

Abstract
The human leukocyte antigen (HLA) genes on chromosome 6 are instrumental in many innate and adaptive immune responses. The HLA genes/haplotypes can also be involved in immune dysfunction and autoimmune diseases. It is now becoming apparent that many of the non-antigen-presenting HLA genes make significant contributions to autoimmune diseases. Interestingly, it has been reported that autism subjects often have associations with HLA genes/haplotypes, suggesting an underlying dysregulation of the immune system mediated by HLA genes. Genetic studies have only succeeded in identifying autism-causing genes in a small number of subjects suggesting that the genome has not been adequately interrogated. Close examination of the HLA region in autism has been relatively ignored, largely due to extraordinary genetic complexity. It is our proposition that genetic polymorphisms in the HLA region, especially in the non-antigen-presenting regions, may be important in the etiology of autism in certain subjects.
1. Autism
Leo Kanner first described autism in 1943 [1] after finding 11 children with common symptoms of obsessiveness, stereotypy, and echolalia at Johns Hopkins University. Autism remained an esoteric disorder for several decades until physicians and parents connected these symptoms with an increasing number of patients. It is important to note that the diagnostic criteria have been modified over the years to include a broader category of symptoms, thus increasing the number of children diagnosed with the disorder, now referred to as Autism Spectrum Disorder (ASD) [2]. Currently, the Centers for Disease Control and Prevention (CDC) states that the incidence of ASD is 1 out of 110 children in the United States [3]. The severity of ASD varies greatly with the most severe forms, much like Kanner autism, displaying language regression, seizures, and lower IQ. Altevogt et al. [4] have suggested that autism, or more properly ASD, is not a single disorder, but a collection of similar disorders each with different characteristics and perhaps etiologies.
Even after several decades of research, there is much debate around the world on the etiology of ASD. It is clear that ASD results from abnormal brain development in either the prenatal period or infancy stage of life. Exposure to mercury, maternal viral infections, autoimmune disorders, and the inheritance of certain gene combinations have been implicated in the etiology. Unfortunately, none of these areas have given clear answers as to the etiology. Fortunately, psychologists have made significant strides in treating children and it appears the earlier behavioral treatment starts, the better the outcome. Nevertheless, medical researchers continue to search for the cause(s) of ASD. This paper discusses a possible role for the immune system, and in particular immune function genes in the human leukocyte antigen region (HLA), as a research area that should be more closely investigated.
2. Infections
One of the first areas of interest in the 1960s and 1970s was the search for an infectious agent that might be involved in the etiology of ASD [5]. During this time, there were many case reports in the literature that suggested an association between congenital rubella infection and resultant autistic behaviors. However, after decades of research no definite role for infectious agents in autism etiology has been confirmed. On the other hand, these endeavors have led to observations that perhaps the immune system was involved in autism, and evidence continues to mount that immune abnormalities are indeed associated with ASD.
3. Familial Studies
Studies indicating familial clustering and the increases of ASD in twins have been interpreted by many as an indication of genetic predisposition. Twin studies show the concordance rates of monozygotic twins at 36–96%, whereas dizygotic twins are 0–24% concordant, resulting in an estimated heritability of autism at >90% [6–8]. Additionally, family studies have shown autism to have familial aggregation with 3–8% of subsequently born siblings either being autistic or showing some form of pervasive developmental disorder (PDD) [9, 10]. This is a 3- to 8-fold increase in risk for siblings over the general population. A more recent study gave an estimate of 18.7% sibling recurrence risk for ASD, a 20-fold increase over the general population [11]. It is important to note, however, that family data should be looked at with great caution, as individuals living in the same household will have similar exposures to microorganisms and environmental chemicals. Taking this into account, a recent paper based on data from twin pairs estimates the genetic heritability for ASD to be 14–67% [12]. However, this idea is somewhat controversial as many in the research community continue to feel that the results from family studies are indicative of a strong genetic etiology [13, 14].
4. Genetics
Many of the early genetic studies involved the examination of microsatellites throughout the human genome in an attempt to find genomic regions that would associate with autism. Overall, this approach was not very fruitful and researchers quickly started to examine single-nucleotide polymorphisms (SNPs) as techniques advanced [15]. With this approach many researchers proposed that multiple candidate genes were associated with ASD. Unfortunately, studies proposing candidate genes were often contradictory and proved to be unreliable [15]. One of the most interesting genetic findings in ASD is the association of autism with the MET receptor tyrosine kinase gene located on chromosome 7q31 as MET signaling participates in gastrointestinal repair, immune function, neocortical, and cerebellar growth [16]. It is important to mention that the autism MET associations have been replicated by other research groups [17]. This autism association showed a relative risk of 2.27 which is much lower than the relative risk for HLA gene/allele associations discussed below.
It is very reasonable to believe that deletions or duplications of genetic regions, which can cause lower or higher levels of gene expression, could produce pathological phenotypes. Consequently, newer approaches examining copy number variation (CNV) and microarray analyses of 500,000 SNPs or more have been in vogue for several years in the study of ASD. These newer approaches have identified CNV mutation differences in genes involved in neuronal cell adhesion and ubiquitin degradation as being associated with ASD [18]; however, these results have yet to be replicated by other researchers.
The neurexin-1 gene has been associated with a variety of developmental disorders including ASD [19]. The neurexin-1 gene (2p16.3) is one of the largest genes in the human genome with 24 exons in 1.1 Mb. With two independent promotors there can be over 1,000 neurexin isoforms generated from the 24 exons in different cells or tissue. Another gene of interest is the contactin-associated protein-like 2 (CNTNAP2) gene that was shown to be associated in Old-Order Amish subjects with intractable epilepsy and ASD [20]. Three other groups have now confirmed the involvement of CNTNAP2 in ASD [21–23].
Both the neurexin-1 and the CNTNAP2 genes are involved in synaptic function. Although these approaches have shown a strong association of certain genes with ASD, only a small percentage of subjects with ASD have these mutations. For example, the neurexin-1gene is found in only about 0.5% of autism cases and 0.2% of controls. The 90% missing inheritance may be largely due to marked genetic heterogeneity, suggesting that different ASD phenotypes should be examined separately [24–26]. Recent genetic research has also associated numerous immune function genes with autism [27–30]. A large study that examined SNP data from several genomewide scans on 3,130 subjects with schizophrenia found that the 5 most significant SNP markers are found across the HLA region [31]. It appears that some of missing inheritance, at least in schizophrenia, was uncovered in the HLA region and we suggest a similar finding will be confirmed/uncovered in autism.
5. Immune Abnormalities in Autism
It is no surprise to see immune gene associations in ASD, as numerous researchers have reported immune abnormalities in autism for over 20 years. It has become increasingly obvious that inflammatory processes are associated with autism. Blood levels of the inflammatory cytokines IL-6, INF-γ, and TNF-α were shown to be elevated in autistic individuals compared to controls [32, 33]. Later, seminal work by Vargas et al. [34] utilized direct morphological analysis and immunohistologic techniques to show an active neuroinflammatory process in the cerebral cortex, white matter, and in particular the cerebellum of ASD patients that was dependent on activation of microglia and astroglia. Cytokine profiling demonstrated that neuroinflammation was accompanied by upregulation of the macrophage chemoattractant protein (MCP-1) and TGF-beta in brain tissue, and that MCP-1 was also upregulated in cerebral spinal fluid [34]. More recent work has directly demonstrated that aside from blood, IL-6, TNF-α, and INF-γ are elevated in ASD brains, along with the other inflammatory cytokines GM-CSF and IL-8 [35]. Most recently, it has been reported that the important inflammatory-associated transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is upregulated in both blood [36] and brain tissue [37] of autistic individuals. Other immune abnormalities such as autoantibodies started to be reported in 1993 [38] (Table 1).
Table 1.
