced by BBB Dysfunction
In a previous study where BBB was disrupted and a chronic epileptic focus induced, an early dysfunction of astrocytes was observed prior to the occurrence of neuronal hypersynchronization [37, 43]. These studies raised the possible involvement of serum albumin in the regulation of astrocytic calcium signaling and gene expression. The hypothesis was supported by experiments in cultured astrocytes showing increased intracellular calcium following exposure to albumin [55]. Changes in astrocytic gene expression, morphology, and functions following insults to the brain are well documented [56], and the potential role of astrocytes in seizure generation and epilepsy has been described in both experimental animals and epileptic patients [38, 57–59]. Furthermore, recent studies have indicated novel physiological roles for glial cells in the CNS, such as modulation of synaptic transmission, synaptogenesis, and plasticity [60–62]. It is thus plausible that reactive (“transformed” or “activated”) glial cells play a significant role in reorganization of the neuronal network, leading to hypersynchronicity and hyperexcitability. Changes in astrocytic functions which are potentially involved in increased neuronal excitability and epileptogenesis include the following.
(1) Downregulation of Potassium Inward Rectifying Channels —
(Kir4.1) and water channels (aquaporin 4, AQP4) [37, 41, 63]. Both channels are colocalized most abundantly in astrocytic endfeet and considered crucial for the regulation of the brain's extracellular potassium ([K+]o) and water content. Indeed, down-regulation of Kir4.1 and AQP4 in genetic-engineered mice resulted in impaired [K+]o buffering and spontaneous seizures [64–66]. Down-regulation of Kir4.1 channels and the consequential impaired [K+]o buffering have also been reported in the hippocampus of pilocarpine-treated epileptic rats [67] and in sclerotic hippocampi of TLE patients [68]. Impaired [K+]o homeostasis is specifically significant when the network is activated repetitively. Activity-dependent increase in [K+]o results in altered extracellular volume, lowering of firing threshold, slower neuronal repolarization, increased transmitter release, and facilitated activation of N-methyl-d-aspartate (NMDA) receptors during neuronal depolarization, thus potentially contributing to aberrant plasticity and hyper-synchronization [41].
(2) Reduced Gap Junctions —
Gap junctions, consisting of connexin proteins, are functional channels between cells. Astrocytes are coupled via gap junctions to form large cellular networks, facilitating the spatial buffering of small molecules (e.g., [K+]o). Connexin knockout mice show a mild decrease in the spatial buffering of [K+]o and a decreased seizure threshold, although most buffering capacity is maintained [69].
(3) Impaired Glutamate Metabolism —
Glia cells are important for glutamate release, uptake, and metabolism and may express glutamatergic receptors. Additionally, upregulation of TNFα (together with microglial activation) in epileptic tissue can enhance glutamate release [70]. In hippocampal slice preparations from healthy mice, astrocytic glutamate release has been implicated to underwrite the slow, TTX resistant, NMDA sensitive neuronal inward current (possibly representing the typical “paroxysmal depolarization shift” (PDS) underlining interictal bursts) [71]. However, it is not clear if such release can contribute to epileptogenesis or to the propagation of seizure activity in an epileptic brain. Furthermore, there is no direct evidence of increased glutamate release from astrocytes in tissue from chronic epileptic patients or animals. Decreased uptake and metabolism by transformed astrocytes may also affect extracellular glutamate levels. Astrocytes express two specific glutamate transporters, the EAAT1 (AKA GLAST in rodents) and the EAAT2 (GLT-1) which are responsible for most glutamate uptake in the brain. There is strong evidence to support the assumption that significant impairment of astrocytic glutamate transporters is associated with the development of seizures [59, 70], but to what extent is it sufficient to induce epileptogenesis is controversial [41, 70]. It is also unclear to what extent glutamate transporters are indeed downregulated in epileptic tissue from the temporal lobe, with reports of reduced, normal, or increased levels [59, 70]. Astrocytes are also responsible for the metabolism of the transported glutamate into glutamine by glutamine synthetase. Glutamine is transported back into neurons where it is recycled back to glutamate by mitochondrial glutaminase. Sclerotic brain tissue from TLE patients shows approximately a 40% reduction in the levels of astrocytic glutamine synthetase [59, 72]. Moreover, in experimental animals, inhibition of glutamine synthetase (80–90% inhibition) causes seizures [73].