A list of proteins against which autoantibodies have been found in the serum of subjects with autism.
| Protein | Reference |
|---|---|
| Transglutaminase 2 | [39] |
| 45 and 62 kDa proteins in cerebellum | [40, 41] |
| Voltage dependent anion channel (VDAC) | [42] |
| Hexokinase-1 | [42] |
| Mitochondrial protein | [43] |
| Antimitochondrial DNA auto-antibodies | [43] |
| Nuclear proteins | [44] |
| 52 kDa protein in hypothalamus and thalamus | [45] |
| 43–48 kDa protein in the hypothalamus | [45] |
| Folate receptor | [46] |
| Brain-derived neuro trophic factor | [47] |
| HSP90 | [48] |
| Myelin basic protein (MBP) | [38, 49] |
| Myelin-associated glycoprotein (MAG) | [50] |
| Myelin oligodendrocyte glycoprotein (MOG) | [50] |
| Neuron-axon filament protein | [51] |
| Glial fibrillary acidic protein | [51] |
Autoantibodies to myelin basic protein have been noted by at least a couple of researchers [38, 52]. An increase in autoantibody reactivity has been reported against other brain proteins in ASD including nerve growth factor [53], brain endothelium [54], cerebellar proteins [40], and serotonin 5-HT receptors [55] and transglutaminase-2, a protein important in synaptic stabilization [39]. Croonenberghs et al. [56] noted a significant increase in gamma globulin especially of the IgG2 and IgG4 subclasses in children with autism over a control population. Autoantibodies to several uncharacterized brain-specific proteins have been reported in the plasma of autistic individuals. In particular, western blot analysis has shown the presence of IgG autoantibodies targeting a protein of approximately 52 kDA located in the hypothalamus and thalamus of adult brain [45]. Other autoantibodies targeting three brain proteins of 43–48 kDA located in the hypothalamus have also been observed in the serum of autistic individuals [45]. Autoantibodies targeting cerebellar proteins of 45 and 62 kDA have been associated with ASD [40]. These autoantibodies may be specific to cerebellar Golgi cells, which are GABAergic interneurons [41]. Autoantibodies reactive to human brain proteins in the 36–39 and 61 kDA range have also been found in the sera of mothers of autistic children. [57, 58].
It is important to note that while autoantibodies associated with ASD may be biomarkers, they may not necessarily be pathologic in and of themselves. Central tolerance refers to the process whereby immature lymphocytes are negatively selected based on the ability of their antigen receptors (the B cell receptor or BCR for B cells and the T-cell receptor or TCR for T cells) to recognize self-antigens [59–61]. Until fairly recently, it was believed that most immature lymphocytes recognizing self-antigens (autoimmune repertoire) were normally neutralized by virtue of central tolerance before they could mature, and that peripheral tolerance would insure the removal of any self-reactive lymphocyte escaping central tolerance. Peripheral tolerance refers to the process whereby mature self-reactive lymphocytes which have escaped central tolerance are eliminated, largely by CD95-mediated apoptosis [62]. Thus responses to foreign antigens were viewed as normal, while anti-self responses were considered necessarily pathologic. However, with the realization that many self-reactive lymphocytes survive central and peripheral tolerance, this view has had to be modified [49]. Limited immune responses to self-antigens (autoimmunity) are now understood to be normal and not necessarily pathologic [63].
Upon stimulation of the system with a pathogen, cognate lymphocyte clones representative of the antiforeign repertoire normally expand and mature, providing a protective immune response. On the other hand, it is the expansion and maturation of those clones representative of the autoimmune repertoire that leads to autoimmune disease. In other words, the developing immune system can be characterized as balancing production between antiforeign (protective) and antiself (autoimmune) repertoires. While a beneficial function of naturally occurring, low level autoimmune antibodies, also referred to as natural antibodies (NAs), remains a matter of debate, it appears as if the repertoire of NA is reflective of the susceptibility to develop specific autoimmune diseases [64, 65]. Because central tolerance of T cells depends to a large extent upon the strength of the TCR interaction with an autoantigen or an HLA class I or II complex [59], the NA repertoire will to a large extent depend upon the HLA haplotype, with some haplotypes favoring autoantibodies targeting one antigen over another. For instance, in the case of ASD we have found an association with low level antibody responses to tissue transglutaminase, and that this response appears linked to the HLA-DR3/DQ2 and DR7/DQ2 haplotypes [39]. It is likely that the other autoantibodies noted above as being associated with ASD may be linked to different haplotypes.
Aside from autoantibodies and altered cytokine levels there appear to be other immune abnormalities associated with ASD. It was noted that there were decreased numbers of T-lymphocytes and an altered ratio of suppressor T-lymphocytes to helper T-lymphocytes [66, 67] and altered T-lymphocytes responses in children with autism [68]. Warren et al. [69] reported that subjects with autism had reduced NK cell killing in the standard K562 target cell cytotoxicity assay. This observation of decreased NK cell killing has been repeated by at least a couple of other research teams [28, 70]. One research group [71] observed that monocyte counts and neopterin levels were increased in autistic children compared to gender and age-matched healthy controls suggesting that the immune system was overactivated in the ASD group. Another elegant set of experiments involved the stimulation of cultured monocytes with several toll-like-receptor (TLR) ligands. The monocytes from subjects with ASD had significant increases or decreases in proinflammatory cytokines depending on the particular TLR ligand added to the cell culture media [72].
One large-scale study found that the frequency of autoimmune disorders in the families with autistic children was found to be higher than in control subjects, especially mothers of autistic children [73]. Another group demonstrated that autoimmune diseases were increased significantly in families with ASD compared with those of healthy control subjects [74], suggesting a link between the disorders. Croen et al. [75] showed that maternal psoriasis diagnosed around the time of pregnancy is significantly associated with a subsequent diagnosis of autism in the child. Additionally, they showed a 2-fold increase in risk for a child having ASD if the mother was diagnosed and with asthma or allergies during pregnancy. An association between a family history of type 1 diabetes mellitus (T1DM) and infantile autism as well as a significant association between maternal histories of either rheumatoid arthritis (RA) or celiac disease and ASDs was noted by Atladóttir et al. [76].
6. Autism HLA Genetics
HLA is the name for the major histocompatibility complex (MHC) in humans and HLA and MHC are often used interchangeably in the literature. The HLA region on chromosome 6p21 (about 4 × 106 bp) is of major interest in basic research as well as medicine as genes/proteins in this region are involved in many biological processes such as histocompatibility, inflammation, ligands for immune cell receptors, and the complement cascade. The HLA region has 20 typical HLA genes and 112 nontypical HLA genes (Table 2) that are inherited together as frozen blocks of DNA called ancestral or extended haplotypes. Complete DNA sequences have been published for 8 of the more common ancestral haplotypes in an effort to expedite basic and disease research [77]. It should be mentioned that smaller haplotypes can also be constructed for genes that are linked. HLA genes also play a role in reproduction, pregnancy maintenance, mate selection, and even kin recognition [78, 79] and have been associated with over 100 diseases/disorders including autism. The proteins encoded by HLA genes are ligands, receptors, cytokines, signaling factors, heat shock proteins, transcription regulators, and so forth. Current research is increasingly demonstrating a role for HLA proteins in neural cell interactions, synaptic function, cerebral hemispheric specialization, central nervous system (CNS) development [80–84], and even neurological disorders [85]. The genes of the HLA region are shown in Figure 1. Shiina et al. [86, 87] have published two excellent reviews on the HLA super-locus. Not only is there extraordinary complexity in the HLA genes, there are extensive haplotype-related transcriptional differences [88]. It has been shown that genetic mechanisms outside of the non-antigen-binding HLA genes in the ancestral haplotype 8.1 (also referred to as COX) are associated with susceptibility to many autoimmune diseases [89]. It is our proposition that HLA genes/proteins should be more carefully examined due to increasing evidence of autoimmune type associations in autism.
Table 2.
Genes and alleles in the HLA region.
| HLA genes | Number | HLA alleles | Number |
|---|---|---|---|
| HLA class I genes | 6 | HLA class I alleles | 2215 |
| HLA class II genes | 12 | HLA class II alleles | 986 |
| HLA class I-like genes | 2 | HLA class I-like alleles | 94 |
| Non-HLA genes | 112 | ||
|
| |||
| Total genes | 132 | ||
|
| |||
| HLA class I genes | A/B/C/E/F/G | ||
| HLA class I-like genes | MICA/MICB | ||
| HLA class II genes | DRA/DRB/DQ/DP/DM/DO | ||
Figure 1.