(4) Increased Release of Inflammatory Mediators by Transformed Astrocytes —
As mentioned above, astrocytes can produce different pro- and anti-inflammatory molecules, and these can be pro- and antiepileptogenic. Accumulating experimental evidence support the role of IL-1β in reducing seizure threshold [74], exacerbating BBB dysfunction [75], as well as promoting epileptogenesis in the kinate SE model of epilepsy [76, 77].
4. From BBB Dysfunction to Neuronal Dysfunction and Hyperexcitability
The brain neuropil demands an adequate extracellular environment for the regulation of synaptic function. Following BBB dysfunction, there is a decrease in nutrient supply, faulty clearance of toxic molecules, secretion of cytokines, and disturbance in the ionic balance, all of which affect neuronal activity.
As mentioned above, changes in [K+]o buffering may directly affect synaptic release and postsynaptic responses, including the activation of NMDA receptors due to associated depolarization; in addition, BBB dysfunction is expected to equilibrate the brain's extracellular environment with serum concentrations of electrolytes, resulting in increased Ca2+ and lower Mg2+ levels in the neuronal microenvironment [78], thus further unblocking of NMDA receptors and enhancing excitability.
The astrocytic and microglial-mediated release of inflammatory mediators has also been shown to directly affect neuronal excitability. Recent data suggest that the release of inflammatory cytokines, such as the aforementioned IL-1β (but also cyclooxygenase-2, TNF-α, IL-6, and TGF-β), may directly affect synaptic signaling and plasticity [79]. IL-1 receptor 1 (IL-1R1) has been recently shown to colocalize on hippocampal pyramidal neurons with the NMDA receptor. Furthermore, activation of neuronal IL-1R1 enhances Ca2+ influx via NMDA receptors thus promoting hyperexcitability, epileptogenesis [74], and excitotoxicity [80]. IL1-β appears also to stabilize NMDA receptor localization on the membrane by preventing endocytosis and protecting it from degradation [81]; finally, IL-1β inhibits GABA-mediated Cl− fluxes, thus potentially reducing inhibitory transmission [82].
In human and animal models of TLE aberrant neurogenesis and dispersion of neurons in the dentate granule cell (DSG) layer is found [83]. Findings suggest that hilar ectopic DGCs in TLE integrate abnormally and can lead to network dysfunction. In-vitro studies have shown that pro inflammatory cytokines, including IL-6, IL1-β and TNF-α play a pivot role in suppressing neurogenesis [84, 85]. Interestingly over expression of TGF-β has been shown to enhance neural stem cells differentiation to a neuronal phenotype in the adult dentate gyrus [86, 87], Suggesting a required homeostatic balance between pro and anti-neurogenic cytokines. Thus, BBB dysfunction and the consequential albumin activation of TGF-β receptors may “push” stem cells in to neuronal fate, inducing the aberrant neurogenesis seen in TLE.
5. Implications to Human Epilepsy
Is there any evidence in human studies for the involvement of pathology at the BBB and epileptogenesis? As mentioned above, pathological and immunohistochemical studies in human epileptic tissue consistently demonstrated structural evidence for abnormal BBB and, serum albumin within the neuropil and cellular elements; as functional evidence for abnormal vessels permeability for large hydrophilic molecules [30, 43]. Imaging BBB permeability in patients with postraumatic epilepsy clearly shows BBB abnormalities in more than 50% of the patients [88]. Interestingly, in most of these patients there was a close spatial correlation between the BBB lesion and the suspected epileptic focus, based on inter-ictal EEG and the use of source localization methods. Moreover, the spatial extent of cortical dysfunction as measured by quantitative EEG analyses correlated with the size of the BBB disrupted region, but not with that of the anatomical lesion. However, prospective clinical followup is still awaited to question directly the risk to develop epilepsy in patients with BBB abnormalities. Such a study may clarify why some patients with dysfunctional BBB do not develop epilepsy while others do. It is yet unclear whether location, duration or extent of BBB dysfunction are critical risk factors for epileptogenesis and influence the time of onset, location or the severity of the disease. An interesting question is to what extent BBB dysfunction is related to drug refractoriness in epileptic patients; such resistance to antiepileptic drugs may hint of upregulation of drug transporters on the dysfunctional endothelial cells [89–91].
To summarize, accumulating experimental evidence points to a common vascular origin underlying insult-induced epileptogenesis of the temporal lobe (Figure 1). The significant changes occurring in the vascular system after different insults lead to exposure of the neuropil to serum-derived albumin inducing activation of specific signaling pathways, astroglial activation and release of cytokines, eventually causing hyperexcitability and synchronicity of the neuronal network. Future research into identification of the involved signaling pathways may prove to be the first milestone in developing novel therapeutics for the prevention and treatment of TLE.
Acknowledgments
This paper is supported by the Sonderforschungsbereich TR3 (AF and UH), the Israeli Science Foundation (566/07, AF), the Binational US-Israel Foundation (BSF 2007185, AF), and the National Institute of Health (NIH RO1 NS066005, AF).
References
- 1.Dubé C, Richichi C, Bender RA, Chung G, Litt B, Baram TZ. Temporal lobe epilepsy after experimental prolonged febrile seizures: prospective analysis. Brain. 2006;129(4):911–922. doi: 10.1093/brain/awl018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee PN, Hauser WA. Incidence and prevalence. In: Engel J Jr., Pedley TA, editors. Epilepsy a Comprehensive Textbook. 2nd edition. Philadelphia, Pa, USA: Lippincot Williams & Wilkins; 2008. pp. 45–56. [Google Scholar]
- 3.Hauser WA, Annegers JF, Rocca WA. Descriptive epidemiology of epilepsy: contributions of population-based studies from Rochester, Minnesota. Mayo Clinic Proceedings. 1996;71(6):576–586. doi: 10.4065/71.6.576. [DOI] [PubMed] [Google Scholar]
- 4.Stoll G, Kleinschnitz C, Meuth SG, et al. Transient widespread blood-brain barrier alterations after cerebral photothrombosis as revealed by gadofluorine M-enhanced magnetic resonance imaging. Journal of Cerebral Blood Flow and Metabolism. 2009;29(2):331–341. doi: 10.1038/jcbfm.2008.129. [DOI] [PubMed] [Google Scholar]
- 5.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 6.Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35(9):2220–2225. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]
- 7.Fabene PF, Mora GN, Martinello M, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nature Medicine. 2008;14(12):1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith SL, Hall ED. Mild pre- and posttraumatic hypothermia attenuates blood-brain barrier damage following controlled cortical impact injury in the rat. Journal of Neurotrauma. 1996;13(1):1–9. doi: 10.1089/neu.1996.13.1. [DOI] [PubMed] [Google Scholar]
- 9.Grillon E, Provent P, Montigon O, Segebarth C, Rémy C, Barbie EL. Blood-brain barrier permeability to manganese and to Gd-DOTA in a rat model of transient cerebral ischaemia. NMR in Biomedicine. 2008;21(5):427–436. doi: 10.1002/nbm.1206. [DOI] [PubMed] [Google Scholar]
- 10.Nagaraja TN, Keenan KA, Fenstermacher JD, Knight RA. Acute leakage patterns of fluorescent plasma flow markers after transient focal cerebral ischemia suggest large openings in blood-brain barrier. Microcirculation. 2008;15(1):1–14. doi: 10.1080/10739680701409811. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surgical Neurology. 2006;66(3):232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 12.Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Molecular Neurobiology. 2001;24(1–3):169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt OI, Morganti-Kossmann MC, Heyde CE, et al. Tumor necrosis factor-mediated inhibition of interleukin-18 in the brain: a clinical and experimental study in head-injured patients and in a murine model of closed head injury. Journal of Neuroinflammation. 2004;1, article 13 doi: 10.1186/1742-2094-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan W, Xiang S, Tu H, Kastin AJ. Cytokines Interact with the Blood-Brain Barrier. Wiley-VCH Verlag GmbH & Co. KGaA; 2006. [Google Scholar]
- 15.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nature Reviews Cancer. 2002;2(10):795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 16.Plate KH, Breier G, Weich HA, Mennel HD, Risau W. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. International Journal of Cancer. 1994;59(4):520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 17.Croll SD, Ransohoff RM, Cai N, et al. VEGF-mediated inflammation precedes angiogenesis in adult brain. Experimental Neurology. 2004;187(2):388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Jain RK, Di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nature Reviews Neuroscience. 2007;8(8):610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 19.Dore-Duffy P, LaManna JC. Physiologic angiodynamics in the brain. Antioxidants & Redox Signaling. 2007;9(9):1363–1371. doi: 10.1089/ars.2007.1713. [DOI] [PubMed] [Google Scholar]