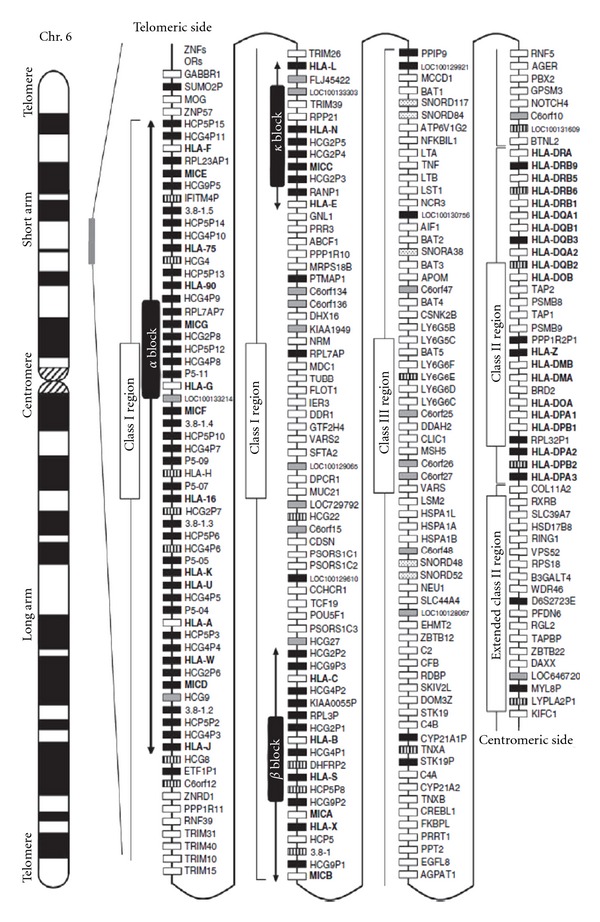
Gene map of the human leukocyte antigen (HLA) region. The major histocompatibility complex (MHC) gene map corresponds to the genomic coordinates of 29677984 (GABBR I) to 33485635 (KIFC1) in the human genome build 36.3 of the National Center for Biotechnology Information (NCBI) map viewer. The regions separated by arrows show the HLA subregions such as extended class I, classical class I, class III, classical class II, and extended class II regions from telomere (left and top side) to centromere (right and bottom side). White, gray, striped and black boxes show expressed genes, gene candidates, noncoding genes and pseudogenes, respectively. The location of the alpha, beta, and kappa blocks containing the cluster of duplicated HLA class I genes in the class I region are indicated. (Reprinted with permission from the Journal of Human Genetics [87].)
7. HLA Associations in ASD
It was suggested over 30 years ago by Stubbs and Magenis [90] that the HLA region might be important in autism. Warren et al. [91] first reported that the HLA ancestral haplotype 44.1 (B44-SC30-DR4) was associated with autism with a relative risk of 7.9. That result was confirmed in a separate case/control population [92]. Interestingly, the individual components of AH 44.1 (A2-B44-SC30-DR4) include a deleted C4B gene and DRβ1*0401, both of which have been shown independently to be significantly associated with ASD [93, 94]. Examination with different genetic markers than those used by Warren suggested that certain HLA haplotypes are associated with autism in Sardinian and Italian families [95, 96].
Warren et al. [97] reported that the shared epitope-binding pocket (DRβ1*0401, *0404, and *0101) in the third hypervariable region of DRβ1 has a strong association with autism. A relative risk of 19.8 for autism was reported for subjects with one of the two extended HLA haplotypes. Both of these haplotypes have many allelic similarities especially the DRβ1*0401.
AH 44.1 (HLA-A2, Cw5, B44, Bf*S, C2*C, C4A3, C4BQ0, DRβ1*0401, DQB1*0301).
AH 62.1 (HLA-A2, Cw3, B15, Bf*S, C2*C, C4A3, C4B3, DRβ1*0401, DQB1*0302).
The shared epitope has been associated with several autoimmune diseases such as rheumatoid arthritis, psoriatic arthritis, and systemic lupus erythematosus [98]. Torres et al. [93] confirmed the association of the HLA-DR4 allele and also found that the DR13 and DR14 alleles occurred less often in subjects with autism, suggesting a possible protective mechanism. Interestingly, the DR13 allele was inherited less frequently than expected from the mothers. Associations with autism and the DR4 allele have since been confirmed in three additional research groups. Lee et al. [99] demonstrated that boys with autism and their mothers had a significantly higher frequency of DR4 than normal control subjects (odds ratios 4.20 and 5.54, resp.), suggesting that a maternal-fetal immune interaction could be involved in autism. Johnson et al. [100] reported significant transmission disequilibrium for HLA-DR4 (odds ratio 4.67) from maternal grandparents to mothers of children with autism which also suggests a maternal-fetal interaction for HLA-DR4. It has been recently shown in Han Chinese that the HLA-DRβ1 allele frequencies including DR4 are different in subjects with autism versus control subjects [101].
It was reported 20 years ago that subjects with autism had a significant increase in the C4B null allele (C4B gene deletion) compared to control subjects [91]. After this observation, it was also noted that subjects with autism had a significant deficiency in the plasma C4B protein [102]. These initial findings of an increase in the deletion of the C4B gene was supported by examining a new population of subjects with autism [94]. Mostafa and Shehab [103] have recently reported a significant increase in the deletion of the C4B gene in the Egyptian population. They also reported an increased risk when there was a family history of autoimmune diseases in the autism population. Descriptions of several non-antigen-binding HLA genes that are associated with autoimmune diseases are discussed below and listed in Table 3.
Table 3.
Non-classical HLA genes associated with autoimmune diseases.
| Gene | HGNC gene number | Autoimmune disease | References | |
|---|---|---|---|---|
| Extended class I | OR2H2 | 8253 | SLE | [104] |
|
| ||||
| Class I | RNF39 | 18064 | Behçet's disease | [105] |
| TRIM39 | 10065 | Behçet's disease | [105] | |
| PSORS1 locus | 9573 | Systemic sclerosis, Psoriasis | [106–111] | |
| MICA | 7090 | T1DM, AD, SLE | [112–114] | |
| MICB | 7091 | SLE | [104] | |
|
| ||||
| Class III | BAT1–BAT5 | 13917–21 | Alzheimer's, AIDS | [115, 116] |
| NFKBIL1 | 7800 | Sjögren's syndrome, SLE, RA | [117, 118] | |
| TNF Block | 11892 | Alzheimer's, Psoriasis, Autoimmune hepatitis, Sarcoidosis | [115, 119–121] | |
| AIF1 | 352 | T1DM | [122] | |
| HSP70 genes | 5232–4 | MS | [123] | |
| Complement genes | 1248, 1324, 1323 | SLE, myasthenia gravis, T1DM | [124–126] | |
| SKIV2L | 10898 | SLE | [127] | |
| ATF6B (CREBL1) | 2349 | SLE | [104] | |
| NOTCH4 | 7884 | Systemic sclerosis | [128] | |
| C6orf10 | 13922 | SLE | [104] | |
| BTNL2 | 1142 | Ulcerative colitis, Sarcoidosis | [129, 130] | |
|
| ||||
| Class II | TAP2 | 44 | Psoriasis | [131] |
| PSMB8 | 9545 | Hypersensitivity pneumonitis | [132] | |
| Psoriasis | [133] | |||
| TAP1 | 43 | Vitiligo | [134] | |
| PSMB9 | 9546 | Psoriasis, Vitiligo | [133, 134] | |
| HLA-DM | 4934, 4935 | Psoriasis, Antiphospholipid auto-antibodies, RA, SLE, T1DM | [131, 135–138] | |
| HLA-DO | 4936, 4937 | common variable immunodeficiency | [139] | |
|
| ||||
| Class II Extended | HSD17B8 | 3554 | SLE | [104] |
| DAXX | 2681 | MS | [140] | |
Abbreviations: (HGNC) HUGO Gene Nomenclature Committee (http://www.genenames.org/); (SLE) systemic lupus erythematosus; (AIDS) acquired immune deficiency syndrome; (T1DM) type 1 diabetes mellitus; (AD) Addison's disease; (RA) rheumatoid arthritis; (MS) multiple sclerosis, PSORS1 psoriasis locus genes (CDSN, PSORS1C1, PSORS1C2, CCHCR1, POU5F1, PSORS1C3), TNF Block genes (LTA, TNF, LTB, LST1), HSP70 genes (HSPA1L, HSPA1A, HSPA1B), Complement genes (C2, C4B, C4A).
8. Non-Antigen-Binding HLA Genes in Class I
Although MICA/MICB genes are structurally similar to classical antigen-binding HLA class I genes, they encode proteins that interact with different T-cell receptors in response to stress as infection or neoplastic transformation. The two genes are located on the centromeric end of the class I region near HLA-B at the border of the class III region (Figure 1) [87]. MICA/MICB proteins are ligands for NKG2D receptors on NK cell and gamma/delta T-cell receptors. Adaptive immunity involves three lymphocyte populations (B cells, alpha/beta T cells, and gamma/delta T cells). Gamma/delta T cells represent a small population of T cells that possess a T-cell receptor that is distinct from the typical alpha/beta T cell. They are concentrated in the intestinal mucosa and appear to have a prominent role in recognizing small bacterial phosphoantigens and undefined antigens presented by MICA/MICB proteins. Gamma/delta T cells have potent cytotoxic activity and have been considered a link between innate and adaptive immunity. Polymorphisms in MICA/MICB genes have been associated with T1DM, AD, and SLE independent of DRβ1 alleles. There are several genes associated with psoriasis (PSORSI locus). It is unclear how the risk is spread among these genes as they are closely linked. Two genes RNF39 and TRIM39 in the class I region have been associated with Behçet's disease.
9. Non-Antigen-Binding HLA Genes in Class III
Tumor necrosis factor-alpha (TNF-α) is a proinflammatory, multifunctional cytokine that plays important roles in cell physiology. It is synthesized in numerous cells including macrophages, NK cells, T cells, mast cells, osteoblasts, granulocytes, smooth muscle cells, fibroblasts, and keratinocytes. In the CNS, TNF-α is made in microglia cells, astrocytes, and neurons [141]. In the normal state, TNF-α drives acute and chronic inflammatory responses that leads to the removal of injurious stimuli and the restoration of homeostasis. TNF-α is necessary for neural cell differentiation and neuron maturation and there is evidence that it is critical in the normal brain for proper synaptic function.
There is extensive research that implicates TNF-α as a key mediator in disease progression, inflammation, blood-brain-barrier deterioration, and even cell death [142]. Elevated levels are present in numerous neurological disorders including multiple sclerosis, Alzheimer's disease, Parkinson's disease, ischemia, traumatic brain injury, and as mentioned above, ASD [141]. It is unclear if TNF-α contributes to the disease state or the higher concentrations limit neuronal injury. There are three adjacent genes: lymphotoxin alpha and beta (LTA and LTB) and leukocyte-specific transcript-1 (LST1) in the TNF-α block. LTA and LTB are proinflammatory cytokines like TNF-α, and LST1 plays a role in inflammatory and infectious diseases. It has been suggested that vaccine-induced immunity changes with certain haplotypes in these genes [143]. Two other genes important in immune function are adjacent to the TNF-α block. Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor-like 1(NF-κBIL1) on the telomeric side of TNA-α is an inhibitor of NF-κB, an important pleiotropic immune system transcription factor that is upregulated in ASD [144, 145]. In addition, NF-κBIL1 has been associated with several autoimmune diseases (Table 3). The natural cytotoxicity trigger receptor three (NCR3) gene on the centromeric side encodes proteins that are ligands for activating receptors on NK cells that recognize tumor cells [146]. Typical HLA-B and -C proteins are also ligands for KIR receptors on NK cells.
There are numerous heat shock proteins (HSPs), also referred to as chaperones, that assist the folding of newly synthesized proteins as well as those that have been unfolded or denatured [147]. HSPs are also important in cell protective functions and in inhibiting the apoptosis cascade. They are named by their molecular weights (HSP100, HSP90, HSP70, HSP60, and smaller HSPs). Although the proteins appear in normal cellular functions, they are induced to higher levels in trauma, epilepsy, neurodegenerative diseases, and other injuries. An underlying feature among AD, Parkinson's disease, spinocerebellar ataxia, and other neurodegenerative diseases is the accumulation of misfolded proteins and HSPs are being studied because of their role in folding and refolding proteins [147]. There is intense interest in HSPs as they appear to protect neurons [148] and one must remember that postmitotic neurons are unable to dilute misfolded or aggregated proteins through division.
There are three HSP70 genes (HSPA1L, HSPA1A, and HSPA1B) in the class III HLA region that are adjacent but separate genes. HSP70 proteins have been demonstrated to stimulate IL-6 and TNF-α production, activate microglial cells, and stimulate phagocytosis [148]. HSP70 proteins are also important in autoimmunity by enhancing antigen presentation in both HLA class I and HLA class II systems [147, 149]. Peptides that are associated with HSP70 at the time of T-cell presentation have been shown to be more antigenic and therefore involved in autoimmunity [149, 150].
Another gene that is independently associated with autoimmune diseases outside of non-antigen-binding HLA alleles is allograft inflammatory factor-1 (AIF1). The SNP rs2269475 C > T in AIF1 has been associated with RA. The T allele was significantly higher in the RA patients and there was no significant linkage disequilibria between the AIF1 SNP and DRβ1 alleles. Anticyclic citrullinated peptide antibodies commonly used to monitor RA were significantly increased in carriers with the T allele [151] but not the C allele.
10. Non-Antigen-Binding HLA Genes in Class II
Although HLA-DM and -DQ proteins have structures like classical antigen-binding HLA proteins, they work in the cytoplasm and not at the cell surface antigen-presenting HLA proteins. DM stabilizes and edits the peptide repertoire presented by DQ proteins by catalyzing CLIP release. The associations of DQ2 with T1DM and celiac disease have been known for several decades; however, the biochemistry behind these associations has not been elucidated. It is now known that DQ2 is a poor substrate for DM and it has been proposed that antigen presentation in the thymus and periphery can be affected by impaired DQ-DM interactions so as to promote autoimmune disease [152]. HLA-DO is another nonclassical class II HLA protein that is involved in the loading of peptides to HLA-DR proteins by modulating the function of HLA-DM [153].
There are two genes in the class II region that encode proteins involved in the transport of antigen from the cytoplasm to the endoplasmic reticulum for binding to class I HLA proteins: transporter associated with antigen-processing 1 and 2 (TAP1 and TAP2). Several research groups have reported associations of TAP2 genes with SLE independent of classical HLA alleles [154]. This interaction is very interesting as it means that class II genes can affect class I antigen binding.
There are two other genes in the extended class II region that have been associated with autoimmune diseases (Table 3). Hydroxysteroid 17-beta dehydrogenase 8 (HSD17β8) is important in regulating the concentrations of active estrogens and androgens and high levels of estrogen are well known to be associated with SLE and other autoimmune diseases. The second gene, death-associated protein 6 (DAXX), is a very important protein that interacts with a variety of proteins in the nucleus and cytoplasm. Perhaps most importantly, it interacts with the death receptor Fas. Engstrom et al. [155] published a paper describing decreased expression of Fas on CD4 + T lymphocytes but higher serum levels of soluble Fas in ASD.
11. Summary
There is mounting evidence that the immune system plays a role in the pathogenesis of ASD in certain individuals. This evidence comes from several research areas including an increase in proinflammatory cytokines in blood and brain, autoantibodies to numerous antigens, and HLA associations.
Autism HLA associations have been observed across the entire HLA region. For example, in the class I region HLA-A2 has been associated with autism by at least two research groups [156, 157]. In the class II region several researchers have reported autism associations with the DRβ1*04 allele [93, 99, 100]. Strong associations also appear in the class III region where the C4B null allele has been associated with autism with relative risks of 4.3 [94] and 4.6 [97], and an odds ratio of 6.3 [103]. The HLA-associated risk is the highest for autism (19.8) when combining two ancestral haplotypes (44.1 and 62.1). Both of these haplotypes have HLA-A2 and DRβ1*0401 as well as other genetic similarities; however, these two alleles cannot account for all of the 19.8 risk. Compared to other genetic associations with autism, the HLA associations may be more important than realized, as they have the highest genetically associated risk, that we are aware of for autism. For example the MET gene, one of the most studies genetic regions in autism, has a relative risk of 2.27 [17].
It is our premise that some of the autism missing inheritance may be hidden in the HLA region, both in classical HLA alleles and nonclassical HLA genes, as seen in schizophrenia [31]. For example, the HLA class III region contains clusters of genes such as TNF-α, HSP70, C4A/C4B, and NF-κBIL1 that are seminal in cellular function and are also associated with numerous autoimmune diseases (Table 3).
Acknowledgment
This report was supported in part by NIH Grant (RO1ES016669) and the Utah Autism Foundation (Salt Lake City, Utah).