- 20.Hara A, Okayasu I. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: correlation with angiogenesis and prognostic significance. Acta Neuropathologica. 2004;108(1):43–48. doi: 10.1007/s00401-004-0860-0. [DOI] [PubMed] [Google Scholar]
- 21.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends in Pharmacological Sciences. 2001;22(4):201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 22.Gerwins P, Skoldenberg E, Claesson-Welsh L. Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Critical Reviews in Oncology/Hematology. 2000;34(3):185–194. doi: 10.1016/s1040-8428(00)00062-7. [DOI] [PubMed] [Google Scholar]
- 23.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407(6801):242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 24.MacIntyre A, Hammond CJ, Little CS, Appelt DM, Balin BJ. Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiology Letters. 2002;217(2):167–172. doi: 10.1111/j.1574-6968.2002.tb11470.x. [DOI] [PubMed] [Google Scholar]
- 25.Verma S, Lo Y, Chapagain M, et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology. 2009;385(2):425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hauser WA. Status epilepticus: epidemiologic considerations. Neurology. 1990;40(5):9–13. [PubMed] [Google Scholar]
- 27.Dubé CM, Brewster AL, Richichi C, Zha Q, Baram TZ. Fever, febrile seizures and epilepsy. Trends in Neurosciences. 2007;30(10):490–496. doi: 10.1016/j.tins.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scantlebury MH, Heida JG. Febrile seizures and temporal lobe epileptogenesis. Epilepsy Research. 2010;89(1):27–33. doi: 10.1016/j.eplepsyres.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Rigau V, Morin M, Rousset MC, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130(7):1942–1956. doi: 10.1093/brain/awm118. [DOI] [PubMed] [Google Scholar]
- 30.van Vliet EA, Araújo SDC, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130(2):521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- 31.De Simoni MG, Perego C, Ravizza T, et al. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. European Journal of Neuroscience. 2000;12(7):2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- 32.Folbergrová J, Ingvar M, Siesjö BK. Metabolic changes in cerebral cortex, hippocampus, and cerebellum during sustained bicuculline-induced seizures. Journal of Neurochemistry. 1981;37(5):1228–1238. doi: 10.1111/j.1471-4159.1981.tb04673.x. [DOI] [PubMed] [Google Scholar]
- 33.Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47(11):1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- 34.Marchi N, Fan Q, Ghosh C, et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiology of Disease. 2009;33(2):171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchi N, Oby E, Batra A, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48(10):1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman A, Kaufer D, Heinemann U. Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Research. 2009;85(2-3):142–149. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivens S, Kaufer D, Flores LP, et al. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130(2):535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 38.Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nature Reviews Neuroscience. 2006;7(3):194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 39.Tomkins O, Friedman O, Ivens S, et al. Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiology of Disease. 2007;25(2):367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Cacheaux LP, Ivens S, David Y, et al. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. Journal of Neuroscience. 2009;29(28):8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.David Y, Cacheaux LP, Ivens S, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? Journal of Neuroscience. 2009;29(34):10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchi N, Angelov L, Masaryk T, et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48(4):732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marchi N, Teng Q, Ghosh C, et al. Blood-brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Research. 2010;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorter JA, Gonçalves Pereira PM, van Vliet EA, Aronica E, Lopes da Silva FH, Lucassen PJ. Neuronal cell death in a rat model for mesial temporal lobe epilepsy is induced by the initial status epilepticus and not by later repeated spontaneous seizures. Epilepsia. 2003;44(5):647–658. doi: 10.1046/j.1528-1157.2003.53902.x. [DOI] [PubMed] [Google Scholar]