References
- 1.Kanner L. Autistic disturbances of affective contact. The Nervous Child. 1943;2(2):217–250. [Google Scholar]
- 2.Volkmar FR, State M, Klin A. Autism and autism spectrum disorders: diagnostic issues for the coming decade. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2009;50(1-2):108–115. doi: 10.1111/j.1469-7610.2008.02010.x. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorders-Autism and developmental disabilities monitoring network, United States, 2006. CDC—Morbidity and Mortality Weekly Report. 2009;58(10):1–20. [PubMed] [Google Scholar]
- 4.Altevogt BM, Hanson SL, Leshner AI. Autism and the environment: challenges and opportunities for research. Pediatrics. 2008;121(6):1225–1229. doi: 10.1542/peds.2007-3000. [DOI] [PubMed] [Google Scholar]
- 5.Chess S. Autism in children with congenital rubella. Journal of Autism and Childhood Schizophrenia. 1971;1(1):33–47. doi: 10.1007/BF01537741. [DOI] [PubMed] [Google Scholar]
- 6.Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. Journal of Child Psychology and Psychiatry and Allied Disciplines. 1977;18(4):297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 7.Steffenburg S, Gillberg C, Hellgren L, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. Journal of Child Psychology and Psychiatry and Allied Disciplines. 1989;30(3):405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 8.Bailey A, le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychological Medicine. 1995;25(1):63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 9.Bolton P, Macdonald H, Pickles A, et al. A case-control family history study of autism. Journal of Child Psychology and Psychiatry and Allied Disciplines. 1994;35(5):877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 10.Ritvo ER, Jorde LB, Mason-Brothers A, et al. The UCLA-University of Utah epidemiologic survey of autism: recurrence risk estimates and genetic counseling. American Journal of Psychiatry. 1989;146(8):1032–1036. doi: 10.1176/ajp.146.8.1032. [DOI] [PubMed] [Google Scholar]
- 11.Ozonoff S, Young GS, Carter A, et al. Recurrence risk for autism spectrum disorders: a baby siblings research consortium study. Pediatrics. 2011;128(3):e488–e495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry. 2011;68(11):1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Molecular Psychiatry. 2007;12(1):2–22. doi: 10.1038/sj.mp.4001896. [DOI] [PubMed] [Google Scholar]
- 14.Rutter M. Genetic studies of autism: from the 1970s into the millennium. Journal of Abnormal Child Psychology. 2000;28(1):3–14. doi: 10.1023/a:1005113900068. [DOI] [PubMed] [Google Scholar]
- 15.State MW. The genetics of child psychiatric disorders: focus on autism and tourette syndrome. Neuron. 2010;68(2):254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell DB, Sutcliffe JS, Ebert PJ, et al. A genetic variant that disrupts MET transcription is associated with autism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Judson MC, Eagleson KL, Levitt P. A new synaptic player leading to autism risk: met receptor tyrosine kinase. Journal of Neurodevelopmental Disorders. 2011;3(3):282–292. doi: 10.1007/s11689-011-9081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ching MS, Shen Y, Tan WH, et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. American Journal of Medical Genetics B. 2010;153(4):937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. The New England Journal of Medicine. 2006;354(13):1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 21.Alarcón M, Abrahams BS, Stone JL, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. American Journal of Human Genetics. 2008;82(1):150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arking DE, Cutler DJ, Brune CW, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. American Journal of Human Genetics. 2008;82(1):160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakkaloglu B, O’Roak BJ, Louvi A, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. American Journal of Human Genetics. 2008;82(1):165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein DB. Common genetic variation and human traits. The New England Journal of Medicine. 2009;360(17):1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 26.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 27.Gregg JP, Lit L, Baron CA, et al. Gene expression changes in children with autism. Genomics. 2008;91(1):22–29. doi: 10.1016/j.ygeno.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Enstrom AM, Lit L, Onore CE, et al. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain, Behavior, and Immunity. 2009;23(1):124–133. doi: 10.1016/j.bbi.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 30.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. Activation of the inflammatory response system in autism. Neuropsychobiology. 2002;45(1):1–6. doi: 10.1159/000048665. [DOI] [PubMed] [Google Scholar]
- 33.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, van de Water J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. Journal of Neuroimmunology. 2011;232(1-2):196–199. doi: 10.1016/j.jneuroim.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Chauhan A, Sheikh AM, et al. Elevated immune response in the brain of autistic patients. Journal of Neuroimmunology. 2009;207(1-2):111–116. doi: 10.1016/j.jneuroim.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naik US, Gangadharan C, Abbagani K, Nagalla B, Dasari N, Manna SK. A study of nuclear transcription factor-κ B in childhood autism. PLoS ONE. 2011;6(5):p. e19488. doi: 10.1371/journal.pone.0019488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young AM, Campbell E, Lynch S, Suckling J, Powis SJ. Aberrant NF-κB expression in autism spectrum condition: a mechanism for neuroinflammation. Frontiers in Psychiatry. 2011;2(27):1–8. doi: 10.3389/fpsyt.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh VK, Warren RP, Odell JD, Warren WL, Cole P. Antibodies to myelin basic protein in children with autistic behavior. Brain, Behavior, and Immunity. 1993;7(1):97–103. doi: 10.1006/brbi.1993.1010. [DOI] [PubMed] [Google Scholar]
- 39.Rosenspire A, Yoo W, Menard S, Torres AR. Autism spectrum disorders are associated with an elevated autoantibody response to tissue transglutaminase-2. Autism Research. 2011;4(4):242–249. doi: 10.1002/aur.194. [DOI] [PubMed] [Google Scholar]
- 40.Goines P, Haapanen L, Boyce R, et al. Autoantibodies to cerebellum in children with autism associate with behavior. Brain, Behavior, and Immunity. 2011;25(3):514–523. doi: 10.1016/j.bbi.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wills S, Rossi CC, Bennett J, et al. Further characterization of autoantibodies to GABAergic neurons in the central nervous system produced by a subset of children with autism. Molecular Autism. 2011;2:p. 5. doi: 10.1186/2040-2392-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez-Gronow M, Cuchacovich M, Francos R, et al. Antibodies against the voltage-dependent anion channel (VDAC) and its protective ligand hexokinase-I in children with autism. Journal of Neuroimmunology. 2010;227(1-2):153–161. doi: 10.1016/j.jneuroim.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Angelidou A, Alysandratos KD, et al. Mitochondrial DNA and anti-mitochondrial antibodies in serum of autistic children. Journal of Neuroinflammation. 2010;7:p. 80. doi: 10.1186/1742-2094-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mostafa GA, Kitchener N. Serum anti-nuclear antibodies as a marker of autoimmunity in Egyptian autistic children. Pediatric Neurology. 2009;40(2):107–112. doi: 10.1016/j.pediatrneurol.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Cabanlit M, Wills S, Goines P, Ashwood P, van de Water J. Brain-specific autoantibodies in the plasma of subjects with autistic spectrum disorder. Annals of the New York Academy of Sciences. 2007;1107:92–103. doi: 10.1196/annals.1381.010. [DOI] [PubMed] [Google Scholar]
- 46.Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics. 2007;38(6):276–281. doi: 10.1055/s-2008-1065354. [DOI] [PubMed] [Google Scholar]
- 47.Connolly AM, Chez M, Streif EM, et al. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biological Psychiatry. 2006;59(4):354–363. doi: 10.1016/j.biopsych.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Evers M, Cunningham-Rundles C, Hollander E. Heat shock protein 90 antibodies in autism. Molecular Psychiatry. 2002;7(supplement 2):S26–S28. doi: 10.1038/sj.mp.4001171. [DOI] [PubMed] [Google Scholar]
- 49.Brickman CM, Shoenfeld Y. The mosaic of autoimmunity. Scandinavian Journal of Clinical and Laboratory Investigation. 2001;61(235):3–15. [PubMed] [Google Scholar]
- 50.Vojdani A, Campbell AW, Anyanwu E, Kashanian A, Bock K, Vojdani E. Antibodies to neuron-specific antigens in children with autism: possible cross-reaction with encephalitogenic proteins from milk, Chlamydia pneumoniae and Streptococcus group A. Journal of Neuroimmunology. 2002;129(1-2):168–177. doi: 10.1016/s0165-5728(02)00180-7. [DOI] [PubMed] [Google Scholar]
- 51.Singh VK, Warren R, Averett R, Ghaziuddin M. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatric Neurology. 1997;17(1):88–90. doi: 10.1016/s0887-8994(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 52.Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. Journal of Neuroimmunology. 2001;120(1-2):170–179. doi: 10.1016/s0165-5728(01)00421-0. [DOI] [PubMed] [Google Scholar]
- 53.Bashina VM, Kozlova IA, Kliushnik TP. An elevation in the level of autoantibodies to nerve-growth factor in the blood serum of schizophrenic children. Zhurnal Nevrologii i Psikhiatrii Imeni S.S. Korsakova. 1997;97(1):47–51. [PubMed] [Google Scholar]
- 54.Connolly AM, Chez MG, Pestronk A, Arnold ST, Mehta S, Deul RK. Serum autoantibodies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. The Journal of Pediatrics. 1999;134(5):607–613. doi: 10.1016/s0022-3476(99)70248-9. [DOI] [PubMed] [Google Scholar]
- 55.Todd RD, Ciaranello RD. Demonstration of inter- and intraspecies differences in serotonin binding sites by antibodies from an autistic child. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(2):612–616. doi: 10.1073/pnas.82.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Croonenberghs J, Wauters A, Devreese K, et al. Increased serum albumin, γ globulin, immunoglobulin IgG, and IgG2 and IgG4 in autism. Psychological Medicine. 2002;32(8):1457–1463. doi: 10.1017/s0033291702006037. [DOI] [PubMed] [Google Scholar]
- 57.Singer HS, Morris CM, Gause CD, Gillin PK, Crawford S, Zimmerman AW. Antibodies against fetal brain in sera of mothers with autistic children. Journal of Neuroimmunology. 2008;194(1-2):165–172. doi: 10.1016/j.jneuroim.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Croen LA, Braunschweig D, Haapanen L, et al. Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biological Psychiatry. 2008;64(7):583–588. doi: 10.1016/j.biopsych.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nature Reviews Immunology. 2005;5(10):772–782. doi: 10.1038/nri1707. [DOI] [PubMed] [Google Scholar]
- 60.Sprent J. Central tolerance of T cells. International Reviews of Immunology. 1996;13(2):95–105. doi: 10.3109/08830189509061740. [DOI] [PubMed] [Google Scholar]
- 61.Basten A, Brink R, Peake P, et al. Self tolerance in the B-cell repertoire. Immunological Reviews. 1991;(122):5–19. doi: 10.1111/j.1600-065x.1991.tb00593.x. [DOI] [PubMed] [Google Scholar]
- 62.Baumann S, Krueger A, Kirchhoff S, Krammer PH. Regulation of T cell apoptosis during the immune response. Current Molecular Medicine. 2002;2(3):257–272. doi: 10.2174/1566524024605671. [DOI] [PubMed] [Google Scholar]
- 63.Leng Q, Bentwich Z. Beyond self and nonself: fuzzy recognition of the immune system. Scandinavian Journal of Immunology. 2002;56(3):224–232. doi: 10.1046/j.1365-3083.2002.01105.x. [DOI] [PubMed] [Google Scholar]
- 64.Meffre E, Salmon JE. Autoantibody selection and production in early human life. Journal of Clinical Investigation. 2007;117(3):598–601. doi: 10.1172/JCI31578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quintana FJ, Weiner HL. Understanding natural and pathological autoimmunity. Journal of Neuroimmunology. 2006;174(1-2):1–2. doi: 10.1016/j.jneuroim.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Warren RP, Margaretten NC, Pace NC, Foster A. Immune abnormalities in patients with autism. Journal of Autism and Developmental Disorders. 1986;16(2):189–197. doi: 10.1007/BF01531729. [DOI] [PubMed] [Google Scholar]
- 67.Denney DR, Frei BW, Gaffney GR. Lymphocyte subsets and interleukin-2 receptors in autistic children. Journal of Autism and Developmental Disorders. 1996;26(1):87–97. doi: 10.1007/BF02276236. [DOI] [PubMed] [Google Scholar]
- 68.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, van de Water J. Altered T cell responses in children with autism. Brain, Behavior, and Immunity. 2011;25(5):840–849. doi: 10.1016/j.bbi.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Warren RP, Foster A, Margaretten NC. Reduced natural killer cell activity in autism. Journal of the American Academy of Child and Adolescent Psychiatry. 1987;26(3):333–335. doi: 10.1097/00004583-198705000-00008. [DOI] [PubMed] [Google Scholar]
- 70.Vojdani A, Mumper E, Granpeesheh D, et al. Low natural killer cell cytotoxic activity in autism: the role of glutathione, IL-2 and IL-15. Journal of Neuroimmunology. 2008;205(1-2):148–154. doi: 10.1016/j.jneuroim.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 71.Sweeten TL, Posey DJ, McDougle CJ. High blood monocyte counts and neopterin levels in children with autistic disorder. American Journal of Psychiatry. 2003;160(9):1691–1693. doi: 10.1176/appi.ajp.160.9.1691. [DOI] [PubMed] [Google Scholar]
- 72.Enstrom AM, Onore CE, van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain, Behavior, and Immunity. 2010;24(1):64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Comi AM, Zimmerman AW, Frye VH, Law PA, Peeden JN. Familial clustering of autoimmune disorders and evaluation of medical risk factors in autism. Journal of Child Neurology. 1999;14(6):388–394. doi: 10.1177/088307389901400608. [DOI] [PubMed] [Google Scholar]
- 74.Sweeten TL, Bowyer SL, Posey DJ, Halberstadt GM, McDougle CJ. Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics. 2003;112(5):e420–e424. doi: 10.1542/peds.112.5.e420. [DOI] [PubMed] [Google Scholar]
- 75.Croen LA, Grether JK, Yoshida CK, Odouli R, van de Water JV. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case-control study. Archives of Pediatrics and Adolescent Medicine. 2005;159(2):151–157. doi: 10.1001/archpedi.159.2.151. [DOI] [PubMed] [Google Scholar]
- 76.Atladóttir HO, Pedersen MG, Thorsen P, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics. 2009;124(2):687–694. doi: 10.1542/peds.2008-2445. [DOI] [PubMed] [Google Scholar]
- 77.Horton R, Gibson R, Coggill P, et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC haplotype project. Immunogenetics. 2008;60(1):1–18. doi: 10.1007/s00251-007-0262-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knapp LA. The ABCs of MHC. Evolutionary Anthropology. 2005;14(1):28–37. [Google Scholar]
- 79.Ziegler A, Kentenich H, Uchanska-Ziegler B. Female choice and the MHC. Trends in Immunology. 2005;26(9):496–502. doi: 10.1016/j.it.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 80.Xiao BG, Link H. Immune regulation within the central nervous system. Journal of the Neurological Sciences. 1998;157(1):1–12. doi: 10.1016/s0022-510x(98)00049-5. [DOI] [PubMed] [Google Scholar]
- 81.Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290(5499):2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boulanger LM, Shatz CJ. Immune signalling in neural development, synaptic plasticity and disease. Nature Reviews Neuroscience. 2004;5(7):521–531. doi: 10.1038/nrn1428. [DOI] [PubMed] [Google Scholar]
- 83.Cullheim S, Thams S. The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Research Reviews. 2007;55(1):89–96. doi: 10.1016/j.brainresrev.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 84.Ohtsuka M, Inoko H, Kulski JK, Yoshimura S. Major histocompatibility complex (Mhc) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genomics. 2008;9:p. 178. doi: 10.1186/1471-2164-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bailey SL, Carpentier PA, McMahon EJ, Begolka WS, Miller SD. Innate and adaptive immune responses of the central nervous system. Critical Reviews in Immunology. 2006;26(2):149–188. doi: 10.1615/critrevimmunol.v26.i2.40. [DOI] [PubMed] [Google Scholar]
- 86.Shiina T, Inoko H, Kulski JK. An update of the HLA genomic region, locus information and disease associations. Tissue Antigens. 2004;64(6):631–649. doi: 10.1111/j.1399-0039.2004.00327.x. [DOI] [PubMed] [Google Scholar]
- 87.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. Journal of Human Genetics. 2009;54(1):15–39. doi: 10.1038/jhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 88.Vandiedonck C, Taylor MS, Lockstone HE, et al. Pervasive haplotypic variation in the spliceo-transcriptome of the human major histocompatibility complex. Genome Research. 2011;21(7):1042–1054. doi: 10.1101/gr.116681.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Candore G, Campagna IC, Cuppari I, di Carlo D, Mineo C, Caruso C. Genetic control of immune response in carriers of the 8.1 ancestral haplotype. Annals of the New York Academy of Sciences. 2007;1110:151–158. doi: 10.1196/annals.1423.017. [DOI] [PubMed] [Google Scholar]