- 45.Fujikawa DG. Prolonged seizures and cellular injury: understanding the connection. Epilepsy & Behavior. 2005;7:S3–S11. doi: 10.1016/j.yebeh.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Gekle M, Knaus P, Nielsen R, et al. Transforming growth factor-β reduces megalin- and cubilin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. The Journal of Physiology. 2003;552(2):471–481. doi: 10.1113/jphysiol.2003.048074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siddiqui SS, Siddiqui ZK, Malik AB. Albumin endocytosis in endothelial cells induces TGF-β receptor II signaling. American Journal of Physiology. 2004;286(5):L1016–L1026. doi: 10.1152/ajplung.00356.2003. [DOI] [PubMed] [Google Scholar]
- 48.Luo J, Lin AH, Masliah E, Wyss-Coray T. Bioluminescence imaging of Smad signaling in living mice shows correlation with excitotoxic neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18326–18331. doi: 10.1073/pnas.0605077103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ata KA, Lennmyr F, Funa K, Olsson Y, Terént A. Expression of transforming growth factor-β1, 2, 3 isoforms and type I and II receptors in acute focal cerebral ischemia: an immunohistochemical study in rat after transient and permanent occlusion of middle cerebral artery. Acta Neuropathologica. 1999;97(5):447–455. doi: 10.1007/s004010051013. [DOI] [PubMed] [Google Scholar]
- 50.Matz PG, Lewén A, Chan PH. Neuronal, but not microglial, accumulation of extravasated serum proteins after intracerebral hemolysate exposure is accompanied by cytochrome c release and DNA fragmentation. Journal of Cerebral Blood Flow and Metabolism. 2001;21(8):921–928. doi: 10.1097/00004647-200108000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Hooper C, Pinteaux-Jones F, Fry VAH, et al. Differential effects of albumin on microglia and macrophages; implications for neurodegeneration following blood-brain barrier damage. Journal of Neurochemistry. 2009;109(3):694–705. doi: 10.1111/j.1471-4159.2009.05953.x. [DOI] [PubMed] [Google Scholar]
- 52.Maggio N, Shavit E, Chapman J, Segal M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. Journal of Neuroscience. 2008;28(3):732–736. doi: 10.1523/JNEUROSCI.3665-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seiffert E, Dreier JP, Ivens S, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. Journal of Neuroscience. 2004;24(36):7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pitsch J, Schoch S, Gueler N, Flor PJ, van der Putten H, Becker AJ. Functional role of mGluR1 and mGluR4 in pilocarpine-induced temporal lobe epilepsy. Neurobiology of Disease. 2007;26(3):623–633. doi: 10.1016/j.nbd.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 55.Nadal A, Sul JY, Valdeolmillos M, McNaughton PA. Albumin elicits calcium signals from astrocytes in brain slices from neonatal rat cortex. The Journal of Physiology. 1998;509(3):711–716. doi: 10.1111/j.1469-7793.1998.711bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. In: Weber JT, Mass AIR, editors. Neurotrauma: New Insights into Pathology and Treatment. Vol. 161. New York, NY, USA: Elsevier; 2007. pp. 61–79. (Progress in Brain Research). [DOI] [PubMed] [Google Scholar]
- 57.Heinemann U, Gabriel S, Jauch R, et al. Alterations of glial cell function in temporal lobe epilepsy. Epilepsia. 2000;41(6):S185–S189. doi: 10.1111/j.1528-1157.2000.tb01579.x. [DOI] [PubMed] [Google Scholar]
- 58.Jabs R, Seifert G, Steinhäuser C. Astrocytic function and its alteration in the epileptic brain. Epilepsia. 2008;49(2):3–12. doi: 10.1111/j.1528-1167.2008.01488.x. [DOI] [PubMed] [Google Scholar]
- 59.Wetherington J, Serrano G, Dingledine R. Astrocytes in the Epileptic Brain. Neuron. 2008;58(2):168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eroglu Ç, Allen NJ, Susman MW, et al. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filosa A, Paixo S, Honsek SD, et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nature Neuroscience. 2009;12(10):1285–1292. doi: 10.1038/nn.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010;463(7278):232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K+ homeostasis and altered electrophysiological properties of post-traumatic hippocampal gila. Journal of Neuroscience. 1999;19(18):8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53(6):631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- 65.Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. Increased seizure threshold in mice lacking aquaporin-4 water channels. NeuroReport. 2004;15(2):259–262. doi: 10.1097/00001756-200402090-00009. [DOI] [PubMed] [Google Scholar]
- 66.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of K4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. Journal of Neuroscience. 2007;27(42):11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabriel S, Eilers A, Kivi A, et al. Effects of barium on stimulus induced changes in extracellular potassium concentration in area CA1 of hippocampal slices from normal and pilocarpine-treated epileptic rats. Neuroscience Letters. 1998;242(1):9–12. doi: 10.1016/s0304-3940(98)00012-3. [DOI] [PubMed] [Google Scholar]