- 90.Stubbs EG, Magenis RE. HLA and autism. Journal of Autism and Developmental Disorders. 1980;10(1):15–19. doi: 10.1007/BF02408429. [DOI] [PubMed] [Google Scholar]
- 91.Warren RP, Singh VK, Cole P, et al. Increased frequency of the null allele at the complement C4b locus in autism. Clinical and Experimental Immunology. 1991;83(3):438–440. doi: 10.1111/j.1365-2249.1991.tb05657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Daniels WW, Warren RP, Odell JD, et al. Increased frequency of the extended or ancestral haplotype B44-SC30-DR4 in autism. Neuropsychobiology. 1995;32(3):120–123. doi: 10.1159/000119223. [DOI] [PubMed] [Google Scholar]
- 93.Torres AR, Maciulis A, Stubbs EG, Cutler A, Odell D. The transmission disequilibrium test suggests that HLA-DR4 and DR13 are linked to autism spectrum disorder. Human Immunology. 2002;63(4):311–316. doi: 10.1016/s0198-8859(02)00374-9. [DOI] [PubMed] [Google Scholar]
- 94.Odell D, Maciulis A, Cutler A, et al. Confirmation of the association of the C4B null allelle in autism. Human Immunology. 2005;66(2):140–145. doi: 10.1016/j.humimm.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 95.Guerini FR, Bolognesi E, Manca S, et al. Family-based transmission analysis of HLA genetic markers in Sardinian children with autistic spectrum disorders. Human Immunology. 2009;70(3):184–190. doi: 10.1016/j.humimm.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 96.Guerini FR, Bolognesi E, Chiappedi M, et al. HLA polymorphisms in Italian children with autism spectrum disorders: results of a family based linkage study. Journal of Neuroimmunology. 2011;230(1-2):135–142. doi: 10.1016/j.jneuroim.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 97.Warren RP, Odell JD, Warren WL, et al. Strong association of the third hypervariable region of HLA-DR β1 with autism. Journal of Neuroimmunology. 1996;67(2):97–102. doi: 10.1016/0165-5728(96)00052-5. [DOI] [PubMed] [Google Scholar]
- 98.de Almeida DE, Ling S, Holoshitz J. New insights into the functional role of the rheumatoid arthritis shared epitope. FEBS Letters. 2011;585(23):3619–3626. doi: 10.1016/j.febslet.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee LC, Zachary AA, Leffell MS, et al. HLA-DR4 in families with autism. Pediatric Neurology. 2006;35(5):303–307. doi: 10.1016/j.pediatrneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 100.Johnson WG, Buyske S, Mars AE, et al. HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Archives of Pediatrics and Adolescent Medicine. 2009;163(6):542–546. doi: 10.1001/archpediatrics.2009.74. [DOI] [PubMed] [Google Scholar]
- 101.Chien YL, Wu YY, Chen CH, et al. Association of HLA-DRB1 alleles and neuropsychological function in autism. Psychiatric Genetics. 2012;22(1):46–49. doi: 10.1097/YPG.0b013e32834915ae. [DOI] [PubMed] [Google Scholar]
- 102.Warren RP, Burger RA, Odell D, Torres AR, Warren WL. Decreased plasma concentrations of the C4B complement protein in autism. Archives of Pediatrics and Adolescent Medicine. 1994;148(2):180–183. doi: 10.1001/archpedi.1994.02170020066011. [DOI] [PubMed] [Google Scholar]
- 103.Mostafa GA, Shehab AA. The link of C4B null allele to autism and to a family history of autoimmunity in Egyptian autistic children. Journal of Neuroimmunology. 2010;223(1-2):115–119. doi: 10.1016/j.jneuroim.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 104.Barcellos LF, May SL, Ramsay PP, et al. High-density SNP screening of the major histocompatibility complex in systemic lupus erythematosus demonstrates strong evidence for independent susceptibility regions. PLoS Genetics. 2009;5(10) doi: 10.1371/journal.pgen.1000696. Article ID e1000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kurata R, Nakaoka H, Tajima A, et al. TRIM39 and RNF39 are associated with Behçet’s disease independently of HLA-B∗51 and -A∗26. Biochemical and Biophysical Research Communications. 2010;401(4):533–537. doi: 10.1016/j.bbrc.2010.09.088. [DOI] [PubMed] [Google Scholar]
- 106.Allanore Y, Saad M, Dieudé P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genetics. 2011;7(7) doi: 10.1371/journal.pgen.1002091. Article ID e1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gonzalez S, Martinez-Borra J, del Río JS, et al. The OTF3 gene polymorphism confers susceptibility to psoriasis independent of the association of HLA-Cw∗0602. Journal of Investigative Dermatology. 2000;115(5):824–828. doi: 10.1046/j.1523-1747.2000.00133.x. [DOI] [PubMed] [Google Scholar]
- 108.Asumalahti K, Veal C, Laitinen T, et al. Coding haplotype analysis supports HCR as the putative susceptibility gene for psoriasis at the MHC PSORS1 locus. Human Molecular Genetics. 2002;11(5):589–597. doi: 10.1093/hmg/11.5.589. [DOI] [PubMed] [Google Scholar]
- 109.Holm SJ, Carlén LM, Mallbris L, Ståhle-Bäckdahl M, O’Brien KP. Polymorphisms in the SEEK1 and SPR1 genes on 6p21.3 associate with psoriasis in the Swedish population. Experimental Dermatology. 2003;12(4):435–444. doi: 10.1034/j.1600-0625.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 110.Holm SJ, Sánchez F, Carlén LM, Mallbris L, Ståhle M, O’Brien KP. HLA-Cw∗0602 associates more strongly to psoriasis in the Swedish population than variants of the novel 6p21.3 gene PSORS1C3. Acta Dermato-Venereologica. 2005;85(1):2–8. doi: 10.1080/00015550410023527. [DOI] [PubMed] [Google Scholar]
- 111.Orrù S, Giuressi E, Carcassi C, Casula M, Contu L. Mapping of the major psoriasis-susceptibility locus (PSORS1) in a 70-Kb interval around the corneodesmosin gene (CDSN) American Journal of Human Genetics. 2005;76(1):164–171. doi: 10.1086/426948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gambelunghe G, Brozzetti A, Ghaderi M, Candeloro P, Tortoioli C, Falorni A. MICA gene polymorphism in the pathogenesis of type 1 diabetes. Annals of the New York Academy of Sciences. 2007;1110:92–98. doi: 10.1196/annals.1423.011. [DOI] [PubMed] [Google Scholar]
- 113.Falorni A, Brozzetti A, Torre DL, Tortoioli C, Gambelunghe G. Association of genetic polymorphisms and autoimmune Addison’s disease. Expert Review of Clinical Immunology. 2008;4(4):441–456. doi: 10.1586/1744666X.4.4.441. [DOI] [PubMed] [Google Scholar]
- 114.Gambelunghe G, Gerli R, Bocci EB, et al. Contribution of MHC class I chain-related A (MICA) gene polymorphism to genetic susceptibility for systemic lupus erythematosus. Rheumatology. 2005;44(3):287–292. doi: 10.1093/rheumatology/keh459. [DOI] [PubMed] [Google Scholar]
- 115.Gnjec A, D’Costa KL, Laws SM, et al. Association of alleles carried at TNFA -850 and BAT1 -22 with Alzheimer’s disease. Journal of Neuroinflammation. 2008;5:p. 36. doi: 10.1186/1742-2094-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Limou S, le Clerc S, Coulonges C, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS genomewide association study 02) Journal of Infectious Diseases. 2009;199(3):419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 117.Castiblanco J, Anaya JM. The IκBL gene polymorphism influences risk of acquiring systemic lupus erythematosus and Sjögren’s syndrome. Human Immunology. 2008;69(1):45–51. doi: 10.1016/j.humimm.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 118.Tamiya G, Shinya M, Imanishi T, et al. Whole genome association study of rheumatoid arthritis using 27039 microsatellites. Human Molecular Genetics. 2005;14(16):2305–2321. doi: 10.1093/hmg/ddi234. [DOI] [PubMed] [Google Scholar]
- 119.Reich K, Hüffmeier U, König IR, et al. TNF polymorphisms in psoriasis: association of psoriatic arthritis with the promoter polymorphism TNF∗-857 independent of the PSORS1 risk allele. Arthritis and Rheumatism. 2007;56(6):2056–2064. doi: 10.1002/art.22590. [DOI] [PubMed] [Google Scholar]
- 120.Oliveira LC, Porta G, Marin MLC, Bittencourt PL, Kalil J, Goldberg AC. Autoimmune hepatitis, HLA and extended haplotypes. Autoimmunity Reviews. 2011;10(4):189–193. doi: 10.1016/j.autrev.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 121.Kieszko R, Krawczyk P, Chocholska S, Dmoszyńska A, Milanowski J. TNF-α and TNF-β gene polymorphisms in Polish patients with sarcoidosis. Connection with the susceptibility and prognosis. Sarcoidosis Vasculitis and Diffuse Lung Diseases. 2010;27(2):131–137. [PubMed] [Google Scholar]
- 122.Eike MC, Olsson M, Undlien DE, et al. Genetic variants of the HLA-A, HLA-B and AIF1 loci show independent associations with type 1 diabetes in Norwegian families. Genes and Immunity. 2009;10(2):141–150. doi: 10.1038/gene.2008.88. [DOI] [PubMed] [Google Scholar]