- 68.Schröder W, Hinterkeuser S, Seifert G, et al. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41(6):S181–S184. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- 69.Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. Journal of Neuroscience. 2006;26(20):5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bezzi P, Domercq M, Brambilla L, et al. CXCR4-activated astrocyte glutamate release via TNFa: amplification by microglia triggers neurotoxicity. Nature Neuroscience. 2001;4(7):702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 71.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nature Medicine. 2005;11(9):973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eid T, Thomas MJ, Spencer DD, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. The Lancet. 2004;363(9402):28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- 73.Eid T, Ghosh A, Wang Y, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008;131(8):2061–2070. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vezzani A, Baram TZ. New roles for interleukin-1 beta in the mechanisms of epilepsy. Epilepsy Currents. 2007;7:45–50. doi: 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Argaw AT, Zhang Y, Snyder BJ, et al. IL-1β regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. Journal of Immunology. 2006;177(8):5574–5584. doi: 10.4049/jimmunol.177.8.5574. [DOI] [PubMed] [Google Scholar]
- 76.Vezzani A, Moneta D, Conti M, et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11534–11539. doi: 10.1073/pnas.190206797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vezzani A, Conti M, De Luigi A, et al. Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. Journal of Neuroscience. 1999;19(12):5054–5065. doi: 10.1523/JNEUROSCI.19-12-05054.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature Reviews Neuroscience. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 79.Yu B, Shinnick-Gallagher P. Interleukin-1β inhibits synaptic transmission and induces membrane hyperpolarization in amygdala neurons. Journal of Pharmacology and Experimental Therapeutics. 1994;271(2):590–600. [PubMed] [Google Scholar]
- 80.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. Journal of Neuroscience. 2003;23(25):8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. In: Bagetta G, Corasaniti MT, Lipton SA, editors. International Review of Neurobiology Neuroinflammation in Neuronal Death and Repair. Vol. 82. New York, NY, USA: Academic Press; 2007. pp. 247–263. [DOI] [PubMed] [Google Scholar]
- 82.Wang S, Cheng Q, Malik S, Yang J. Interleukin-1β inhibits γ-aminobutyric acid type A (GABAA) receptor current in cultured hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics. 2000;292(2):497–504. [PubMed] [Google Scholar]
- 83.Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Annals of Neurology. 2006;59(1):81–91. doi: 10.1002/ana.20699. [DOI] [PubMed] [Google Scholar]
- 84.Ben-Hur T, Ben-Menachem O, Furer V, Einstein O, Mizrachi-Kol R, Grigoriadis N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Molecular and Cellular Neuroscience. 2003;24(3):623–631. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- 85.Koo JW, Duman RS. IL-1β is an essential mediator of the antineurogenic and anhedonic effects of stress. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(2):751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor β increases neurogenesis in the adult dentate gyrus. European Journal of Neuroscience. 2006;23(1):83–93. doi: 10.1111/j.1460-9568.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- 87.Mathieu P, Piantanida AP, Pitossi F. Chronic expression of transforming growth factor-beta enhances adult neurogenesis. NeuroImmunoModulation. 2010;17(3):200–201. doi: 10.1159/000258723. [DOI] [PubMed] [Google Scholar]
- 88.Tomkins O, Shelef I, Kaizerman I, et al. Blood-brain barrier disruption in post-traumatic epilepsy. Journal of Neurology, Neurosurgery & Psychiatry. 2008;79(7):774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- 89.Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Löscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007;52(2):333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 90.Marchi N, Gonzalez-Martinez J, Nguyen MT, Granata T, Janigro D. Transporters in drug-refractory epilepsy: clinical significance. Clinical Pharmacology and Therapeutics. 2010;87(1):13–15. doi: 10.1038/clpt.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nature Reviews Neuroscience. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]