- 123.Cwiklinska H, Mycko MP, Szymanska B, Matysiak M, Selmaj KW. Aberrant stress-induced Hsp70 expression in immune cells in multiple sclerosis. Journal of Neuroscience Research. 2010;88(14):3102–3110. doi: 10.1002/jnr.22476. [DOI] [PubMed] [Google Scholar]
- 124.Pickering MC, Walport MJ. Links between complement abnormalities and systemic lupus erythematosus. Rheumatology. 2000;39(2):133–141. doi: 10.1093/rheumatology/39.2.133. [DOI] [PubMed] [Google Scholar]
- 125.Franciotta D, Cuccia M, Dondi E, Piccolo G, Cosi V. Polymorphic markers in MHC class II/III region: a study on Italian patients with myasthenia gravis. Journal of the Neurological Sciences. 2001;190(1-2):11–16. doi: 10.1016/s0022-510x(01)00573-1. [DOI] [PubMed] [Google Scholar]
- 126.Jenhani F, Bardi R, Gorgi Y, Ayed K, Jeddi M. C4 polymorphism in multiplex families with insulin dependent diabetes in the Tunisian population: standard C4 typing methods and RFLP analysis. Journal of Autoimmunity. 1992;5(2):149–160. doi: 10.1016/0896-8411(92)90196-w. [DOI] [PubMed] [Google Scholar]
- 127.Fernando MM, Stevens CR, Sabeti PC, et al. Identification of two independent risk factors for lupus within the MHC in United Kingdom families. PLoS Genetics. 2007;3(11):p. e192. doi: 10.1371/journal.pgen.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gorlova O, Martin JE, Rueda B, et al. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genetics. 2011;7(7) doi: 10.1371/journal.pgen.1002178. Article ID e1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pathan S, Gowdy RE, Cooney R, et al. Confirmation of the novel association at the BTNL2 locus with ulcerative colitis. Tissue Antigens. 2009;74(4):322–329. doi: 10.1111/j.1399-0039.2009.01314.x. [DOI] [PubMed] [Google Scholar]
- 130.Milman N, Svendsen CB, Nielsen FC, Hansen TVO. The BTNL2 A allele variant is frequent in Danish patients with sarcoidosis. Clinical Respiratory Journal. 2011;5(2):105–111. doi: 10.1111/j.1752-699X.2010.00206.x. [DOI] [PubMed] [Google Scholar]
- 131.Pyo CW, Hur SS, Kim YK, Kim TY, Kim TG. Association of TAP and HLA-DM genes with psoriasis in Koreans. Journal of Investigative Dermatology. 2003;120(4):616–622. doi: 10.1046/j.1523-1747.2003.12091.x. [DOI] [PubMed] [Google Scholar]
- 132.Camarena Á, Aquino-Galvez A, Falfán-Valencia R, et al. PSMB8 (LMP7) but not PSMB9 (LMP2) gene polymorphisms are associated to pigeon breeder’s hypersensitivity pneumonitis. Respiratory Medicine. 2010;104(6):889–894. doi: 10.1016/j.rmed.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 133.Krämer U, Illig T, Grune T, Krutmann J, Esser C. Strong associations of psoriasis with antigen processing LMP and transport genes TAP differ by gender and phenotype. Genes and Immunity. 2007;8(6):513–517. doi: 10.1038/sj.gene.6364404. [DOI] [PubMed] [Google Scholar]
- 134.Casp CB, She JX, McCormack WT. Genes of the LMP/TAP cluster are associated with the human autoimmune disease vitiligo. Genes and Immunity. 2003;4(7):492–499. doi: 10.1038/sj.gene.6364016. [DOI] [PubMed] [Google Scholar]
- 135.Sanchez ML, Katsumata K, Atsumi T, et al. Association of HLA-DM polymorphism with the production of antiphospholipid antibodies. Annals of the Rheumatic Diseases. 2004;63(12):1645–1648. doi: 10.1136/ard.2003.015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Morel J, Roch-Bras F, Molinari N, Sany J, Eliaou JF, Combe B. HLA-DMA∗0103 and HLA-DMB∗0104 alleles as novel prognostic factors in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2004;63(12):1581–1586. doi: 10.1136/ard.2003.012294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Morel J, Cda CS, Avinens O, Sany J, Combe B, Eliaou JF. Polymorphism of HLA-DMA and DMB alleles in patients with systemic lupus erythematosus. Journal of Rheumatology. 2003;30(7):1485–1490. [PubMed] [Google Scholar]
- 138.Sang YM, Yan C, Zhu C, Ni GC, Hu YM. Association of human leukocyte antigen non-classical genes with type 1 diabetes. Chinese Journal of Pediatrics. 2003;41(4):260–263. [PubMed] [Google Scholar]
- 139.Chen X, Jensen PE. MHC class II antigen presentation and immunological abnormalities due to deficiency of MHC class II and its associated genes. Experimental and Molecular Pathology. 2008;85(1):40–44. doi: 10.1016/j.yexmp.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Satoh JI, Nakanishi M, Koike F, et al. Microarray analysis identifies an aberrant expression of apoptosis and DNA damage-regulatory genes in multiple sclerosis. Neurobiology of Disease. 2005;18(3):537–550. doi: 10.1016/j.nbd.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 141.Montgomery SL, Bowers WJ. Tumor necrosis factor-α and the roles it plays in homeostatic and degenerative processes within the central nervous system. doi: 10.1007/s11481-011-9287-2. Journal of Neuroimmune Pharmacology. In press. [DOI] [PubMed] [Google Scholar]
- 142.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of Neuroinflammation. 2008;5, article 45 doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ovsyannikova IG, Vierkant RA, Pankratz VS, Jacobson RM, Poland GA. Extended LTA, TNF, LST1 and hla gene haplotypes and their association with rubella vaccine-induced immunity. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011806. Article ID e11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Naik US, Gangadharan C, Abbagani K, Nagalla B, Dasari N, Manna SK. A study of nuclear transcription factor-κ B in childhood autism. PLoS ONE. 2011;6(5) doi: 10.1371/journal.pone.0019488. Article ID e19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Young AM, Campbell E, Lynch S, Suckling J, Powis SJ. Aberrant NF-κB expression in autism spectrum condition: a mechanism for neuroinflammation. Front Psychiatry. 2011;2(27):1–8. doi: 10.3389/fpsyt.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. Journal of Biological Chemistry. 2003;278(46):46081–46086. doi: 10.1074/jbc.M308491200. [DOI] [PubMed] [Google Scholar]
- 147.Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochemistry Research International. 2011;2011:18 pages. doi: 10.1155/2011/618127. Article ID 618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kakimura JI, Kitamura Y, Takata K, et al. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. The FASEB Journal. 2002;16(6):601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- 149.Mycko MP, Cwiklinska H, Walczak A, Libert C, Raine CS, Selmaj KW. A heat shock protein gene (Hsp70.1) is critically involved in the generation of the immune response to myelin antigen. European Journal of Immunology. 2008;38(7):1999–2013. doi: 10.1002/eji.200737661. [DOI] [PubMed] [Google Scholar]
- 150.Millar DG, Garza KM, Odermatt B, et al. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo . Nature Medicine. 2003;9(12):1469–1476. doi: 10.1038/nm962. [DOI] [PubMed] [Google Scholar]
- 151.Pawlik A, Kurzawski M, Szczepanik T, et al. Association of allograft inflammatory factor-1 gene polymorphism with rheumatoid arthritis. Tissue Antigens. 2008;72(2):171–175. doi: 10.1111/j.1399-0039.2008.01086.x. [DOI] [PubMed] [Google Scholar]
- 152.Hou T, Macmillan H, Chen Z, et al. An insertion mutant in DQA1∗0501 restore susceptibility to HLA-DM: implications for disease associations. Journal of Immunology. 2011;187(5):2442–2452. doi: 10.4049/jimmunol.1100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Deshaies F, Diallo DA, Fortin JS, et al. Evidence for a human leucocyte antigen-DM-induced structural change in human leucocyte antigen-DOβ . Immunology. 2009;127(3):408–417. doi: 10.1111/j.1365-2567.2008.02984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ramos PS, Langefeld CD, Bera LA, Gaffney PM, Noble JA, Moser KL. Variation in the ATP-binding cassette transporter 2 gene is a separate risk factor for systemic lupus erythematosus within the MHC. Genes and Immunity. 2009;10(4):350–355. doi: 10.1038/gene.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Engstrom HA, Ohlson S, Stubbs EG, et al. Decreased expression of CD95 (FAS/APO-1) on CD4+ T-lymphocytes from participants with autism. Journal of Developmental and Physical Disabilities. 2003;15(2):155–163. [Google Scholar]
- 156.Ferrante P, Saresella M, Guerini FR, Marzorati M, Musetti MC, Cazzullo AG. Significant association of HLA A2-DR11 with CD4 naive decrease in autistic children. Biomedicine and Pharmacotherapy. 2003;57(8):372–374. doi: 10.1016/s0753-3322(03)00099-4. [DOI] [PubMed] [Google Scholar]
- 157.Torres AR, Sweeten TL, Cutler A, et al. The association and linkage of the HLA-A2 class I allele with autism. Human Immunology. 2006;67(4-5):346–351. doi: 10.1016/j.humimm.2006.01.001. [DOI] [PubMed] [Google Scholar]